

Abstract
Abnormal activation of CXCR4 during inflammatory/infectious states may lead to neuronal dysfunction or damage. The major goal of this study was to determine the coupling of CXCR4 to p53-dependent survival pathways in primary neurons. Neurons were stimulated with the HIV envelope protein gp120IIIB or the endogenous CXCR4 agonist, SDF-1α. We found that gp120 stimulates p53 activity and induces expression of the p53 pro-apoptotic target Apaf-1 in cultured neurons. Inhibition of CXCR4 by AMD3100 abrogates the effect of gp120 on both p53 and Apaf-1. Moreover, gp120 neurotoxicity is markedly reduced by the p53-inhibitor, pifithrin-α. The viral protein also regulates p53 phosphorylation and expression of other p53-responsive genes, such as MDM2 and p21. Conversely, SDF-1α, which can promote neuronal survival, increases p53 acetylation and p21 expression in neurons. Thus, the stimulation of different p53 targets could be instrumental in determining the outcome of CXCR4 activation on neuronal survival in neuroinflammatory disorders.
Introduction
The chemokine receptor CXCR4 and its ligand SDF-1α are constitutively expressed in the brain and play essential roles in the development and function of the CNS (Lazarini et al., 2003; Klein and Rubin, 2004). The role of CXCR4 in neuroinflammatory disorders is gradually getting clearer as it is over-expressed in various neurodegenerative diseases, including neuroAIDS (Glabinski et al., 2000; Petito et al., 2001; Martinez-Caceres et al., 2002)—a progressive neurodegenerative condition that is caused by neuronal damage/death in specific CNS areas (Gonzalez-Scarano and Martin-Garcia, 2005). Host and viral factors are implicated in HIV-1 neurotoxicity (Kaul et al., 2001; Power et al., 2002; Xu et al., 2004). The role of CXCR4 in AIDS neuropathogenesis is still unclear and may include effects on neuronal and non-neuronal cells secondarily related to the infection process. (Gonzalez-Scarano and Martin-Garcia, 2005). SDF-1α expression is up-regulated in the basal ganglia of advanced AIDS patients (Rostasy et al., 2003) and HIV-induced cleavage of SDF-1α, which impairs the interaction of the chemokine with CXCR4, has been suggested as a potential mechanism for neurodegeneration (Zhang et al., 2003). Increased incidence of CXCR4-using strains of HIV-1 has been observed in AIDS patients at the later stages of disease, when neuronal damage is usually more pronounced (Clapham and McKnight, 2001).
The HIV envelope protein gp120 initiates the viral fusion to target cells via its binding to a chemokine receptor and to CD4. CXCR4 along with CCR5 serves as a major co-receptor for the HIV-1 (Bleul et al., 1996; Oberlin et al., 1996). Neurons and glia from different species express chemokine receptors, including CXCR4 and CCR5, and their interaction with gp120 can induce a pro-apoptotic cascade of signaling events leading to neuronal loss (Meucci and Miller, 1996; Meucci et al., 1998; Kaul and Lipton, 1999; Ohagen et al., 1999). Hence, the activation of chemokine receptors by gp120 could be an important factor in AIDS-related neuroencephalopathy (Nath, 2002). Furthermore, the study of gp120/CXCR4 interactions may provide important hints about the coupling of CXCR4 to neurodegenerative pathways (Bodner et al., 2003; Khan et al., 2004). Several studies with rodent or human cultures have shown that the interaction of gp120 with neuronal and glia chemokine receptors results in neuronal death, but the mechanisms of gp120 neurotoxicity are still unclear. For instance, activation of glutamate receptors, of caspase 3, and sphingolipid pathways have all been suggested to play a role in neuroAIDS (Bezzi et al., 2001; Kaul et al., 2001; Jana and Pahan, 2004).
HIV neuropathogenesis might also involve the activity of cell cycle proteins (Khan et al., 2003), such as the transcription factor p53 (Garden et al., 2004). This nuclear protein is well-known for its ability to induce cell cycle arrest and/or apoptosis in cells undergoing various types of stresses (Haupt et al., 2003). Also, p53 regulates neuronal apoptosis in physiological and pathological situations and has been shown to be up-regulated in vivo in various neurodegenerative disorders (Morrison et al., 2003; Silva et al., 2003). Furthermore, increased expression of p53 has been detected in both neurons and glia in the brains of AIDS patients with dementia and gp120 neurotoxicity is reduced in cultures from p53-deficient mice (Garden et al., 2004). These findings suggest that transcriptional activation of p53 may result in the expression of apoptotic genes leading to neuronal death. However, it is not clear whether this is a direct effect on neurons and whether it is mediated by CXCR4.
In the present study, we have used the bilaminar cell culture system (where pure populations of neurons are co-cultured with a glial feeder layer that supports their growth and differentiation) in order to further characterize the coupling of CXCR4 to survival pathways. Our major goal was to identify the role of CXCR4 in stimulating p53-dependent pathways in neurons. We particularly focused on p53 responsive pro-apoptotic genes, namely the apoptotic protease activating factor-1 (Apaf-1), which we found to be up-regulated in gp120-treated neurons in a CXCR4-dependent manner and likely involved in CXCR4-mediated cell death.
Results
Involvement of CXCR4 in the up-regulation of p53 by gp120
Stabilization/up-regulation of the p53 protein is an important first step that takes place in neurons under various kinds of toxic insults (Miller et al., 2000). Treatment of the bilaminar cortical cultures with gp120IIIB (200 pM) increased protein level of p53 in neurons (Fig. 1A), as determined by Western blot analysis. This increase was evident in neuronal nuclei only; there was no detectable p53 in the cytoplasm of both untreated and treated cultures (Fig. 1B). In contrast to gp120IIIB, SDF-1α (20 nM) did not increase p53 protein level, even when treatments lasted as long as 6 h (Fig. 1C). However, in the absence of trophic support, SDF-1α treatment reduced neuronal p53 content (Fig. 1D). Interestingly, SDF-1α reduced transactivation of a p53 luciferase reporter gene in a CXCR4 positive cell line (Fig. 1E).
Fig. 1.
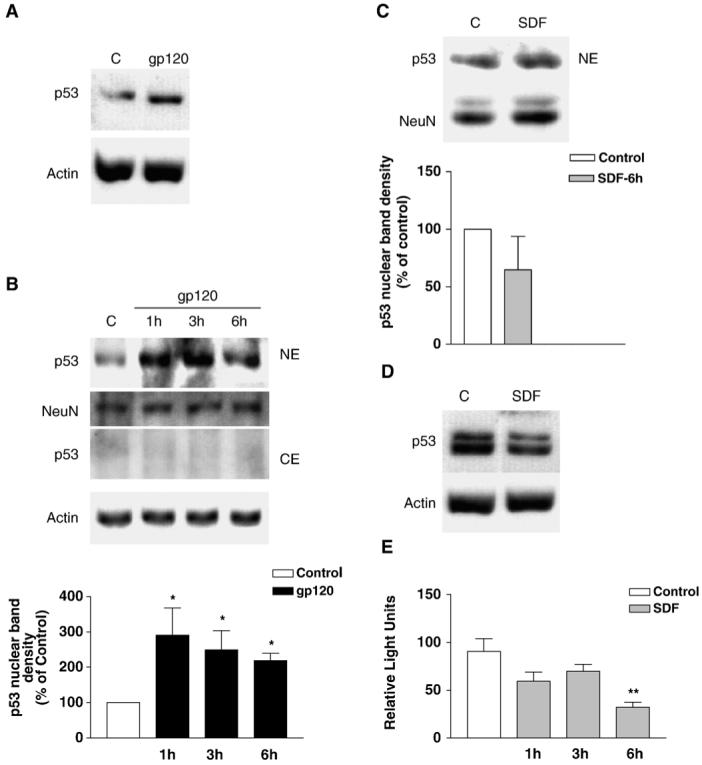
Effect of gp120IIIB and SDF-1α on neuronal p53. gp120IIIB (200 pM) increases total (A) and nuclear (B) protein levels of p53 in primary cortical neurons—as determined by Western blots using polyclonal (A) or monoclonal (B) antibodies against p53. Antibodies against NeuN and actin were used to verify equal protein loading in nuclear and cytosolic extracts, respectively. Treatment with gp120 in panel A lasted 6 h. The graph in panel B reports data from three experiments where neurons were treated with gp120 for 1 to 6 h before collecting nuclear extracts (*P < 0.05 vs. control). Panel C shows the effect of SDF-1α (20 nM, 6 h) on total p53 levels under similar conditions. However, in the absence of all trophic support, i.e. in the absence of glia and glial-conditioned medium (D), SDF-1α decreases p53 protein levels in neurons. Under these experimental conditions, which are unfavorable to neuronal survival, an additional band is observed above the normal p53 band. This smaller upper band may represent the level of phosphorylated p53. (E) In a cell line expressing CXCR4 receptor, SDF-1α reduces p53 transcriptional activity as indicated by the luciferase reporter gene assays reported in panel (E); **P < 0.01 vs. control. The data in each graph (mean ± SEM) are from 3 independent experiments.
To determine the involvement of CXCR4 in the effect of gp120IIIB on p53 protein level, a specific CXCR4 antagonist, the bicyclam AMD3100, was used (Hatse et al., 2002). When the neuronal cultures were treated with AMD3100 (100 ng/ml) 15 min before gp120 treatment, gp120-mediated p53 increase was abolished (Fig. 2). This indicates that p53 up-regulation requires CXCR4 activation by gp120IIIB. As a first step to investigate whether neuronal p53 increase is due to an interaction of gp120IIIB with neuronal receptors, we tested the ability of the gp120 to bind to the membrane of live cultured neurons, taking advantage of nanoprobes, which are more stable and sensitive than traditional fluorophores (Bruchez et al., 1998; Larson et al., 2003). Thus, cortical neurons were incubated with biotin-labeled gp120IIIB and then stained using streptavidin conjugated to quantum dots nanocrystals. Afterwards, neurons were permeabilized and stained with an antibody against the neuronal marker β tubulin III. Using this protocol, only membrane-bound extracellular gp120 was exposed to the nanoprobes. Concentrations of gp120IIIB in the picomolar range (i.e. similar to what we had been using for survival and signal transduction assays) were used for these experiments. We found that most neurons bound gp120IIIB (Fig. 3).
Fig. 2.
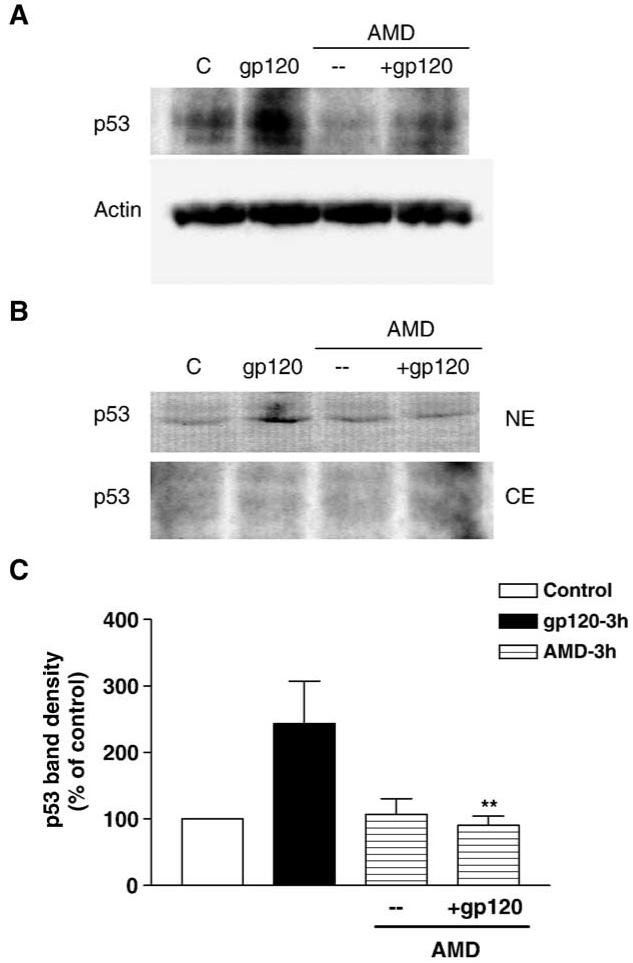
Increase in neuronal p53 expression by gp120 requires CXCR4 activity. A specific CXCR4 antagonist, AMD3100 (100 ng/ml) completely abolishes the increase in p53 protein levels induced by gp120IIIB (200 pM, 3 h); this effect was observed with both total (A) and nuclear cell extracts (B) using monoclonal anti-p53. The graph (C) shows the analysis (mean ± SEM) of 3 experiments with total cell extracts (**P < 0.01 vs. gp120).
Fig. 3.
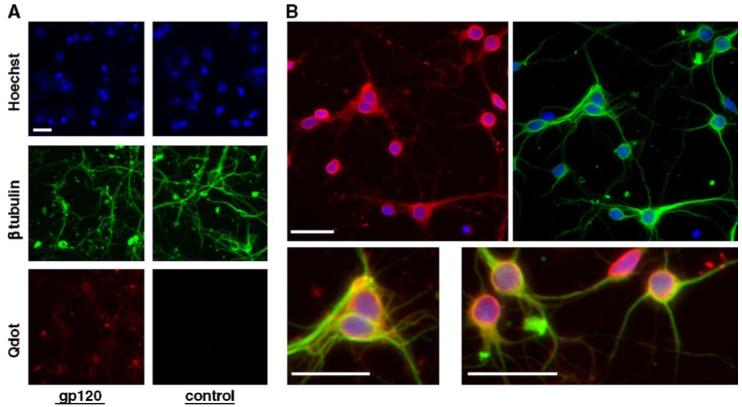
Binding of gp120 to primary cortical neurons. Pure cultures of rat cortical neurons (7 DIV) were incubated with either labeled gp120IIIB (400 pM) or with vehicle (in control cells), fixed and then stained with nanoprobes (Q-Dots). Neurons that bind gp120 appear in red. Hoechst 33342 (blue) was used for nuclear staining and β tubulin III immunostaining was used as a neuronal marker (green). Neurons were permeabilized (in order to perform the staining for the neuronal marker) after treatment with the nanoprobes. Therefore, only the gp120 bound to the plasma membrane was detected. Panel A shows both control and gp120-treated neurons, while in panel B enlarged images of gp120-treated cells are reported. The scale bar represents 20 μm.
Next, we asked whether the increase in neuronal p53 protein level was a result of the direct interaction of gp120IIIB with neurons and removed the glial layer before starting treatments with the gp120. However, removal of the glial feeder layer itself caused upregulation of nuclear p53 protein level (not shown), thus masking gp120 action. Therefore, we focused on direct indicators of p53 transcriptional activity, such as the phosphorylation of its N-terminal domain and the expression of p53 transcriptional targets.
Effect of gp120 and SDF-1α on p53 phosphorylation and expression of p53 transcriptional targets
Phosphorylation of p53 at specific residues of its N-terminal domain is crucial to the activity of this transcription factor and inhibits the binding of p53 to its negative regulator MDM2, which is also a transcriptional target of p53 (Brooks and Gu, 2003). Serine 15 phosphorylation results in increased p53 activity that leads to neuronal apoptosis (Zhu et al., 2002). In our bilaminar cell culture system, we observed that gp120IIIB treatment caused a significant increase in the protein level of phospho-p53 (Ser15) in neuronal extracts, independently of the presence of glia (Figs. 4A, 5B). In addition, the stimulation of p53 phosphorylation was independent of the amount of total p53, as indicated by the low level of phospho-p53 at 6 h of treatment (Fig. 4A), when total p53 levels are still high (Fig. 1B). The viral protein did not affect levels of acetyl-p53 (lysine373 and 382) indicating a specific effect on p53 phosphorylation. Finally, no effect of SDF-1α on phospho-p53 was observed in the same experimental conditions (Fig. 4A) whereas a 3 h SDF-1α treatment increased acetylated p53 content (Fig. 4A). Acetylation of p53 enhances its site-specific DNA binding both in vivo and in vitro (Luo et al., 2004). However, further studies are required to better understand the significance of this effect of SDF.
Fig. 4.
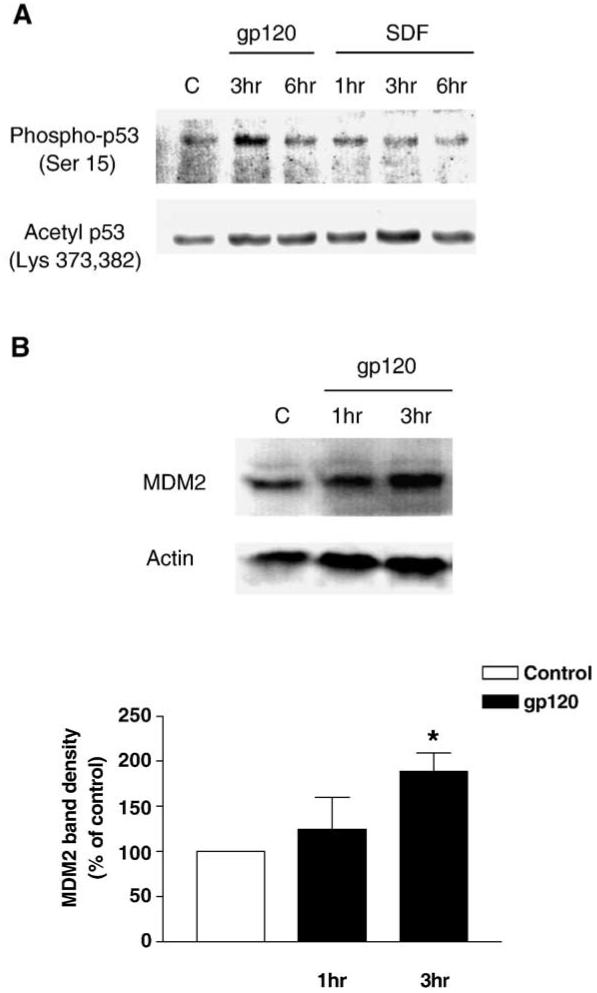
Effect of gp120 on p53 phosphorylation and MDM2 expression. Immunoblots with antibodies that selectively recognize p53 protein phosphorylated on Serine 15 (A, top gel) indicate that gp120 (200 pM) treatment increased p53 phosphorylation in neurons, while SDF-1α (20 nM) did not affect p53 phosphorylation. In the same samples, SDF-1α stimulates acetylation of p53 (A, bottom gel) that was detected with an antibody against the acetylated p53 (Lysine 373/382). Western blot analysis also shows that MDM2 is up-regulated in total neuronal extracts after a 3 h gp120 treatment (B). A monoclonal antibody against the region between the amino acids 154-167 of the MDM2 protein was used here. The bar graph reports the densitometry analysis from three separate experiments (*P < 0.05).
Fig. 5.
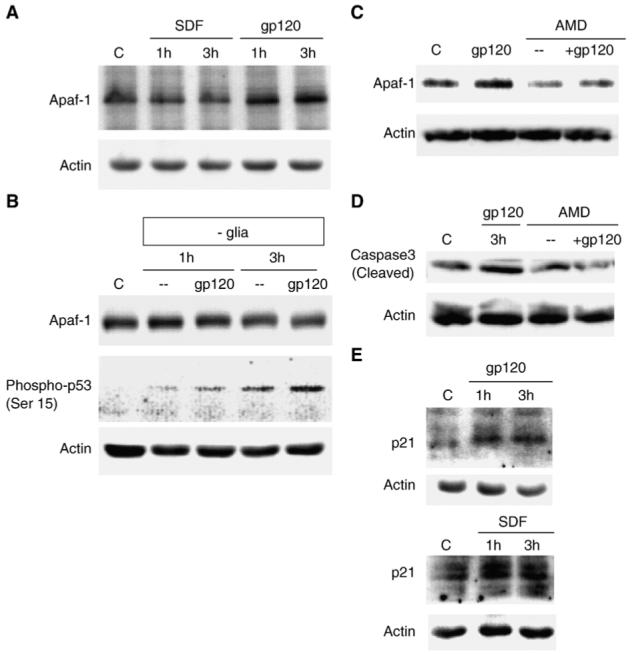
Effect of gp120 and SDF-1α on the pro-apoptotic protein Apaf-1. The protein levels of Apaf-1 are increased in protein extracts obtained from gp120IIIB-treated (200 pM) neurons and immunoblotted with anti-Apaf-1 polyclonal antibody (A). No effect of gp120 on Apaf-1 was observed in the absence of glia using the same antibody (B). However, the Western blot analyses showed that Apaf-1 up-regulation by gp120 is blocked by the CXCR4 antagonist AMD3100 (100 ng/ml) (C). Treatment with AMD3100 also inhibits the increase in the level of active caspase 3 induced by gp120 (D), as assessed by immunoblots with antibodies against the cleaved (i.e. active) form of caspase 3. Both gp120 and SDF-1α (20 nM) increase p21 protein levels in neuronal protein extracts (E).
We then looked at the effect of gp120IIIB on p53 transcriptional targets and found that gp120IIIB increased neuronal MDM2 protein level (approximately 2-fold increase at 3 h) (Fig. 4B). This could reflect the known regulatory feedback between MDM2 and p53 (Li et al., 2003).
As MDM2 is also one of the major regulator of p53 activity (it promotes p53 degradation and regulates p53 nuclear content) (Li et al., 2003), we also tested the ability of gp120IIIB to up-regulate other p53 transcriptional targets that are not directly involved in the regulation of p53, such as the pro-apoptotic protein Apaf-1, a crucial component of the apoptosome complex (Fortin et al., 2001; Ferraro et al., 2003). gp120IIIB was able to increase Apaf-1 protein level in neurons (Fig. 5A). Apaf-1 up-regulation in response to the gp120 treatment was inhibited by AMD3100 (Fig. 5C), indicating the involvement of CXCR4 in this effect. As previously demonstrated by other groups (Garden et al., 2002), gp120 also stimulated caspase activity in neurons (Fig. 5D), which is likely a consequence of the formation of the Apaf-1-mediated apoptosome complex. Indeed, AMD3100 treatment also completely inhibited the increase in cleaved caspase 3 induced by gp120IIIB in the same neuronal preparation (Fig. 5D). In contrast, SDF-1α - which we have previously shown to promote neuronal survival (Meucci et al., 1998; Khan et al., 2003) - did not affect Apaf-1 levels in neurons (Fig. 5A), but increased the level of p21, another p53 target also regulated by gp120 (Fig. 5E). SDF-1α also stimulates MDM2 (Khan et al., 2004). These data suggest that different mechanisms may be involved in regulation of neuronal survival by CXCR4. Thus, the stimulation of different p53 targets may be instrumental in determining the outcome of CXCR4 activation on neuronal survival.
Inhibition of p53 blocks gp120 neurotoxicity
In order to determine whether gp120 neurotoxicity depends on p53 transcriptional activity, cultures were treated with gp120IIIB in the presence or absence of a synthetic p53 inhibitor, pifithrin-α, which specifically inhibits p53 DNA-binding activity (Komarov et al., 1999; Murphy et al., 2004). The p53 inhibitor reduced gp120-mediated neuronal death by 50% (Fig. 6A). However, since pifithrin-α itself is slightly neurotoxic (ref. (Culmsee et al., 2001, 2003) and data not shown), the actual reduction of cell death by p53 inhibition may even be higher than what we observed. Thus, p53 transcriptional activation/repression may account for most, if not all, gp120-neurotoxicity. In fact, the p53 inhibitor reduced the effect of gp120IIIB on Apaf-1 protein level as well (Fig. 6B).
Fig. 6.
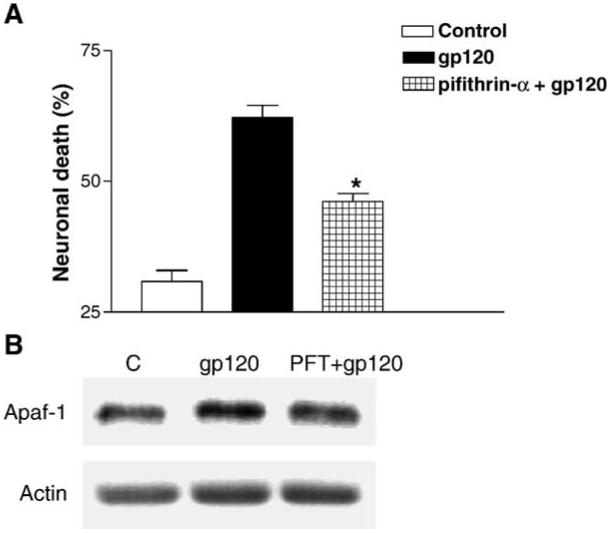
Inhibition of p53 transcriptional activity reduces gp120 neuro-toxicity. The small molecule p53 inhibitor, pifithrin-α (PFT), reduces gp120-induced neuronal death (*P < 0.001 vs. gp120) (A). PFT pretreatment (100 nM) also reduces gp120-induced Apaf-1 up-regulation in neurons as determined by Western blot (B).
Discussion
The principal finding of this study is that activation of CXCR4 by the HIV-1 envelope protein gp120IIIB up-regulates the apoptotic protein Apaf-1 in cultured neurons. This effect is mediated by p53 activation. On the other hand, SDF-1α, the natural CXCR4 ligand, does not increase Apaf-1 levels in neurons, but rather regulates other p53 targets. Thus, modulation of specific p53 responsive genes may determine the final outcome of CXCR4 activation on neuronal survival in neuroinflammatory disorders.
In fact, evidence has been mounting regarding the involvement of p53 in HIV-induced apoptosis. For instance, cell lines expressing lymphotropic HIV-1 envelope protein (Env) were found to undergo apoptosis via syncytial formation when cultured with lymphocytes expressing CD4/CXCR4 (Castedo et al., 2003; Roumier et al., 2003). This process involves the transcriptional activation of p53, up-regulation of certain p53 responsive genes, like BAX and PUMA, as well as mitochondrial membrane permeabilization (Castedo et al., 2002, 2003; Roumier et al., 2003; Perfettini et al., 2004). Other studies have shown that antisense oligonucleotides against p53 inhibit gp120-induced apoptosis of human neuroblastoma cells whereas gp120 neurotoxicity is reduced in cortical cultures from p53-deficient mice (Yeung et al., 1998; Garden et al., 2004). Thus, it is likely that p53 activation is involved in the neuronal damage caused by HIV in vivo—a hypothesis supported by the up-regulation of p53 in brains of HIV demented patients (Silva et al., 2003; Garden et al., 2004).
Although p53 is a well-known inducer of neuronal and non-neuronal apoptosis, the mechanisms involved are still a matter of debate. While initially identified exclusively as a transactivator, recent studies indicate that p53 is a multifunctional protein and it can induce apoptosis by different mechanisms (i.e. which may or may not require transactivation) (Haupt et al., 2003). In post-mitotic neurons, p53 transcriptional activity has been reported to be essential for the induction of p53-mediated cell death (Fortin et al., 2001; Cregan et al., 2004). This is in agreement with our results and underscores the relevance of Apaf-1 as a potential mediator of HIV-induced neuronal injury in vivo. Interestingly, Apaf-1 is one of the p53 responsive genes regulated by either one of the two transactivation domains of p53 and whose expression is remarkably increased in neurons carrying p53 mutants lacking the Mdm2 binding domain (Cregan et al., 2004).
The presence of glia, particularly microglia, plays a major role in gp120 neurotoxicity in vitro, though the viral protein is also known to affect neuronal survival directly (Gonzalez-Scarano and Martin-Garcia, 2005). We found a consistent increase in p53 phosphorylation in neurons treated with gp120 either in the absence or in the presence of glia. However, glia is necessary for gp120 to induce Apaf-1 expression in neurons as no changes in Apaf-1 were observed in the absence of glia. Therefore, the direct effect of gp120 on neurons could involve different mechanisms, such as p53-dependent transcriptional repression of pro-survival genes like Bcl-2 (Ho and Benchimol, 2003), which prevents HIV-induced neurotoxicity in vitro (Chen et al., 2002). Interestingly, the presence of glia also affects SDF-1α-mediated regulation of p53 (Figs. 1D, 5E), which argues for the importance of neuronal-glial interactions in CXCR4 regulation of neuronal survival.
Active p53 increases expression of several pro-apoptotic genes, including Bax, Apaf-1, NOXA, and PUMA among others that are mainly responsible for neuronal apoptosis via the intrinsic pathway involving mitochondrial dysfunction (Cregan et al., 1999, 2002, 2004). Expression of Apaf-1 is of particular interest as it has been shown to be involved in making a polyprotein complex with cytochrome c and ATP/dATP, known as apoptosome (Ferraro et al., 2003). The apoptosome binds pro-caspase 9, which cleaves itself to convert to an activated form. Active caspase 9 would catalytically activate executioner caspases, like caspase 3 and caspase 7. Presence of active caspase 3 in neurons has been observed in the brains of patients with HIV dementia as well as in mixed cultures of neurons and glia treated with gp120 (Garden et al., 2002). In this study, gp120 increased the levels of both Apaf-1 and activated caspase 3 in neurons and these effects were abrogated by the pre-treatment of cultures with AMD3100, a CXCR4 antagonist. Moreover, gp120-mediated neurotoxicity as well as increase in Apaf-1 protein levels were reduced by the p53 inhibitor, pifithrin-α. This p53 inhibitor has been reported to block binding of p53 to DNA, which would interfere with both transactivation and trans-repression (Komarov et al., 1999; Murphy et al., 2004).
In summary, in line with the recently proposed role of CXCR4 in neuronal survival and differentiation (Klein and Rubin, 2004), SDF-1α and gp120IIIB controlled p53 activity in a glia-dependent manner. The HIV-1 protein stimulated p53 expression and phosphorylation, which resulted in elevated transcriptional activity of known apoptotic genes, in contrast to SDF-1α that preferentially regulates pro-survival genes. This supports previous observations on the ability of SDF-1α to promote neuronal survival under various conditions (Meucci et al., 1998; Chalasani et al., 2003; Khan et al., 2003). Indeed, SDF-1α stimulates Akt phosphorylation and promotes nuclear translocation of anti-apoptotic proteins regulated by Akt, while gp120IIIB is unable to activate Akt (despite its effects on other CXCR4-mediated signaling pathways) (Khan et al., 2004). Finally, the two CXCR4 ligands exert different effects on other cell cycle proteins that are known to interact with p53, namely the retinoblastoma protein Rb, and one of its major targets, the transcription factor E2F-1, which indirectly stabilizes p53 (Khan et al., 2003). Based on our current evidence, the opposite action of SDF and gp120 on the Rb/E2F pathway (and possibly p53) may be related to the different involvement of Akt.
In conclusion, p53 activity might be a crucial factor in HIV neuropathogenesis as it represents a converging point of many survival pathways known to be altered in neuroAIDS. Further understanding of the molecular and cellular mechanisms involved in p53 activation and characterization of the full array of p53 downstream targets in neurons would be of utmost importance to develop appropriate therapeutic measures against neuroinflammatory/neurodegenerative disorders and to elucidate the physiological roles of CXCR4 in the CNS.
Experimental methods
Cell cultures
Neurons
Cortical neurons were obtained from the brains of 17-18 day old rat embryos and cultured in serum-free medium using a bilaminar cell culture system, as previously reported for hippocampal neurons (originally described by Banker and Cowan (1977) and modified as in: (Meucci and Miller, 1996; Meucci et al., 1998, 2000). Briefly, neurons are co-cultured with a feeder layer of secondary astrocytes that supports the growth and differentiation of the pure neuronal layer. The secondary glia culture is prepared 2 days before the neuronal primary culture by plating glia on handmade plastic “coverslips” (∼60 mm in diameter; 5 to 7 × 105 cells/each). Cells are maintained in DMEM-10%FBS until the day of the neuronal preparation. The glial coverslips are moved to the dishes containing the neurons 4 h after neuronal plating (1 × 106/dish). At this time, the culture medium is replaced with serum-free high-glucose DMEM containing N2.1 supplement (GIBCO). Neurons were separated from glia immediately before the protein extraction, unless otherwise specified.
HOS cells
HOS.CXCR4 cells were maintained in DMEM containing 10% fetal calf serum and 1 μg/ml puromycin. These cells were obtained through the AIDS Research and Reference Program, Division of AIDS, NIAID, NIH (from Dr. Nathaniel Landau) and are CD4 negative (Landau and Littman, 1992; Deng et al., 1996).
Western blots
After treatments cells were washed with ice-cold balanced salt solution and scraped in lysis buffer [25 mM Tris/150 mM NaCl/5 mM NaF/1 mM EDTA/1 mM DTT/1% Nonidet P-40/5μg each of aprotinin, leupeptin, and pepstatin/1 mM 4-(2-aminoethyl)benzeresulfonyl fluorideHCE (AEBSF)/1 mM vanadate]. A lower concentration of detergent (0.1%) was used for the extraction of cytosolic proteins when they had to be separated from nuclear proteins, and the pellet was further processed with a hyper-osmotic buffer as previously described (Meucci et al., 1998, 2000). Unless indicated in the figure, total cell lysates were used for Western blots. The protein concentration in cell lysates was determined by bicinchoninic acid protein assay from Pierce. Proteins were resolved by SDS-PAGE and transferred to PVDF membranes for immunoblotting. The following primary antibodies were used: anti-p53 (either a polyclonal from Cell Signaling Technology at 1:2000 or a monoclonal from BD-Biosciences at 1:1000), antiphospho p53 (serine 15, 1:1000, Cell Signaling Technology), antiacetylated p53 (lysine 373 and 382, 1:1000, Upstate Biotechnology), anti-MDM2 (SMP14, 1:2000, Santa Cruz Biotechnology), anti-p21 (a monoclonal from BD-Pharmingen at 1:500), anticleaved caspase 3 (a polyclonal from Cell Signaling Technology at 1:1000) and anti-Apaf-1 (a polyclonal from Cell Signaling Technology at 1:1000). Anti-actin (a polyclonal from Sigma at 1:5000) and anti-NeuN (a monoclonal from Chemicon, at 1:1000) were used to check equal protein loading and as cytoplasmic and nuclear markers, respectively. An image acquisition and analysis system, ChemiDoc System (BioRad) as well as the Un-Scan-It software (Silk Scientific) were used for detection of chemiluminescent bands and densitometric analysis. The intensity of the bands reported in each figure shows the relative level of a specific protein as indicated in the figure legend. Generally, only one band in the expected molecular range of the target protein is detected. In a few cases, additional bands close to the main band are reported, which may represent post-translational modifications of the target protein (i.e. phosphorylation). Only the main band is used for the densitometry analysis.
Immunocytochemistry
For gp120 binding experiments, live neurons were treated with biotinylated gp120IIIB (400 pM) for 30 min or overnight, fixed and stained with streptavidin conjugated with Qdot 605 (at 1:250, from Quantum Dot Corp.). Cells were then permeabilized with Triton X-100 in order to stain for a specific neuronal marker, β tubulin III (using a monoclonal antibody at 1:500, from Covance Inc.). A secondary antibody conjugated to Cy2 (at 1:500, from Jackson Immunoresearch Labs) was used for β tubulin III staining. Nuclear counterstaining was obtained with Hoechst 33342 (3 μg/ml). The cells were mounted on a glass slide and observed under an epifluorescent microscope (Olympus IX70) connected to a CCD camera (Micromax), and images were acquired using the software Metamorph (Universal Imaging).
p53 reporter gene assay
Path Detect® p53 cis reporting system (Stratagene) was used along with the Bright-Glo™ Luciferase assay system (Promega) to detect p53 transcriptional activity in accordance with the manufacturers’ protocols. The sequence of the p53-binding enhancer element in the cis-reporter plasmid is as follows; (TGCCTGGACTTGCCTGG)14.
A plasmid expressing the green fluorescent protein (pEGFP-N1, Clontech, BD Biosciences) was used to estimate transfection efficiency in the different groups. The plasmids were transfected into the cells using the standard calcium phosphate method.
Survival assay
Neuronal survival studies were performed as previously described using Hoechst 33342 and an antibody against cleaved caspase 3 (Meucci and Miller, 1996; Meucci et al., 1998, 2000). In order to inhibit endogenous p53, the cultures were treated with pifithrin-α (100 nM or 1 μM) 1 h prior to gp120 treatment (200 pM). Neuronal viability was assessed 18-24 h later.
Statistical analysis
Data are reported as mean±SEM with sample size for each experiment. Each experiment has been repeated at least two more times. One-way ANOVA, followed by Newman-Keuls multiple comparison procedure, has been used for analysis of survival experiments. Paired t test has been used to compare differences in the band densities of immunoblots. Analysis of immunostaining has been performed using the software Metamorph (Universal Imaging).
Materials
Unless otherwise specified, tissue culture media are from Gibco-Invitrogen and other general reagents are from Sigma. SDF-1α was purchased from R&D System. The lyophilized protein was reconstituted (100 μg/ml) in 0.1% BSA/PBS and aliquots stored at -20°C. Recombinant HIV-1 gp120IIIB (both unlabelled and biotinylated from ImmunoDiagnostics, Inc.) was diluted and stored as previously described (Meucci et al., 1998, 2000). AMD3100 and pifithrin-α (cyclic) were obtained from Sigma and dissolved in sterile water and DMSO, respectively.
Acknowledgments
This work has been supported by NIH grants DA15014 and DA19808 (O.M.), GM067892 (A.F.) and by a grant from the W.W. Smith Charitable Trust (O.M., # A0302).
References
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:342–397. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat. Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- Bodner A, Toth PT, Oh SB, Lu M, Tran PB, Chin RK, Ren D, Miller RJ. CD4 dependence of gp120IIIB-CXCR4 interaction is cell-type specific. J. Neuroimmunol. 2003;140:1–12. doi: 10.1016/s0165-5728(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Bruchez M, Jr., Moronne M, Gin P, Weiss S, Alivisatos AP. Semiconductor nanocrystals as fluorescent biological labels. Science. 1998;281:2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- Castedo M, Roumier T, Blanco J, Ferri KF, Barretina J, Tintignac LA, Andreau K, Perfettini JL, Amendola A, Nardacci R, Leduc P, Ingber DE, Druillennec S, Roques B, Leibovitch SA, VilellaBach M, Chen J, Este JA, Modjtahedi N, Piacentini M, Kroemer G. Sequential involvement of Cdk1, mTOR and p53 in apoptosis induced by the HIV-1 envelope. EMBO J. 2002;21:4070–4080. doi: 10.1093/emboj/cdf391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Andreau K, Roumier T, Piacentini M, Kroemer G. Mitochondrial apoptosis induced by the HIV-1 envelope. Ann. N. Y. Acad. Sci. 2003;1010:19–28. doi: 10.1196/annals.1299.004. [DOI] [PubMed] [Google Scholar]
- Chalasani SH, Baribaud F, Coughlan CM, Sunshine MJ, Lee VM, Doms RW, Littman DR, Raper JA. The chemokine stromal cell-derived factor-1 promotes the survival of embryonic retinal ganglion cells. J. Neurosci. 2003;23:4601–4612. doi: 10.1523/JNEUROSCI.23-11-04601.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Sulcove J, Frank I, Jaffer S, Ozdener H, Kolson DL. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J. Virol. 2002;76:9407–9419. doi: 10.1128/JVI.76.18.9407-9419.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham PR, McKnight A. HIV-1 receptors and cell tropism. Br. Med. Bull. 2001;58:43–59. doi: 10.1093/bmb/58.1.43. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J. Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J. Neurosci. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Ferraro E, Corvaro M, Cecconi F. Physiological and pathological roles of Apaf1 and the apoptosome. J. Cell. Mol. Med. 2003;7:21–34. doi: 10.1111/j.1582-4934.2003.tb00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J. Cell Biol. 2001;155:207–216. doi: 10.1083/jcb.200105137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D’Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA. Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J. Neurosci. 2002;22:4015–4024. doi: 10.1523/JNEUROSCI.22-10-04015.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- Glabinski AR, O’Bryant S, Selmaj K, Ransohoff RM. CXC chemokine receptors expression during chronic relapsing experimental autoimmune encephalomyelitis. Ann. N. Y. Acad. Sci. 2000;917:135–144. doi: 10.1111/j.1749-6632.2000.tb05377.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat. Rev., Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by AMD3100 is strictly confined to CXCR4. FEBS Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis—The p53 network. J. Cell Sci. 2003;116:4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- Ho J, Benchimol S. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 2003;10:404–408. doi: 10.1038/sj.cdd.4401191. [DOI] [PubMed] [Google Scholar]
- Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J. Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. U. S. A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Musser BJ, Resue DM, Fatatis A, Meucci O. The chemokine receptor CXCR4 regulates cell-cycle proteins in neurons. J. NeuroVirol. 2003;9:300–314. doi: 10.1080/13550280390201010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Patel JP, Huynh N, Wang J, Huang Z, Fatatis A, Meucci O. Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Res. Hum. Retroviruses. 2004;20:1063–1071. doi: 10.1089/aid.2004.20.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25:306–314. doi: 10.1016/j.it.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285:1733–1737. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- Landau NR, Littman DR. Packaging system for rapid production of murine leukemia virus vectors with variable tropism. J. Virol. 1992;66:5110–5113. doi: 10.1128/jvi.66.8.5110-5113.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–148. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Monoversus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Caceres EM, Espejo C, Brieva L, Pericot I, Tintore M, Saez-Torres I, Montalban X. Expression of chemokine receptors in the different clinical forms of multiple sclerosis. Mult. Scler. 2002;8:390–395. doi: 10.1191/1352458502ms841oa. [DOI] [PubMed] [Google Scholar]
- Meucci O, Miller RJ. gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-beta1. J. Neurosci. 1996;16:4080–4088. doi: 10.1523/JNEUROSCI.16-13-04080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8075–8080. doi: 10.1073/pnas.090017497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell Death Differ. 2000;7:880–888. doi: 10.1038/sj.cdd.4400736. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochem. Res. 2003;28:15–27. doi: 10.1023/a:1021687810103. [DOI] [PubMed] [Google Scholar]
- Murphy PJ, Galigniana MD, Morishima Y, Harrell JM, Kwok RP, Ljungman M, Pratt WB. Pifithrin-alpha inhibits p53 signaling after interaction of the tumor suppressor protein with hsp90 and its nuclear translocation. J. Biol. Chem. 2004;279:30195–30201. doi: 10.1074/jbc.M403539200. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J. Infect. Dis. 2002;186(Suppl 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cellline-adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, Osathanondh R, Gartner S, Shi B, Shaw G, Gabuzda D. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: evidence for a role of the envelope. J. Virol. 1999;73:897–906. doi: 10.1128/jvi.73.2.897-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini JL, Roumier T, Castedo M, Larochette N, Boya P, Raynal B, Lazar V, Ciccosanti F, Nardacci R, Penninger J, Piacentini M, Kroemer G. NF-kappaB and p53 are the dominant apoptosis-inducing transcription factors elicited by the HIV-1 envelope. J. Exp. Med. 2004;199:629–640. doi: 10.1084/jem.20031216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petito CK, Roberts B, Cantando JD, Rabinstein A, Duncan R. Hippocampal injury and alterations in neuronal chemokine co-receptor expression in patients with AIDS. J. Neuropathol. Exp. Neurol. 2001;60:377–385. doi: 10.1093/jnen/60.4.377. [DOI] [PubMed] [Google Scholar]
- Power C, Gill MJ, Johnson RT. Progress in clinical neurosciences: the neuropathogenesis of HIV infection: host-virus interaction and the impact of therapy. Can. J. Neurol. Sci. 2002;29:19–32. doi: 10.1017/s0317167100001682. [DOI] [PubMed] [Google Scholar]
- Rostasy K, Egles C, Chauhan A, Kneissl M, Bahrani P, Yiannoutsos C, Hunter DD, Nath A, Hedreen JC, Navia BA. SDF-1alpha is expressed in astrocytes and neurons in the AIDS dementia complex: an in vivo and in vitro study. J. Neuropathol. Exp. Neurol. 2003;62:617–626. doi: 10.1093/jnen/62.6.617. [DOI] [PubMed] [Google Scholar]
- Roumier T, Castedo M, Perfettini JL, Andreau K, Metivier D, Zamzami N, Kroemer G. Mitochondrion-dependent caspase activation by the HIV-1 envelope. Biochem. Pharmacol. 2003;66:1321–1329. doi: 10.1016/s0006-2952(03)00480-5. [DOI] [PubMed] [Google Scholar]
- Silva C, Zhang K, Tsutsui S, Holden JK, Gill MJ, Power C. Growth hormone prevents human immunodeficiency virus-induced neuronal p53 expression. Ann. Neurol. 2003;54:605–614. doi: 10.1002/ana.10729. [DOI] [PubMed] [Google Scholar]
- Xu Y, Kulkosky J, Acheampong E, Nunnari G, Sullivan J, Pomerantz RJ. HIV-1-mediated apoptosis of neuronal cells: proximal molecular mechanisms of HIV-1-induced encephalopathy. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7070–7075. doi: 10.1073/pnas.0304859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MC, Geertsma F, Liu J, Lau AS. Inhibition of HIV-1 gp120-induced apoptosis in neuroblastoma SK-N-SH cells by an antisense oligodeoxynucleotide against p53. AIDS. 1998;12:349–354. doi: 10.1097/00002030-199804000-00002. [DOI] [PubMed] [Google Scholar]
- Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, Clark-Lewis I, Overall CM, Power C. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat. Neurosci. 2003;6:1064–1071. doi: 10.1038/nn1127. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Mao XO, Sun Y, Xia Z, Greenberg DA. p38 mitogen-activated protein kinase mediates hypoxic regulation of Mdm2 and p53 in neurons. J. Biol. Chem. 2002;277:22909–22914. doi: 10.1074/jbc.M200042200. [DOI] [PubMed] [Google Scholar]