

Abstract
IL-17 alone is a relatively weak inducer of gene expression but cooperates with other cytokines including TNFα to generate a strong response in part via prolongation of mRNA half-life. Because TRAF6 has been reported to be essential for signaling by IL-17 we examined its involvement in IL-17-mediated mRNA stabilization. While over-expression of TRAF6 in HeLa cells activates NFκB, it does not stabilize transfected KC mRNA. Furthermore, a dominant negative TRAF6 abrogates NFκB activation but does not block IL-17-induced chemokine mRNA stabilization. IL-17 can stabilize KC and MIP-2 mRNAs comparably in TNFα-treated MEFs from TRAF6+/+ and TRAF6−/− mice. TRAF6 is known to couple upstream signals with activation of p38 MAP kinase and MAKAP2 (MK2), both of which have been shown to be important for Toll/Interleukin 1 Receptor-mediated mRNA stabilization in various cell types. Inhibition of p38 MAP kinase, however, does not block IL-17-induced KC mRNA stabilization and IL-17 can stabilize KC mRNA equally in MEFs from both wild type and MK2/3 doubly deficient mice. Finally, IL-17 can amplify the levels of multiple TNFα-stimulated mRNAs in wild type and TRAF6-deficient cells but not in cells from Act1−/− mice. Collectively these findings demonstrate the existence of a TRAF6/p38 MAP kinase-independent pathway that couples the IL-17 receptor with enhanced mRNA stability. Because the most potent effects of IL-17 on gene expression are obtained in cooperation with other cytokines such as TNFα, these findings suggest that this pathway is a major contributing mechanism for response to IL-17.
Keywords: signal transduction, gene regulation, cytokines, inflammation
INTRODUCTION
IL-17 is recognized to play a critical role in several forms of inflammation associated with host defense or autoimmunity (1–3). In response to stimulation with IL-17, resident tissue cell populations produce a variety of pro-inflammatory gene products resulting in the influx of neutrophils (2–4). However, the molecular mechanisms by which IL-17 regulates target gene expression are poorly understood. It has been repeatedly observed that while IL-17 alone exerts only modest effect on gene expression, the combination of IL-17 with other inflammatory stimuli (particularly TNFα) leads to a strong, synergistic response (4, 5). Because IL-17 will almost certainly be present in vivo in a microenvironment containing multiple cytokines including TNFα, the ability to amplify responses induced by other stimuli is likely to be physiologically important.
While many genes induced during inflammation exhibit a significant increase in transcription, often dependent upon activation of the transcription factor NFκB, the mature mRNAs frequently exhibit very short half-lives that serve as a check to ensure that the protein is not produced inappropriately (6–10). These remarkably short half-lives are often prolonged via signals from inflammatory stimuli enabling robust gene expression during the response (11–14). In many settings this stimulus-induced stabilization requires the action of the p38 MAP kinase cascade including its downstream kinases MAKAP (MK)2 and 3 (11, 12, 15–17).
A number of reports have indicated that activation of NFκB is a major mechanism by which IL-17 regulates target gene expression (18, 19). However, IL-17-mediated NFκB activation is modest in comparison to that induced by agents such as TNFα and IL-1 (20–22). Several laboratories have shown that IL-17 serves as a strong stimulus to induce stabilization of constitutively unstable mRNAs and this may represent an important mechanism by which this cytokine regulates target gene expression (20, 23, 24). The ability of IL-17 to prolong the half-life of unstable mRNAs provides a mechanistic basis for the synergistic response induced by TNFα and IL-17, as TNFα serves as a strong stimulus for NFκB-dependent transcription, but is unable to prolong the half-life of the mRNA (14, 20, 25).
While the signal transduction pathway(s) initiated in response to IL-17 remain largely undefined, several reports implicate participation of adaptor proteins including NFκB Activator 1 (Act1)2 and TNF receptor associated factor (TRAF)6 (21, 22, 26). IL-17-induced activation of NFκB and JNK and modulation of IL-6 expression were shown to be dependent on TRAF6 using TRAF6-deficient mouse embryo fibroblasts (MEFs) (26). Using mice deficient in Act1, this signaling adaptor was found to be required both for response to IL-17 alone and for the more robust responses seen with the combination of IL-17 and TNFα (21, 22). We subsequently showed that Act1 was both necessary and sufficient for IL-17 to promote stabilization of chemokine mRNA (20).
In the present report we have examined the requirement for TRAF6 in IL-17-induced mRNA stabilization. Surprisingly, using multiple experimental approaches we find that TRAF6 is not required in signaling for stabilization of TNFα-induced chemokine mRNAs. In addition, IL-17-induced mRNA stabilization did not depend upon the action of p38 MAP kinase or downstream kinases MK2 and MAKAP3 MK3. These results further highlight the importance of mRNA stabilization in IL-17-mediated amplification of TNFα-stimulated gene expression and identify a TRAF6/p38/MK2/MK3-independent pathway operating downstream of Act1. Because the potency of response to IL-17 is markedly greater when used in combination with TNFα, this pathway may represent the predominant means through which IL-17 promotes inflammatory gene expression.
MATERIALS AND METHODS
Reagents
G418, formamide, MOPS, salmon sperm DNA, and diethyl-pyrocarbonate were purchased from Sigma-Aldrich. Dulbecco’s modified Eagle’s medium, Dulbecco’s phosphate-buffered saline, penicillin and streptomycin were obtained from Central Cell Services of the Lerner Research Institute. Fetal bovine serum was purchased from BioWhittaker. PolyFect Transfection Reagent was obtained from Qiagen and Tri-Reagent was purchased from Molecular Research Center. Recombinant human and mouse IL-17, TNFα, and IL-1α were purchased from R&D Systems. SB203580 was purchased from Calbiochem. Nylon transfer membrane was purchased from Micron Separation. Luciferase assay buffer and passive lysis buffer were obtained from Promega. Perkin Elmer Life Sciences was the source of [α-32P]-dCTP and Western Lightning Chemiluminescence Reagent Plus. Protein assay reagents were purchased from Bio-Rad. Anti FLAG M2 antibody was purchased from Sigma-Aldrich, antibody against GAPDH was purchased from Chemicon International, and anti mouse IgG HRP linked antibody was purchased from Amersham.
Preparation of Peritoneal Macrophages
C57Bl/6 mice were purchased from Jackson Laboratories and were housed in microisolator cages with autoclaved food and bedding to minimize exposure to viral and microbial pathogens. All procedures were approved by the Institutional Animal Care and Use Committee. TG-elicited peritoneal macrophages were prepared as described previously (27) and cultured overnight in RPMI 1640 medium containing L-glutamine, penicillin, streptomycin, and 5% FBS prior to treatments as described in the text.
Plasmids
Plasmids encoding KC cDNA under control of a tetracycline regulated promoter (pTRE2 KCΔ3) and the NFκB reporter 5X κB luciferase have been previously described as have the plasmids and methods used to prepare radiolabeled CXCL1 (KC), GAPDH, (CXCL2) MIP-2, and IκBζ cDNAs (20, 28–30). Expression plasmids encoding epitope (FLAG)-tagged Act1, TRAF6, and dominant negative (dn) TRAF6 have been previously described (31, 32).
Cell Culture and Transfection
HeLa tet-off cells were purchased from Clontech Laboratories and cultured in DMEM containing 10% FBS, penicillin, and streptomycin and kept under selection with G418. For transfection, HeLa tet-off cells were plated in 100 mm dishes and allowed to grow for 24 hours to 70% confluency. Transfection was performed using PolyFect Transfection Reagent according to the manufacture’s protocol (Qiagen). Wild type, TRAF6−/−, and Act1−/− mouse embryo fibroblasts have been previously described and were cultured in DMEM containing 10% FBS, penicillin, and streptomycin (21, 33). MEFs from MK2/MK3 doubly deficient mice were kindly provided by Dr. Matthias Gaestel (Institute of Biochemistry, Medical School Hannover, Hannover, Germany)(17)
Northern Blot, Real-time PCR, Western Blot, and Luciferase Assay
Total RNA was isolated using TRI-Reagent according to the manufacture’s protocol and northern hybridization was performed as previously described (28). To calculate mRNA half-lives the autoradiographs were quantified using the NIH Image software. mRNA levels for each gene of interest were normalized to GAPDH mRNA levels and plotted as log of the percent remaining mRNA versus time. The best fit to linear decay was determined and the half life calculated from the intersection at the point corresponding to 50% residual RNA. Quantitative real-time PCR was performed as previously described (21). The cDNAs were synthesized with random hexamers using M-MLV reverse transcriptase (Promega) and real time PCR conducted using SYBER Green PCR Master Mix kit (Applied Biosystems). The primer sequences used were obtained from PrimerBank (34). The PrimerBank ID for each gene is as follows: cxcl1 (6680109a1), il6 (26354667a1), nfkbiz (13447398a1), cxcl5 (6677887a1), lcn2 (1019908a1), GAPDH (6679939a1). Western blots were carried out as described previously (35). Luciferase assays were performed according the manufacture’s protocol (Promega).
RESULTS
Over-expression of Act1 but not TRAF6 mediates stabilization of KC mRNA
Robust expression of KC mRNA from the endogenous gene requires both transcription via the activation of NFκB and stabilization of the constitutively unstable mRNA (14, 36). We have employed the tetracycline (tet)-off system to control transcription of transgenic KC mRNA enabling the analysis of mRNA decay in the absence of the requirement for a transcriptional stimulus (28, 37). HeLa cells stably expressing the tet controlled transactivator are transiently transfected with reporter plasmid (pTRE2 KCΔ3) containing the KC 5′UTR, coding region, and a portion of the 3′UTR that confers both instability and stimulus-induced stabilization. In the absence of tet, or its analogue doxycyline (dox), the tet transactivator will drive strong transcription of the KC transgene, but following addition of dox to the culture medium transcription is abolished and the decay of the mRNA can be followed (Fig. 1A). Although KC mRNA rapidly decays following addition of dox, reflecting the constitutive instability of the mRNA, addition of IL-17 along with dox is able to prolong the half-life of the message.
Figure 1. Over-expression of TRAF6 does not induce mRNA stabilization.

A. HeLa tet-off cells were transfected with 2 μg of pTRE2 KCΔ3 and 4 μg of empty vector (pcDNA3) and divided into different treatment groups. Cells were treated with dox (1 μg/μl) alone or in combination with IL-17 (25 ng/ml) for the indicated times. Total RNA was collected and KC and GAPDH levels were determined by northern hybridization. NT represents untreated cells and serves as the zero time point for all treatment conditions. Autoradiographs were quantified using NIH image software and used to calculate half life as described in Materials and Methods (also see C below). B. HeLa tet-off cells were co-transfected with 4 μg of empty vector (pcDNA3) or expression vectors encoding FLAG-tagged Act1 or TRAF6 along with 2 μg of pTRE2 KCΔ3. After overnight culture, dox was added and KC and GAPDH mRNA levels were determined as described in A. C. The autoradiographs from B were quantified and presented graphically as % remaining mRNA versus time. The time at the intercept at 50% remaining RNA was used to determine half life. D. HeLa tet-off cells were co-transfected with 4 μg of empty vector (pcDNA3) or expression vectors encoding FLAG-tagged Act1 or TRAF6 along with 2 μg 5X κB luciferase reporter and .25 μg pTK renilla luciferase (C). Cell lysates were collected 24 hrs after transfection and luciferase activities were determined. Values represent the mean of duplicate samples that have been normalized to renilla. E. Expression levels of Act1 and TRAF6 were determined by western blot using anti-FLAG antibodies. The data are representative of at least 3 independent experiments.
We have previously shown that over-expression of Act1, the adaptor linking directly to the IL-17R, results in enhanced stability of KC mRNA (20). This finding indicates that signals originating from this adaptor are sufficient to initiate the stabilization response. As a first test of the role of TRAF6 in mRNA stabilization we determined if over-expression of this molecule would also be sufficient to engage the mRNA stabilization process. Surprisingly, while over-expression of Act1 stimulates KC mRNA stabilization, over-expression of TRAF6 does not alter the half-life of the message (Fig. 1B,C). Although TRAF6 over-expression did not affect KC mRNA stability it promoted strong activation of NFκB as measured by a co-transfected luciferase reporter (Fig. 1D) demonstrating the functional activity of the over-expressed molecule. Western blot analysis indicated that each of the adaptor proteins was expressed (Fig. 1E). These observations indicate that signals originating from Act1 but not TRAF6 are sufficient for promoting enhanced mRNA stability.
TRAF6 is not required for IL-17- induced mRNA stabilization
Two experimental strategies were employed to determine if TRAF6 was required for IL-17-induced mRNA stabilization. First we tested the ability of a dn version of TRAF6 (31) to interfere with IL-17-induced KC mRNA stabilization. TRAF6 has previously been linked to NFκB activation in response to both IL-17 and IL-1α (26, 33). IL-17 treatment stabilized KC mRNA in HeLa tet-off cells transfected with pTRE2 KC(Δ3) in the presence of either empty vector or an expression vector encoding dnTRAF6 (Fig. 2A). A similar result was obtained in 4 separate experiments, which collectively show that IL-17 prolongs the half life of KC mRNA by a mean value of more than 2 fold in the presence or absence of dnTRAF6 (Fig. 2B). Under the same conditions, even high doses of IL-17 (100 ng/ml) serve as a very weak stimulus for NFκB activation compared to lower doses of IL-1α (10 ng/ml) (Fig. 2C). Consistent with prior reports, however, NFκB activation following IL-17 or IL-1α treatment was diminished in cells expressing the dnTRAF6.
Figure 2. Dominant negative TRAF6 does not block IL-17-induced mRNA stabilization.

A. HeLa tet-off cells were transfected with 2 μg of pTRE2 KCΔ3, 2 μg of 5X κB luciferase reporter, 1 μg of pcDNA3, and 1μg of dn TRAF6 or empty vector (pcDNA3). One set of cultures were treated with dox alone or in combination with IL-17 (25 ng/ml) for the indicated times. Total RNA was collected and KC and GAPDH mRNA levels were determined by northern hybridization. NT represents untreated cells and serves as the zero time point for all treatment conditions. B. The autoradiographs from 4 separate experiments similar to A were quantified as described in the legend to figure 1 and half-lives for each condition determined. The Mean +/− 1 S.D. for each condition were determined and are presented. C. A second set of cultures transfected as in A were treated for 6 hrs with IL-1α (10 ng/ml), IL-17 (100ng/ml) or left untreated. Cell lysates were prepared and luciferase activity was determined. Values represent the mean of duplicate samples.
As a second test for the requirement of TRAF6, we compared the ability of IL-17 to induce mRNA stabilization in wild type and TRAF6-deficient MEFs. We have previously shown that KC mRNA transcribed in response to TNFα is highly unstable and decays rapidly following the addition of actinomycin D (ActD) while the addition of IL-17 along with ActD is able to prolong the half-life of the TNFα-induced KC mRNA (20). The ability of IL-17 treatment to prolong the half-life of KC mRNA remains intact in the TRAF6-deficient cells (Fig 3A, B). Similar results were obtained in 2 additional experiments as demonstrated by comparison of mean half-life values in the two different cell populations either with or without IL-17 treatment (Fig 3C). It is noteworthy that KC mRNA instability was reduced in the TRAF6−/− cells even in the absence of IL-17 but treatment with IL-17 resulted in a quantitatively comparable increase in half-life in both wild type and TRAF6-deficient cells. These results support the conclusion that TRAF6 is not required for IL-17-induced chemokine mRNA stabilization.
Figure 3. TRAF6 is not required for IL-17-induced mRNA stabilization.

A. Wild type and TRAF6 deficient MEFs were treated for 1 hr with TNFα (10 ng/ml). Fresh media was then added containing ActD (5 μg/ml) alone or with IL-17 (10 ng/ml). Total RNA was collected at the indicated times and KC and GAPDH levels were determined by northern hybridization. NT represents cells treated with TNFα for 1 hr and serves as the zero time point for all treatment conditions. B. The autoradiographs in A were quantified as described in the legend to figure 1 and the % remaining KC mRNA relative to GAPDH is shown for each experimental condition. C. Data from three independent experiments were analyzed for KC mRNA half-life and the mean +/− 1 S.D. are shown.
IL-17-induced mRNA stabilization does not require p38 or MK2/MK3
p38 MAP kinase and its downstream kinase MK2 have been shown to play a critical role in stimulus-induced stabilization of pro-inflammatory mRNAs in a variety of settings (11, 12, 15–17) including IL-17. TRAF6 is known to link to p38 activation and thus the finding that TRAF6 was not required for IL-17-induced mRNA stabilization raised questions regarding the role of the p38 MAP kinase cascade in the IL-17 response. As a first test to assess the role of p38 we determined the effect of the p38 MAP kinase inhibitor SB203580 on IL-17-induced KC mRNA stabilization. The presence of the inhibitor did not affect the ability of IL-17 to stabilize KC mRNA in HeLa tet-off cells (Fig. 4A). In contrast, as we have previously reported (38, 39), the ability of LPS to stabilize KC mRNA in mouse macrophages is highly sensitive to the inhibitory effects of SB203580 (Fig 4B).
Figure 4. IL-17-induced mRNA stabilization does not require p38 or MK2 activity.
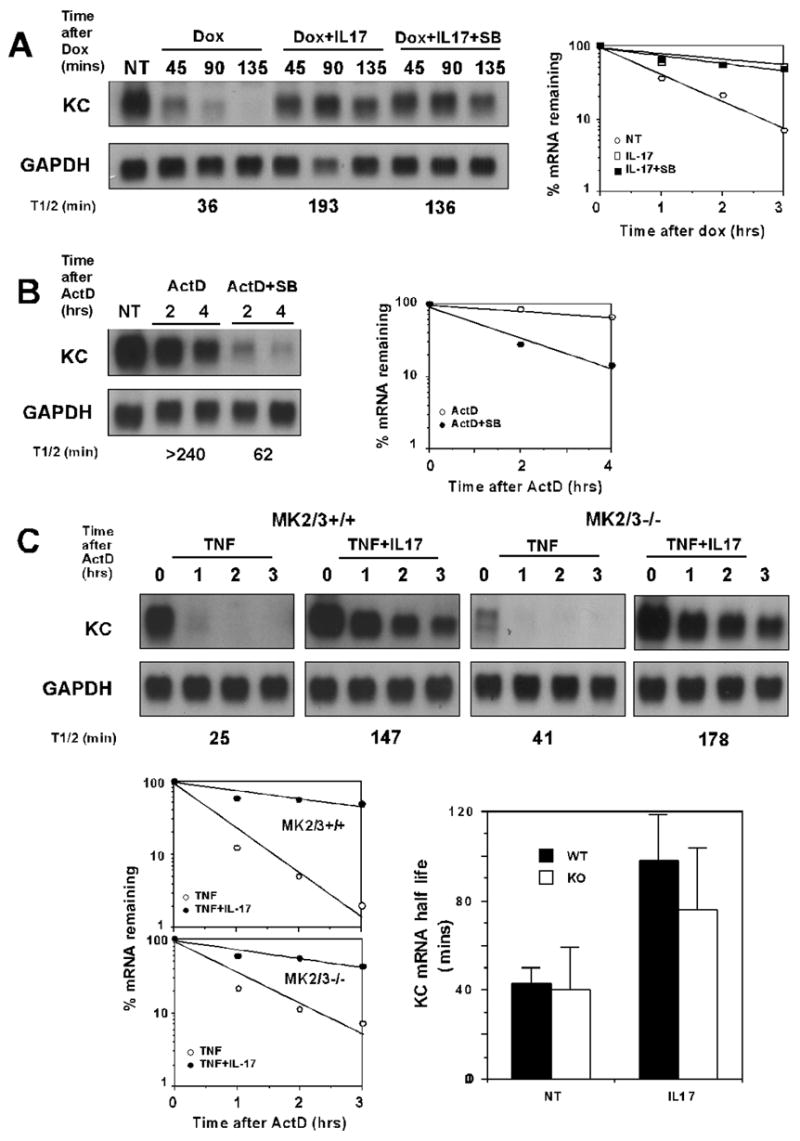
A. HeLa tet-off cells were transfected with 2 μg of pTRE2 KCΔ3 and 4 μg of pcDNA3. Cells were treated with dox alone or with dox plus IL-17 (25 ng/ml) with or without SB203580 (2 μM) for the indicated times. NT represents untreated cells and serves as the zero time point for all treatment conditions. Total RNA was collected and the KC and GAPDH levels were determined by northern hybridization. Blots were quantified as described in the legend to figure 1 and decay curves are presented. B. Thioglycollate elicited peritoneal macrophages were treated with LPS for 3 hrs before the addition of ActD (5 μg/ml) alone or along with SB203580. NT represents cells treated with LPS for 3 hrs and serves as the zero time point for both treatment conditions. Total RNA was prepared at the indicated times and KC and GAPDH mRNA levels were determined by northern hybridization and quantified as above. C. Wild type and MK2/MK3 deficient MEFs were stimulated with TNFα (10 ng/ml) alone or with IL-17 (10 ng/ml) for 1 hr followed by the addition of ActD. Total RNA was collected at the indicated times following addition of ActD and KC and GAPDH mRNA levels were determined by northern hybridization and quantified as above. NT represents cells treated with TNFα for 1 hr and serves as the zero time point for both treatment conditions. The results from three separate experiments were used to determine the mean half-life of KC mRNA +/− 1 S.D. as shown.
To further assess the role of the p38/MK2 pathway in IL-17-induced stabilization we tested the ability of IL-17 to prolong KC mRNA half life in MEFs from MK2/MK3 deficient mice. Wild type and MK2/MK3-deficient MEFs were stimulated with TNFα alone or with IL-17 for 1 hour followed by the addition of ActD. The TNFα-induced KC mRNA was unstable in both cell populations, although the magnitude of response to TNFα was markedly reduced in MK2/3−/− cells (Fig. 4C). However, in both cell populations the half life of KC message induced by the combination of TNFα and IL-17 was significantly and comparably prolonged in comparison to that seen in cells stimulated by TNFα alone (Fig. 4C). Comparable results were obtained in 3 similar experiments as demonstrated by comparison of the mean half-life values (Fig 4C). These findings provide further support for the conclusion that IL-17-induced mRNA stabilization does not require the p38 MAP kinase pathway including MK2 and MK3.
TRAF6 is not required for IL-17 mediated amplification of TNFα-induced gene expression
Prior reports demonstrate that the modest ability of IL-17 alone to induce activation of NFκB and the transcription of select target genes is dependent upon TRAF6 (22, 26). The present study, however, shows that IL-17-induced stabilization of chemokine mRNA transcribed in response to TNFα is TRAF6-independent. The requirement for TRAF6 in NFκB activation but not mRNA stabilization enables a comparison of the relative contribution of the two mechanisms by examining a broader spectrum of IL-17-induced genes in TRAF6-deficient cells. Using quantitative real time PCR we determined the expression a set of 5 genes that have been previously identified as sensitive to IL-17-mediated amplification based upon oligonucleotide microarray analysis of gene expression in fibroblasts stimulated with TNFα alone or with IL-17 (5, 20). Wild type and TRAF6-deficient MEFs were stimulated for 3 or 9 hours with TNFα alone or in combination with IL-17. For comparison the same treatments were also performed on Act1+/− and Act1−/− MEFs since IL-17-induced gene expression has previously been reported to be fully dependent on Act1 (21). In 4 out of the 5 genes studied the response to IL-17 in the TRAF6-deficient MEFs was comparable to that seen in wild type cells (Fig 5). A single dramatic exception was observed for behavior of the gene encoding cxcl5 (LIX). LIX mRNA levels, however, while modestly induced in response to TNFα alone in wild type cells, were below the level of detection in all samples from TNFα-treated TRAF6-deficient cells. Hence there appears to be a deficiency in TNFα-stimulated LIX gene transcription in the TRAF6-deficient cell population rather than a requirement for TRAF6 in IL-17-mediated enhancement of LIX expression. Although IL-6 mRNA levels are reproducibly higher in the TRAF6−/− cells than in wild type cells, it is clear that in both cell populations IL-17 is able to amplify the TNFα induced response. These results are in dramatic contrast to the behavior of the same genes in Act1-deficient MEFs where the response to IL-17 is completely lost. The magnitude of responses in the different MEF cell populations (wild type, TRAF6−/−, Act1+/−, and Act1−/−) show variability but the effects of IL-17 are qualitatively similar for the TRAF6 pair and qualitatively different for the Act1 pair.
Figure 5. TRAF6 is not required for IL-17-mediated amplification of TNFα-induced gene expression.
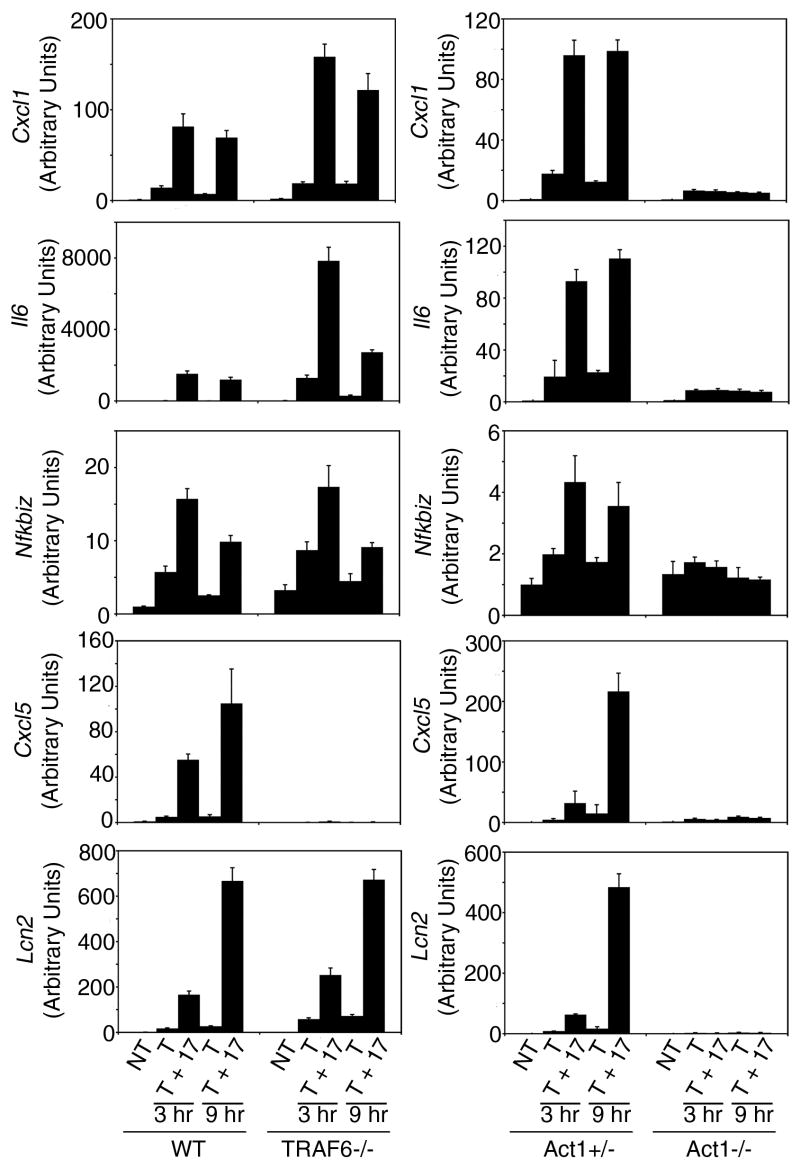
MEFs from wild type, TRAF6−/−, Act1+/−, or Act1−/− mice were left untreated or were stimulated with TNFα (10 ng/ml) alone or in combination with IL-17 (10 ng/ml). Total RNA was isolated following treatment for 3 or 9 hrs and relative mRNA levels for the indicated genes were determined by quantitative real-time PCR. Results are presented as the mean +/− 1/2 the range of duplicate determinations. Similar results were obtained in 2 separate experiments.
To further assess the IL-17-mediated amplification of TNFα-stimulated gene expression in the presence or absence of TRAF6 we compared the half-lives of two additional mRNAs (MIP-2 and IκBζ). As was seen for KC, the half-lives in TNFα-stimulated cells are short and markedly prolonged for both mRNAs in the presence of IL-17 (Fig.6 A and B). As seen with KC mRNA, IL-17 stabilizes the MIP-2 message effectively even when added in the presence of ActD in both cell populations. For IκBζ, mRNA, we compared decay following stimulation with TNFα either alone or in combination with IL-17 because starting mRNA levels were significantly higher. Though the levels of IκBζ mRNA in cells treated with TNFα alone are low and our measure of half-life may be an underestimate, the results demonstrate substantive stabilization of the IκBζ mRNA in the presence of IL-17 and this response is comparable in both cell populations.
Figure 6. Stabilization of MIP-2 and IκBζ mRNAs by IL-17 does not require TRAF6.

A. MEFs from wild type and TRAF6−/− mice were treated with TNFα for 2 hrs prior to addition of ActD (5 μg/ml) or ActD+IL17 (25 ng/ml). MIP-2 and GAPDH mRNA levels were determined after the indicated times by northern hybridization. NT represents cells treated with TNFα for 2 hrs and serves as the zero time point for all treatment conditions. The autoradiographs were quantified as described in the legend to figure 1 and the decay curves for each cell population are shown. B. MEFs from wild type and TRAF6−/− mice were treated with TNFα (10 ng/ml) alone or in combination with IL-17 (25 ng/ml) for 2 hrs. ActD (5 μg/ml) was added to all cultures and the levels of IκBζ and GAPDH mRNAs were determined by northern hybridization after the indicated incubation times. Zero time corresponds to cells treated for 2 hrs with TNFα alone or with IL-17. The autoradiographs were quantified as described in the legend to figure 1 and the decay curves for each cell population are shown. Similar results were obtained in two separate experiments.
DISCUSSION
IL-17-induced inflammatory gene expression has been linked with both transcriptional and post-transcriptional mechanisms though the signaling pathways that couple IL-17 receptor engagement with such responses remain poorly understood. In the present study we undertook to determine if signaling events linked with the activation of NFκB via TRAF6 were also requisite for the prolongation of specific mRNA half life. The data presented establish that the ability of IL-17 to stabilize a selection of TNFα-induced mRNAs does not require the participation of TRAF6. This conclusion is based upon three complementary experimental observations. First, though over-expression of TRAF6 can stimulate the activation of NFκB, it cannot stimulate mRNA stabilization. Second, a dominant negative version of TRAF6 can effectively block activation of NFκB, but does not block mRNA stabilization in response to stimulation with IL-17. Finally, IL-17 remains fully capable to stabilize TNFα-induced KC, MIP-2, and IκBζ mRNAs in TRAF6-deficient cells. Hence the current findings identify a pathway distinct from NFκB activation that leads from the IL-17 receptor to enhanced stability of specific mRNAs.
TRAF6 has been shown to be involved in TLR-mediated activation of MAP kinases, including p38 through the action of TAK1 (33, 40, 41). p38 MAP kinase and its downstream targets MK2 and MK3 have well-established roles in stimulus-induced mRNA stabilization and have been reported to participate in such responses to IL-17 (11, 12, 15–17, 23, 42). The observation that TRAF6, however, was not necessary for the stabilization of KC and MIP-2 mRNAs in response to IL-17 in the present studies raised the possibility that this response did not involve the action p38. More direct tests of this hypothesis using a pharmacologic inhibitor of p38 or cells genetically deficient in MK2 and MK3 further support the conclusion that IL-17-induced stabilization does not require the p38/MK2 pathway. Thus although KC mRNA stabilization in response to TLR ligands requires p38 in mouse macrophages (38, 39), the response to IL-17 in the non-myeloid cell populations studied here is both p38- and MK2/3-independent. The differences between the current findings and prior work both in TLR- and IL-17-induced mRNA stabilization are likely to reflect distinct mechanisms that may function differentially depending upon the cell type. In this regard, we have recently reported that IL-1α-induced stabilization of KC and MIP-2 mRNA is also independent of both TRAF6 and the p38/MK2 pathway in these same cell populations (35). Collectively, these findings suggest a common pathway downstream of Act1 and IRAK1 that mediates mRNA stabilization in non-myeloid cells in response to IL-17 and IL-1α respectively.
The TRAF6/p38 pathway is believed to promote the stabilization of AU rich mRNAs by targeting Tristetraprolin, an RNA binding protein that promotes rapid decay of mRNAs containing multiple copies of the pentameric sequence AUUUA (43–45). Indeed, KC mRNA decay (and LPS-mediated stabilization) in peritoneal macrophages depend upon this mechanism (38). It is noteworthy, however, that IL-17- and IL-1α-mediated stabilization of KC in non-myeloid cells does not require TTP and depends upon a TTP-insensitive sequence in the 3′UTR that contains no AUUUA motifs (Datta et al, manuscript in preparation). This different sequence requirement provides an additional criterion distinguishing this IL-17 signaling pathway.
The variable dependence of responses to IL-17 on TRAF6 appears to relate directly to the mechanism involved; activation of NFκB requires TRAF6 while stabilization of mRNA does not. Thus the assessment of several well recognized IL-17 inducible mRNAs for dependence on TRAF6 provides some measure of the relative contribution of the two mechanistic pathways. It should be noted however, that our study is limited to examining the response to IL-17 in the context of co-stimulation with TNFα. Because TNFα is a potent transcriptional activator but a poor stimulus for mRNA stabilization, this feature of our experimental design focuses attention on the mRNA stabilization pathway by overriding the requirement for transcriptional stimulation. The results demonstrate that many of the IL-17-mediated responses are TRAF6-independent and are, therefore, likely to have some dependence upon the mRNA stabilization pathway. It is, however, worth considering whether transcriptional verus post-transcriptional mechanisms are involved in controlling the expression of each of these mRNAs. Of the 5 TRAF6-independent responses demonstrated in figures 5 and 6, three clearly appear to involve mRNA stabilization: KC (CXCL1), MIP-2, and IκBζ (20, 24). IL-6 is known to be unstable and sensitive to mRNA stabilization (16) but the IL-17 response may also utilize an indirect transcriptional mechanism via the induction of cEBPβ and cEBPδ (46, 47). Lipocalin 2 (Lcn2) has been shown to be inducible by the combination of TNFα and IL-17 (5) and involves transcription but not mRNA stabilization (48). Interestingly, several recent reports demonstrate convincingly that Lcn2 expression is dependent upon both NFκB and the expression of IκBζ (49, 50). This latter finding along with prior work (24) and that reported here, suggests that transcriptional induction of the Lcn2 gene by IL-17 represents a downstream consequence of the stabilization of IκBζ. Thus it appears that multiple mechanisms are contributing to the pattern of enhanced gene expression in IL-17-stimulated cells through both direct and indirect means.
Previous reports showing that the response to IL-17 is dependent upon TRAF6 were focused on the action of IL-17 alone (18, 26) while those reported here reflect the IL-17-mediated amplification of response to TNFα. It is noteworthy that the former responses are, in fact, relatively modest in magnitude while the combination of TNFα and IL-17 often induces robust increases in gene expression that are much greater than the response to either cytokine alone (see fig 5) (5, 20, 21). It seems unlikely that IL-17 will be encountered alone at inflammatory sites in vivo, and hence the cooperative effects seen with TNFα are more likely representative of physiologic responses. The finding that most of the cooperative responses that we assessed were TRAF6-independent suggests that the mRNA stabilization pathway is at least one major mechanism by which IL-17 signals the amplification of inflammatory gene expression. Because this mechanism appears to function in a cell type dependent fashion, there may be circumstances in vivo where TRAF6 dependent, non-cooperative IL-17-mediated responses occur and this remains to be fully explored.
Footnotes
This work was supported by USPHS grants CA39621 and CA62220.
The following abbreviations were used: Act1: NFκB activator 1, TRAF6: TNF receptor associated factor 6, Mouse CXCL1: KC, Mouse CXCL2: MIP-2, MAKAP2/3: MK2/3, Tet: tetracycine, Dox: doxycycline, Lcn2: lipocalin 2
LITERATURE CITED
- 1.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton TA. Molecular Basis of Macrophage Activation: From Gene Expression to Phenotypic Diversity. In: Lewis C, Burke B, editors. The Macrophage. 2. Oxford University Press; Oxford: 2002. pp. 73–102. [Google Scholar]
- 7.Saklatvala J, Dean J, Clark A. Control of the expression of inflammatory response genes. Biochem Soc Symp. 2003:95–106. doi: 10.1042/bss0700095. [DOI] [PubMed] [Google Scholar]
- 8.Fan J, Heller NM, Gorospe M, Atasoy U, Stellato C. The role of post-transcriptional regulation in chemokine gene expression in inflammation and allergy. Eur Respir J. 2005;26:933–947. doi: 10.1183/09031936.05.00120204. [DOI] [PubMed] [Google Scholar]
- 9.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 10.Seko Y, Cole S, Kasprzak W, Shapiro BA, Ragheb JA. The role of cytokine mRNA stability in the pathogenesis of autoimmune disease. Autoimmun Rev. 2006;5:299–305. doi: 10.1016/j.autrev.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 12.Holtmann H, Winzen R, Holland P, Eickemeier S, Hoffmann E, Wallach D, Malinin NL, Cooper JA, Resch K, Kracht M. Induction of Interleukin-8 Synthesis Integrates Effects on Transcription and mRNA Degradation from at Least Three Different Cytokine- or Stress-Activated Signal Transduction Pathways. Mol Cell Biol. 1999;19:6742–6753. doi: 10.1128/mcb.19.10.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoeckle MY. Post-transcriptional regulation of groα, β, γ, and IL-8 mRNAs by IL-1β. Nucleic Acids Res. 1991;19:917–920. doi: 10.1093/nar/19.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tebo J, Datta S, Kishore R, Kolosov M, Major JA, Ohmori Y, Hamilton TA. IL-1-mediated stabilization of mouse KC mRNA depends on sequences in both 5′ and 3′ untranslated regions. J Biol Chem. 2000;275:12987–12993. doi: 10.1074/jbc.275.17.12987. [DOI] [PubMed] [Google Scholar]
- 15.Winzen R, Kracht M, Ritter B. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969– 4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- 17.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337–5344. [PubMed] [Google Scholar]
- 19.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273:27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 20.Hartupee J, Lu C, Novotny M, Li X, Hamilton TA. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 21.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 22.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 23.Faour WH, Mancini A, He QW, Di Battista JA. T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 3′-untranslated region of COX-2 mRNA. J Biol Chem. 2003;278:26897–26907. doi: 10.1074/jbc.M212790200. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki S, Muta T, Matsuo S, Takeshige K. Stimulus-specific induction of a novel nuclear factor-kappaB regulator, IkappaB-zeta, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem. 2005;280:1678–1687. doi: 10.1074/jbc.M409983200. [DOI] [PubMed] [Google Scholar]
- 25.Ohmori Y, Hamilton TA. Cell type and stimulus specific regulation of chemokine gene expression. Biochem Biophys Res Commun. 1994;2:590–596. doi: 10.1006/bbrc.1994.1086. [DOI] [PubMed] [Google Scholar]
- 26.Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1240. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong DA, Major JA, Chudyk A, Hamilton TA. Neutrophil chemoattractant genes KC and MIP-2 are expressed in different cell populations at sites of surgical injury. J Leukoc Biol. 2004;75:641–648. doi: 10.1189/jlb.0803370. [DOI] [PubMed] [Google Scholar]
- 28.Datta S, Novotny M, Li X, Tebo J, Hamilton TA. Toll IL-1 receptors differ in their ability to promote the stabilization of adenosine and uridine-rich elements containing mRNA. J Immunol. 2004;173:2755–2761. doi: 10.4049/jimmunol.173.4.2755. [DOI] [PubMed] [Google Scholar]
- 29.Novotny M, Datta S, Biswas R, Hamilton T. Functionally independent AU-rich sequence motifs regulate KC (CXCL1) mRNA. J Biol Chem. 2005;280:30166–30174. doi: 10.1074/jbc.M502280200. [DOI] [PubMed] [Google Scholar]
- 30.Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276:27657–27662. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 31.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 32.Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartupee J, Li X, Hamilton T. Interleukin 1alpha-induced NFkappaB activation and chemokine mRNA stabilization diverge at IRAK1. J Biol Chem. 2008;283:15689–15693. doi: 10.1074/jbc.M801346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct kappaB sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. J Immunol. 1995;155:3593–3600. [PubMed] [Google Scholar]
- 37.Biswas R, Datta S, Gupta JD, Novotny M, Tebo J, Hamilton TA. Regulation of chemokine mRNA stability by lipopolysaccharide and IL-10. J Immunol. 2003;170:6202–6208. doi: 10.4049/jimmunol.170.12.6202. [DOI] [PubMed] [Google Scholar]
- 38.Datta S, Biswas R, Novotny M, Pavicic P, Herjan T, Mandal P, Hamilton TA. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J Immunol. 2008;180:2545– 2552. doi: 10.4049/jimmunol.180.4.2545. [DOI] [PubMed] [Google Scholar]
- 39.Dai Y, Datta S, Novotny M, Hamilton TA. TGFbeta inhibits LPS-induced chemokine mRNA stabilization. Blood. 2003;102:1178–1185. doi: 10.1182/blood-2002-12-3771. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 41.Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999;13:1297–1308. doi: 10.1101/gad.13.10.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henness S, van Thoor E, Ge Q, Armour CL, Hughes JM, Ammit AJ. IL-17A acts via p38 MAPK to increase stability of TNF-alpha-induced IL-8 mRNA in human ASM. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1283–1290. doi: 10.1152/ajplung.00367.2005. [DOI] [PubMed] [Google Scholar]
- 43.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandler H, Stoecklin G. Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans. 2008;36:491–496. doi: 10.1042/BST0360491. [DOI] [PubMed] [Google Scholar]
- 46.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, Gaffen SL. Functional cooperation between interleukin-17 and tumor necrosis factor-alpha is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 48.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 49.Kayama H, V, Ramirez-Carrozzi R, Yamamoto M, Mizutani T, Kuwata H, Iba H, Matsumoto M, Honda K, Smale ST, Takeda K. Class-specific regulation of pro-inflammatory genes by MyD88 pathways and IkappaBzeta. J Biol Chem. 2008;283:12468–12477. doi: 10.1074/jbc.M709965200. [DOI] [PubMed] [Google Scholar]
- 50.Yamazaki S, Matsuo S, Muta T, Yamamoto M, Akira S, Takeshige K. Gene-specific requirement of a nuclear protein, Ikappa B-zeta, for promoter association of inflammatory transcription regulators. J Biol Chem papers in press. 2008 Sept 29; doi: 10.1074/jbc.M802148200. [DOI] [PubMed] [Google Scholar]