

Since our initial report of metal-catalyzed enyne cycloisomerizations,1 this transformation has become an important reaction class, showing strong potential for simplifying strategies to complex molecules.2 Thusly, developing novel selectivities of this process is a critical objective toward enhancing its utility. Recently, our group has described the utility of ruthenium catalysis in intramolecular cycloisomerizations, providing an efficient, atom-economical method to access an array of 1,4-dienes.3 In an effort to extend this chemistry to endocyclic olefin starting materials, we have observed a remarkable substrate-directing effect that has enabled the highly diastereoselective synthesis of bicyclic structures. Moreover, we have discovered these same substrates can be transformed into analogous 1,4-dienes using palladium catalysis, with the completely opposite sense of diastereoselectivity.
We anticipated that our intramolecular ruthenium-catalyzed cycloisomerizations may provide a convenient method for synthesizing bicyclic compounds from simple enynes such as 1. To that end, we subjected enyne 1 to the standard catalytic conditions (10 mol % CpRu(CH3CN)3PF6, acetone, 23→50 °C, Scheme 1). We were disappointed to observe that no desired 1,4-diene was produced. We rationalized this outcome within our understanding of the reaction mechanism,3 wherein the cycloisomerization is initiated by ruthenacyclopentene formation between the alkyne, alkene, and metal complex. In this intermediate, β-hydride elimination does not occur, likely due to the high energy barrier associated with elimination at a pseudoaxial position.
Scheme 1.

We had observed in our previous studies, however, that differential mechanistic pathways can be operable under ruthenium catalysis depending on the nature of the substrate.3 Specifically, if the alkyne bears an electron-withdrawing group, cycloisomerizations can proceed via an initial allylic C-H activation, followed by alkyne carboruthenation and reductive elimination. Indeed, when analogous substrate 3, featuring a methyl ester at the terminus of the alkyne, was subjected to the same ruthenium-catalyzed cycloisomerization conditions, the desired enyne (4) was afforded in excellent yield as a single observable diastereomer (Scheme 2).
Scheme 2.

Particularly interesting was the sense of stereoselectivity in this transformation. We had initially anticipated generation of the cis-fused decalin system, expecting that a coordinated alkyne would likely direct allylic C-H activation. Carbon-carbon bond formation occurs syn to the ester, however, which implies that the C-H insertion must also occur on the same face. Therefore, it appears that the carbonyl moiety is in fact acting as a coordinating group to stereoselectively direct the formation of the intermediate allyl ruthenium species (Scheme 3). Ligand exchange, subsequent carboruthenation, and reductive elimination produces the observed trans-fused diastereomer (4).4
Scheme 3.

Intrigued by the exceptional yield and high degree of stereoselectivity in the formation of 4, we began to investigate the scope of the reaction. To our delight, the ruthenium-catalyzed cycloisomerization was effective for forming a variety of trans-fused ring systems (Table l).5 Catalyst loadings were decreased to 5 mol %, and could be further lowered to 3 mol %, albeit with slightly diminished yields. In addition to esters, aldehydes, amides, and carboxylic acids can act as directing functional groups. A primary alcohol was also effective, although the reaction times were longer and the reactions required higher catalyst loadings. 7,6-ring systems could also be forged by utilizing cycloheptene precursors (entries 6-8). The alkyne terminus can also be substituted with a ketone or an amide (entries 9 and 10). In all cases, the trans-fused bicyclic systems were accessed in good to excellent yields.
Table 1.
Ruthenium-catalyzed enyne cycloisomerizations
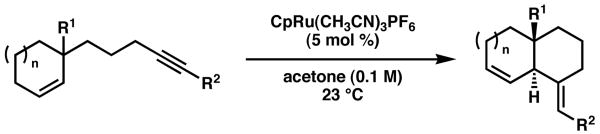 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | n | yield, time (%, h)a | drb |
| 1 | CO2Me | CO2Me | 1 | 90, 3 | > 19:1 |
| 82, 3c | > 19:1 | ||||
| 2 | CHO | CO2Me | 1 | 88, 2 | > 19:1 |
| 3 | CON(OMe)Me | CO2Me | 1 | 79, 3 | > 19:1 |
| 4 | CO2H | CO2Me | 1 | 95, 3 | > 19:1 |
| 5 | CH2OH | CO2Me | 1 | 86, 6d | > 19:1 |
| 6 | CO2Me | CO2Me | 2 | 99, 3 | > 19:1 |
| 7 | CHO | CO2Me | 2 | 87, 5e | > 19:1 |
| 8 | CON(OMe)Me | CO2Me | 2 | 99, 4 | > 19:1 |
| 9 | CO2Me | COi-Pr | 1 | 70, 6e | > 19:1 |
| 10 | CO2Me | CONEt2 | 1 | 70, 3d,f | > 19:1 |
Isolated yield.
Determined by 1H NMR.
Using 3 mol % catalyst.
Using 10 mol % catalyst.
Using 20 mol % catalyst.
Temp: 40 °C.
Other substrates investigated revealed further insights into our understanding of this transformation. Silyl ether 8 and carbonate 9 (see Figure 1), both lacking a similar coordinative ability, were completely unreactive to these enyne cycloisomerizations, even with higher catalyst loadings and elevated temperatures. Nitrile 10 was also unreactive, suggesting that C-H insertion to form the metal allyl species was not solely due to an inductive effect of the key functional group, but a coordinative effect. Cyclooctene 11 also did not react when treated with the ruthenium catalyst, perhaps owing to the conformational rigidity often seen in cyclooctyl systems.
Figure 1.

Substrates unreactive to ruthenium catalysis.
Prior to our investigations into ruthenium-catalyzed cycloisomerizations, we had shown that palladium can catalyze similar transformations to form 1,4-dienes.6 We became curious as to whether similar selectivities would be observed under these differential conditions. When 3 was subjected to our previously described “ligandless” palladium protocol (4 mol % Pd2dba3·CHCl3, 2 equiv HCO2H, DCE),6c a very similar 1,4-diene was afforded as the major product (Scheme 4). Notably, however, the diene was produced with the complete opposite sense of diastereoselectivity, forming exclusively cis-fused bicycle 12. This reaction was plagued, however, by the formation of olefin isomers arising from palladium hydride insertion/elimination pathways.7 Fortunately, this complication could be minimized by the addition of 2 vol % acetonitrile to the reaction mixture, providing 12 in good yield.
Scheme 4.

With the optimal conditions in hand, the substrates utilized in the ruthenium-catalyzed cycloisomerizations were evaluated in this process (Table 2). Gratifyingly, several of the enynes provided the analogous cis-fused 1,4-diene products in good yields and with exclusive stereoselectivities.5 The only unreactive compounds were substrates bearing either a carboxylic acid or an ynamide functional group (entries 4 and 10, respectively).8
Table 2.
Palladium-catalyzed enyne cycloisomerizations
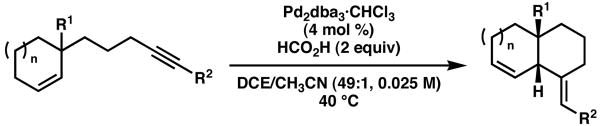 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | n | yield, time (%, h)a | drb |
| 1 | CO2Me | CO2Me | 1 | 92, 4 | > 19:1 |
| 2 | CHO | CO2Me | 1 | 73, 5 | > 19:1 |
| 3 | CON(OMe)Me | CO2Me | 1 | 68, 15 | > 19:1 |
| 4 | CO2H | CO2Me | 1 | NR | -- |
| 5 | CH2OH | CO2Me | 1 | 83, 2.5 | > 19:1 |
| 6 | CO2Me | CO2Me | 2 | 95, 3 | > 19:1 |
| 7 | CHO | CO2Me | 2 | 92, 4 | > 19:1 |
| 8 | CON(OMe)Me | CO2Me | 2 | 95, 2.5 | > 19:1 |
| 9 | CO2Me | COi-Pr | 1 | 67, 12 | > 19:1 |
| 10 | CO2Me | CONEt2 | 1 | NR | -- |
Isolated yield.
Determined by 1H NMR.
Our mechanistic rationale is depicted in Scheme 5, in accordance with our previously proposed mechanism. In this case, olefin insertion occurs selectively syn to the vinyl palladium species in intermediate 15 to provide 16. β-hydride elimination affords olefin complex 17, which readily dissociates in the presence of acetonitrile. In the absence of this solvent additive, reinsertion occurs and subsequently leads to olefin scrambling.
Scheme 5.

The current state of the art in metal-catalyzed enyne cycloisomerizations features transformations focusing almost exclusively on either chemoselectivity or enantioselectivity. This work describes a successful strategy toward achieving high levels of diastereocontrol that can be readily explained through our mechanistic understanding. Simply by switching the metal catalyst and consequently the mechanism, we can now access an array of both trans-fused and cis-fused bicycles in excellent yields via this methodology. Current efforts are directed toward expanding the scope of these transformations, as well as toward synthetic applications.
Supplementary Material
Experimental procedures, compound characterization data, and spectra. This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
We thank the American Cancer Society for a postdoctoral fellowship for E.M.F. We also thank the NSF and NIH (GM13598) for the support of our programs, and Johnson Matthey for the generous donation of metal salts.
References
- 1.Trost BM, Lautens M. J Am Chem Soc. 1985;107:1781–1783. [Google Scholar]
- 2.For reviews on enyne cycloisomerizations, see: Trost BM, Krische MJ. Synlett. 1998:1–16.Michelet V, Toullec PY, Genêt JP. Angew Chem, Int Ed. 2008;47:4268–4315. doi: 10.1002/anie.200701589.
- 3.Trost BM, Toste FD. J Am Chem Soc. 2002;124:5025–5036. doi: 10.1021/ja012450c. [DOI] [PubMed] [Google Scholar]
- 4.Recently, Furstner et al. have described iron-catalyzed stereoselective enyne cycloisomerizations to form either cis- or trans-fused bicycles depending on the nature of the bicycle. See: Furstner A, Majima K, Martín R, Krause H, Kattnig E, Goddard R, Lehmann CW. J Am Chem Soc. 2008;130:1992–2004. doi: 10.1021/ja0777180.
- 5.Relative configuration was determined by NOE experiments. See Supporting Information for details.
- 6.a) Trost BM, Tanoury GJ, Lautens M, Chan C, MacPherson DT. J Am Chem Soc. 1994;116:4255–4267. [Google Scholar]; b) 4278 Trost BM, Romero DL, Rise F. J Am Chem Soc. 1994;116:4268. [Google Scholar]; c) Trost BM, Li Y. J Am Chem Soc. 1996;118:6625–6633. [Google Scholar]
- 7.This phenomenon is frequently seen in analogous palladium-catalyzed intramolecular Heck reactions of cyclic olefins, and is generally suppressed by the addition of silver or thallium salts. These salts were ineffective as additives here. See, Link JT. Org React. 2002;60:157–534.
- 8.The carboxylic acid moiety is likely exchanging with the formate ligand of the palladium complex, which is potentially preventing the olefin insertion from occurring. In entry 10, the highly coordinative nature of the ynamide functional group may be preventing the reaction from proceeding.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, compound characterization data, and spectra. This material is available free of charge via the internet at http://pubs.acs.org.