

Abstract
The A-type potassium channel subunit Kv4.2 influences hippocampal function through regulation of dendritic excitability, and changes in Kv4.2 surface expression alter synaptic plasticity. Recent data from our laboratory demonstrate that EGFP (enhanced green fluorescent protein)-tagged Kv4.2 channels located in dendritic spines are internalized in an activity-dependent manner after synaptic stimulation and during chemically induced long-term potentiation. However, the molecular trigger for Kv4.2 internalization remains unknown. Here we examined the role of protein kinase A (PKA) in Kv4.2 activity-dependent trafficking. In hippocampal neurons, PKA activation with forskolin or 8-Br-cAMP induced Kv4.2 internalization from dendritic spines, whereas PKA inhibition with H89 prevented AMPA-induced internalization. Furthermore, introduction of a point mutation at the C-terminal PKA phosphorylation site of Kv4.2 (S552A) prevented the AMPA-induced internalization of Kv4.2. Together, these data demonstrate that Kv4.2 activity-dependent internalization requires PKA phosphorylation of Kv4.2 at serine 522.
Keywords: potassium channel, IA, Kv4.2, trafficking, hippocampus, excitability, PKA
Introduction
Potassium channels play an important role in regulating neuronal excitability and synaptic plasticity in the hippocampus. In particular, voltage-gated Kv4.2 channels underlie a transient A-type K+ current (IA) that modulates dendritic function (Jerng et al., 2005; Kim and Hoffman, 2008). These channels are localized to the somatodendritic compartment of neurons, with Kv4.2 channel density increasing with distance from the soma (Hoffman et al., 1997; Varga et al., 2000). Activation of A-type K+ channels limits the spread of backpropagating action potentials (bp-APs) into the distal dendrites (Hoffman et al., 1997). This Kv4.2 function limits spike-timing-dependent plasticity, which requires the coincidence of an EPSP and a bp-AP (Markram et al., 1997; Bi and Poo, 1998). Consistently, genetic knockdown of IA boosts dendritic Ca2+ transients from bp-APs (Kim et al., 2005; Chen et al., 2006), and the pharmacological downregulation of IA enhances spike-timing-dependent long-term potentiation (LTP) (Watanabe et al., 2002). Therefore, Kv4.2 channels are key modulators of intrinsic excitability and synaptic plasticity, and downregulation of Kv4.2 currents enhances these processes.
Activity-dependent internalization is one common mechanism for channel downregulation. In fact, after depolarization with AMPA treatment, Kv4.2 channels are trafficked out of dendritic spines in cultured neurons (Kim et al., 2007). Furthermore, Kv4.2 internalization occurs during chemically induced LTP in vitro, suggesting that Kv4.2 trafficking could be an important mechanism for the coregulation of intrinsic excitability and synaptic plasticity. These findings have raised a number of questions about the mechanisms and function of Kv4.2 channel trafficking in hippocampal neurons.
Although Kv4.2 internalization after AMPA or KCL stimulation is NMDA receptor (NMDAR) and Ca2+ dependent (Kim et al., 2007), the downstream pathways mediating internalization are unknown. One possibility is that direct phosphorylation of Kv4.2 α-subunits by protein kinase A (PKA) stimulates channel internalization during synaptic potentiation. Multiple studies have demonstrated a role for PKA in hippocampal synaptic plasticity (Blitzer et al., 1995; Otmakhova et al., 2000; Nguyen and Woo, 2003). In cultured neurons, activation of PKA with forskolin (FSK) produces LTP of evoked EPSCs (Sokolova et al., 2006) through multiple postsynaptic mechanisms, including the synaptic insertion of AMPA receptors (Esteban et al., 2003) and the internalization of small-conductance Ca2+-activated potassium channels (Lin et al., 2008). In addition, PKA downregulates Kv4.2 currents (Schrader et al., 2002), and increases the amplitude of bp-APs in hippocampal dendrites (Hoffman and Johnston, 1998). Therefore, it is possible that during synaptic potentiation, membrane-bound Kv4.2 channels are phosphorylated by PKA and internalized to enhance dendritic excitability and synaptic plasticity. Here, we tested this hypothesis pharmacologically and with site-directed mutagenesis of the Ser552 PKA phosphorylation site. We found that PKA phosphorylation of Kv4.2 at Ser552 is necessary for the activity-dependent internalization of Kv4.2.
Materials and Methods
Neuronal cultures and Sindbis viral expression system.
All constructs were expressed in primary hippocampal neurons cultured from embryonic day 18 Sprague Dawley rat embryos as by Kim et al. (2007) using a modified Sindbis viral expression system (Kim et al., 2004). The Kv4.2g construct and virus was made previously (Kim et al., 2007) and was generated by attaching an enhanced green fluorescent protein (EGFP) tag onto the intracellular C terminus of the wild-type Kv4.2 channel. Extracellularly myc-tagged Kv4.2 (Kv4.2myc) construct was obtained from Rivera et al. (2003) and inserted into the Sinrep(nsP2S726) viral vector using its XbaI and XhoI restriction sites. Mutation of the C-terminal PKA phosphorylation site on the Kv4.2g construct (to produce the Kv4.2S552A construct) was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). Primers 5′-GCCATAGAGGCGCTGTGCAAGAACTC and 5′-GAGTTCTTGCACAGCGCCTCTATGGC were used to mutagenize serine to alanine at site 552. Mutations were confirmed by sequencing analysis. Kv4.2myc and Kv4.2S552A Sindbis virus were produced as by Kim et al. (2005). Neurons [15–18 d in vitro (DIV)] were infected by incubating with Kv4.2g, Kv4.2myc, or Kv4.2gS552A Sinrep(nsP2S726) virus for 1 h at 37°C to generate ∼10–20% infection efficiency.
Immunofluorescence staining.
Twenty-four hours after infection with Kv4.2g or Kv4.2gS552A, neurons were treated with 50 μm AMPA (Sigma) or 10 μm forskolin (EMD Biosciences) for 15 min at 37°C. Cells were fixed with PBS containing 4% paraformaldehyde, 0.1% gluteraldehyde, and 0.12 m sucrose for 10 min, and permeabilized with 0.1% Triton X-100 in PBS for 5 min. Neurons were stained for F-actin using 0.5 μm tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin (Sigma) in PBS + 0.1% BSA for 15 min at room temperature, then washed three times in PBS and mounted on glass slides with Mowiol.
Immunofluorescence internalization assay.
Surface-expressed Kv4.2myc channels were labeled with anti-myc antibody (9E10; 1:200; Santa Cruz Biotechnology) for 1 h at 37°C. Kv4.2myc internalization was stimulated with 100 μm AMPA (Sigma), 10 μm forskolin (EMD Biosciences), or 100 μm 8-Br-cAMP (Sigma) for 15 min at 37°C. Surface-remaining Kv4.2myc channels were labeled with Alexa 555-conjugated (red) anti-mouse antibody (1:100; Invitrogen) for 30 min at 37°C. Cells were washed for 5 min in PBS at 37°C, then fixed and permeabilized as described above. Internalized Kv4.2myc channels were labeled with Alexa 488-conjugated (green) anti-mouse antibody (1:500; Invitrogen) in PBS containing 5% NGS, 0.05% Triton X-100, and 450 mm NaCl, for 90 min at 37°C. Cells were washed three times in PBS and mounted onto glass slides with Mowiol.
Microscopy and image analysis.
Images were collected on a Leica TCS SP2 RS laser-scanning confocal microscope with a 40× oil-immersion objective and acquired using Leica confocal software version 2.6. All image analysis was performed with MetaMorph version 6.3 (Universal Imaging) on z-compressed image stacks. Kv4.2g-containing spines were determined as by Kim et al. (2007). To analyze Kv4.2myc internalization, integrated intensities of green and red fluorescence were measured within the somatic region (demarcated by hand for each image), and there were no differences in somatic area between groups for each experiment (p > 0.05). The intensity ratio was calculated as the integrated intensity of green signal/the total integrated intensity (red + green signals). All data reported are mean ± SEM, and n values are representative of the number of neurons per group unless otherwise stated. All data were analyzed with ANOVA and Tukey's posttest comparisons using Igor Pro version 5.0 Carbon software (WaveMetrics).
Biotinylation assay.
A standard biotinylation assay was used to detect surface-remaining proteins after treatment. Biotinylation assays were performed either on cultured hippocampal neurons infected with Kv4.2g or on acute hippocampal slices from Sprague Dawley rats (postnatal day 14–16) as by Kim et al. (2007). For both experiments, after treatment, surface proteins were biotinylated with 1.5 mg/ml sulfo-NHS-SS-biotin reagent (Pierce) in PBS for 30 min at 4°C. Unbound biotin was quenched with cold 50 mm glycine in PBS. Neurons were lysed with ice-cold lysis buffer [150 mm NaCl, 20 mm Tris-HCl, 1% NP-40, and protease inhibitor mixture (Roche)], sonicated, and centrifuged at 12,000 × g for 10 min. Cell lysates were incubated overnight at 4°C with immobilized streptavidin agarose beads (Pierce). The bound proteins were eluted with SDS sample buffer. Surface proteins were separated by electrophoresis on 10% Tris-bis SDS polyacrylamide gels (Invitrogen) and transferred to nitrocellulose membranes. Western blots were probed with mouse anti-Kv4.2 (1:2000; NeuroMab) and rabbit anti-GAPDH (1:1000; EMD Biosciences) primary antibodies and with secondary antibodies conjugated to infrared dyes (Rockland Immunochemicals). The Odyssey infrared imaging system (LI-COR Biotechnology) was used for signal detection. Quantification of results was performed using Odyssey software.
Electrophysiology.
Coverslips containing cultured 7–10 DIV rat hippocampal neurons were bathed in artificial CSF containing the following (in mm): 145 NaCl, 5 KCl, 10 glucose, 2 CaCl2, 1.3 MgCl2, and 10 HEPES, pH 7.4, and bubbled with 5% CO2/95% O2. Thick-walled patch electrodes with 3–6 MΩ tip resistances were filled with the following (in mm): 140 KCl, 2 MgCl2, 10 HEPES, 5 EGTA, 5 ATP, 1 CaCl2, and 10 d-glucose, pH 7.3 with KOH. Series resistance ranged between 8 and 18 MΩ. No electronic compensation for series resistance was used. Recordings were performed using a Multiclamp 700B amplifier (Molecular Devices), digitized at 10 kHz, filtered at 2 kHz, and acquired and analyzed with IGOR Pro (Wavemetrics). Peak K+ currents were recorded before and 5 min after treatment with AMPA (50 μm, 5 min), forskolin (10 μm, 5 min), or 8-Br-cAMP (50 μm, 5 min) at +120 mV after a 400 ms prepulse to either −120 mV or +30 mV. For the PKA inhibition experiment, H89 (10 μm) was applied to neurons in culture 1 h before and during recordings. For Kv4.2g and Kv4.2S552A recordings, rat hippocampal neurons were infected with Kv4.2g or Kv4.2S552A Sindbis virus at 6–9 DIV and recorded 24 h later, either in the presence or absence of 10 μm forskolin. Voltage-gated Na+ currents were blocked with 1 μm TTX except during treatment. Transient and sustained K+ currents were digitally separated using the prepulse protocol after the subtraction of leak currents. During and after treatments, resting membrane potential was monitored. Cells that did not recover to near the original resting potential of treatment were not analyzed.
Results
PKA activation is sufficient for Kv4.2 internalization
Activation of PKA in hippocampal neurons downregulates IA and boosts dendritic APs (Hoffman and Johnston, 1998). Recently, we showed that AMPA stimulation of neurons causes redistribution of EGFP-tagged Kv4.2 channels (Kv4.2g, with the EGFP tag on the intracellular C terminus) out of dendritic spines (Kim et al., 2007). To determine the key pathways involved in this process, in the present study we examined the role of PKA activation in activity-dependent Kv4.2 internalization. Activation of PKA with 10 μm forskolin induced Kv4.2g redistribution in hippocampal neurons (Fig. 1). Redistribution was measured as a 40.26 ± 5.96% decrease in green fluorescence intensity within spine regions of interest (demarcated using F-actin staining; p < 0.001), as well as a reduction in the percentage of spines containing Kv4.2g (Fig. 1) (p < 0.001). These effects were similar to those observed after AMPA treatment previously (Kim et al., 2007) and here (Fig. 1). These data demonstrate that, similar to AMPA stimulation, PKA activation initiates the redistribution of Kv4.2g away from spines.
Figure 1.
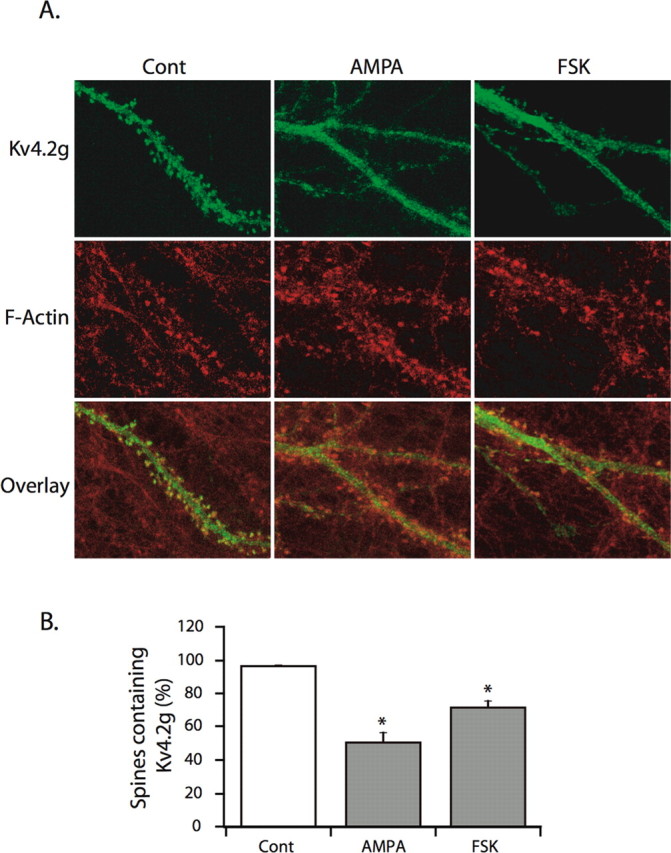
PKA activation induces Kv4.2g redistribution in hippocampal neurons. A, Representative images show that AMPA (50 μm) or FSK (10 μm) treatment causes Kv4.2 redistribution from dendritic spines. B, Summary plot shows a significant decrease in the percentage of spines containing Kv4.2g after AMPA (n = 10 neurons, 1296 spines) or FSK (n = 19 neurons, 2372 spines) treatment relative to control (n = 18 neurons, 2163 spines). All data are mean ± SE. *p < 0.001.
To directly measure internalization of Kv4.2 channels located on the cell surface, we expressed a Kv4.2 construct with an extracellular myc tag (Kv4.2myc) in cultured neurons. Previous characterization of this construct demonstrated that Kv4.2myc is expressed on the surface membrane of COS7 cells, produces a transient K+ current in HEK293 cells, and is localized to the somatodendritic compartment in neurons (Rivera et al., 2003). Here, live neurons expressing Kv4.2myc were surface labeled with anti-myc primary antibody, and internalization was stimulated with AMPA (100 μm) treatment or PKA activation (10 μm forskolin or 100 μm 8-Br-cAMP). Surface remaining channels were then labeled with a red fluorescent secondary antibody. Neurons were then fixed and permeabilized, and internalized channels were labeled with a green fluorescent secondary antibody. As expected, AMPA stimulation triggered Kv4.2myc internalization, measured as an increase in the intensity ratio [calculated as green integrated intensity/total integrated intensity (Fig. 2A,B) (p < 0.001)]. In addition, the PKA agonists forskolin (p < 0.003) and 8-Br-cAMP (p < 0.001) also significantly increased the intensity ratio, indicating that the activation of PKA leads to Kv4.2 internalization (Fig. 2A,B).
Figure 2.

PKA activation elicits Kv4.2myc internalization in hippocampal neurons. A, Representative images show internalized Kv4.2myc channels (green) and surface-remaining Kv4.2myc channels (red) after AMPA (100 μm), FSK (10 μm), or 8-Br-cAMP (100 μm) treatment. B, Summary plot shows a significant increase in the intensity ratio (green integrated intensity/total integrated intensity) after AMPA (n = 11), FSK (n = 16), or 8-Br-cAMP (n = 10) treatments, relative to control (n = 29). C, In cultured hippocampal neurons, endogenous IA peak amplitude is reduced after AMPA (50 μm; n = 12), FSK (10 μm; n = 14), or 8-Br-cAMP (50 μm; n = 18) treatment, and no changes in sustained current peak amplitudes after any treatment were observed (p > 0.30). Open bars, Pretreatment recordings; gray bars, posttreatment recordings. Calibration: 50 pA, 100 ms. D, The cell-surface biotinylation experiment in acute hippocampal slices demonstrates that surface expression of endogenous Kv4.2 is reduced 37 ± 0.09% after FSK (10 μm; n = 3) treatment compared with control (Cont; n = 3). All data are mean ± SE. *p < 0.05.
Evidence that PKA activation induces endogenous Kv4.2 internalization was observed in whole-cell voltage-clamp recordings of cultured neurons. AMPA stimulation reduced IA (p < 0.001) without affecting the sustained K+ current (p = 0.35), consistent with Kv4.2 internalization (Fig. 2C). Similarly, activation of PKA with either forskolin (p < 0.001) or 8-Br-cAMP (p < 0.001) reduced IA without affecting sustained currents (p > 0.30), indicating that PKA activation results in Kv4.2 internalization (Fig. 2C). Biochemical analysis further demonstrated that PKA activation induces internalization of endogenous Kv4.2. A biotinylation assay prepared from acute hippocampal slices revealed a 37 ± 0.09% decrease in surface Kv4.2 after 15 min of forskolin (10 μm) treatment compared with control (Fig. 2D) (p < 0.05). This effect was also observed in cultured hippocampal neurons expressing Kv4.2g (data not shown; p < 0.05). Together, these results show conclusively that PKA activation leads to Kv4.2 internalization in hippocampal neurons.
PKA phosphorylation is necessary for Kv4.2 internalization
To determine whether PKA activation is required for the activity-dependent internalization of Kv4.2, neurons expressing Kv4.2myc were pretreated with the PKA antagonist H89 (10 μm) for 1 h before and during AMPA stimulation (100 μm, 15 min). Treatment with AMPA alone increased the intensity ratio (p < 0.001), indicating Kv4.2 internalization (Fig. 3). However, pretreatment with H89 prevented this increase (p = 0.69) (Fig. 3). Furthermore, intensity ratios between untreated, H89-treated, or H89 + AMPA-treated neurons did not differ (p > 0.05) (Fig. 3), suggesting that PKA activation is necessary for AMPA-induced Kv4.2 internalization. In whole-cell voltage-clamp recordings, pretreatment with H89 (10 μm, n = 3) also prevented the AMPA-induced reduction of endogenous IA (normalized peak amplitude mean ± SE; pre-AMPA, 1 ± 0.18; post-AMPA, 1.29 ± 0.29; p = 0.29; data not shown), and did not affect sustained K+ currents (normalized peak amplitude mean ± SE; pre-AMPA, 1 ± 0.51; post-AMPA, 1.22 ± 0.29; p = 0.57; data not shown). Together with Figure 2, these data indicate that PKA activation is necessary for the activity-dependent internalization of Kv4.2 channels.
Figure 3.
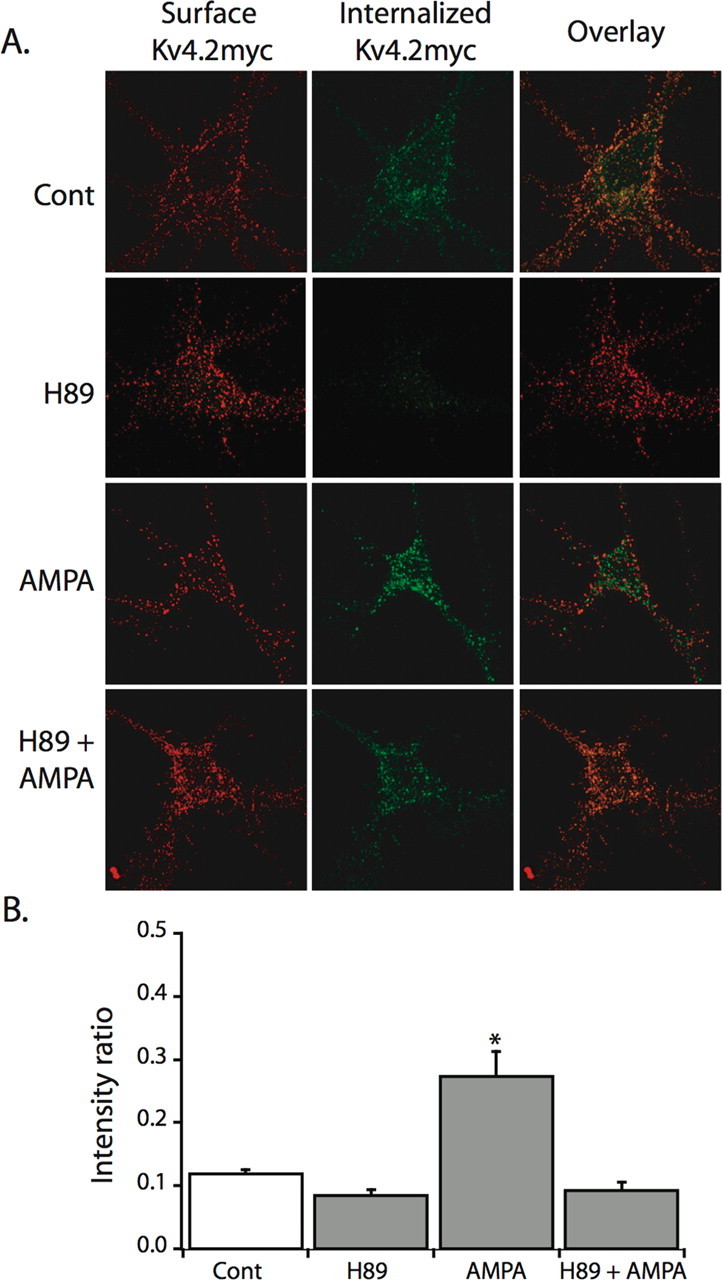
PKA inhibition blocks Kv4.2myc activity-dependent internalization. A, Representative images showing internalized Kv4.2myc channels (green) and surface-remaining Kv4.2myc channels (red) after H89 (10 μm) pretreatment and AMPA (50 μm) stimulation in hippocampal neurons. B, Summary plot shows that the increase in intensity ratio (green integrated intensity/total integrated intensity) after AMPA treatment (n = 14) is prevented by pretreatment with H89 (n = 19). No differences (p > 0.05) were found between control (n = 23), H89 + AMPA (n = 19), and H89 alone (n = 13). All data are mean ± SE. *p < 0.001.
Because PKA phosphorylates multiple postsynaptic targets, we wanted to determine whether the direct phosphorylation of Kv4.2 by PKA is the mechanism of Kv4.2 internalization. Two PKA phosphorylation sites on the Kv4.2 α-subunit have been experimentally confirmed, one N-terminal at Thr38 and one C-terminal at Ser552 (Anderson et al., 2000). Mutagenesis of Ser552 but not Thr38 prevents the forskolin-induced decrease in Kv4.2-mediated K+ currents in oocytes (Schrader et al., 2002). Therefore, we tested whether phosphorylation of Kv4.2 at Ser552 was necessary for activity-dependent Kv4.2 redistribution. Hippocampal neurons expressing EGFP-tagged Kv4.2 with a serine-to-alanine point mutation at site 552 (Kv4.2gS552A) were treated with AMPA for 15 min. No redistribution of Kv4.2gS552A was observed after AMPA stimulation (Fig. 4A), as measured by the percentage of spines containing Kv4.2gS552A (mean ± SEM: control, 93.89 ± 0.87; AMPA, 92.17 ± 1.49; p = 0.33). Compared with the marked redistribution of Kv4.2g observed in Figure 1, these data clearly indicate that the activity-dependent internalization of Kv4.2 channels requires PKA phosphorylation of Kv4.2 at Ser552.
Figure 4.

Kv4.2 internalization requires PKA phosphorylation at Ser 552. A, Representative images show that AMPA (50 μm) does not induce redistribution of Kv4.2gS552A from dendritic spines. AMPA treatment (n = 23 neurons, 1908 spines) had no effect on the percentage of spines containing Kv4.2gS552A (92.17 ± 1.49%), compared with control (93.89 ± 0.87%; n = 25 neurons, 3247 spines; p = 0.33). B, FSK (10 μm; n = 6) significantly reduces the peak current density of IA compared with untreated cultured neurons expressing Kv4.2g (Cont; n = 8; *p < 0.05). This effect was not observed in cultured neurons expressing Kv4.2S552A (Cont, n = 6; FSK, n = 8; p = 0.65). All data are mean ± SE.
To further test this hypothesis, we also recorded IA from hippocampal neurons expressing either Kv4.2g or Kv4.2S552A. Peak current densities of IA were significantly reduced in Kv4.2g-expressing neurons treated with 10 μm forskolin, compared with untreated control neurons expressing Kv4.2g (Fig. 4C) (p < 0.05). However, in neurons expressing Kv4.2S552A (in addition to endogenous Kv4.2), forskolin treatment did not significantly reduce IA density (p = 0.65). Although endogenous Kv4.2 currents are present and caused a detectable forskolin-induced decrease in IA in neurons expressing Kv4.2S552A, this decrease was nonsignificant and was proportionally smaller than the FSK-induced decrease observed in neurons expressing Kv4.2g, indicating that PKA phosphorylation of Kv4.2 at Ser552 is necessary for the activity-dependent internalization of Kv4.2 channels.
Discussion
In the present study, we used an extracellularly myc-tagged Kv4.2 construct expressed in hippocampal neurons to examine the role of PKA in activity-dependent Kv4.2 internalization. Kv4.2 internalization was elicited with PKA activation, and blocked by PKA inhibition. Furthermore, PKA activation reduced IA in hippocampal neurons, consistent with channel internalization. The PKA-induced reduction in IA has also been associated with the boosting of bp-APs (Yuan et al., 2002). In this study, Yuan et al. (2002) suggest that the downregulation of IA is a consequence of Kv4.2 phosphorylation by an extracellular regulated kinase (ERK)-specific pathway downstream of PKA activation. In fact, we have observed that inhibition of ERK/mitogen-activated protein kinase (MAPK) with 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)butadiene (U0126) and 2′-amino-3′-methoxyflavone (PD98059) reduces, but does not prevent, 8-Br-cAMP-induced Kv4.2myc internalization. Using the Kv4.2myc internalization assay, we observed an increase in intensity ratio (indicating Kv4.2myc internalization) after 100 μm 8-Br-cAMP with (n = 14; mean ± SE = 0.21 ± 0.02; p = 0.049) or without (n = 13; mean ± SE = 0.30 ± 0.05; p < 0.001) pretreatment with ERK inhibitors 10 μm U0126 + 10 μm PD98059, compared with control (n = 23; mean ± SE = 0.11 ± 0.01; data not shown). Most likely, ERK signaling is involved in, but not necessary for, Kv4.2 internalization. This may also explain why qualitatively more internalization is observed after AMPA treatment (activating both PKA and ERK/MAPK cascades independently) than after forskolin or 8-Br-cAMP treatment alone (Figs. 1B, 2B).
We have demonstrated here that PKA mediates Kv4.2 trafficking directly through phosphorylation at residue S552 (Fig. 4). This is consistent with Schrader et al. (2002), who found that PKA activation reduces the amplitude of Kv4.2 currents when coexpressed with KChIP3 in oocytes. Consistent with these data and Figure 2D, we also observed that forskolin (10 μm, 15 min) induces the internalization of Kv4.2g, but not Kv4.2S552A (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) when expressed in COS7 cells. Interestingly, these data suggest that KChIP3 may not be involved in PKA-mediated Kv4.2 internalization, because KChIP3 was not coexpressed in this experiment.
Although our data clearly indicate a direct role for PKA in Kv4.2 activity-dependent trafficking, many questions still remain about the precise mechanisms of internalization. To address this, we also examined the role of the Kv4.2 C-terminal dileucine internalization motif in PKA-mediated Kv4.2 internalization. Multiple membrane proteins contain similar C-terminal dileucine motifs that serve as signals for protein targeting and endocytic sorting. For example, the dileucine motif of β2 adrenergic receptors does not mediate basal “default” recycling but is necessary for sequence-directed recycling (Hanyaloglu and von Zastrow, 2008). The dileucine motif on Kv4.2 is important for dendritic targeting of Kv4.2 but does not affect the basal rate of Kv4.2 endocytosis (Rivera et al., 2003). To address whether the Kv4.2 dileucine motif mediates activity-dependent Kv4.2 internalization, we mutated the Kv4.2 C-terminal dileucine residues 481–482 to alanines (Kv4.2LLAA). When expressed in COS7 cells, Kv4.2g but not Kv4.2LLAA is internalized after forskolin application (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Because mutation of the dileucine motif blocked PKA-induced Kv4.2 internalization, these residues are likely to mediate Kv4.2 activity-dependent internalization, although the precise mechanism by which these residues are involved remains unknown.
One possibility is that Kv4.2 channels are tethered to the plasma membrane by associations with scaffolding proteins, and PKA phosphorylation of Kv4.2 disrupts this association. For example, stimulation of PKA causes the rapid dissociation of Kir2.3 K+ channels from PSD-95 (Cohen et al., 1996). To test whether PKA activation similarly disrupts the association between Kv4.2 and PSD-95, we performed coimmunoprecipitation experiments using COS7 cells cotransfected with Kv4.2g and PSD-95. Results indicate that Kv4.2 and PSD-95 are associated, but this link is unchanged after PKA activation (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Although these data do not exclude the possibility that PKA phosphorylation of Kv4.2 disrupts associations with other scaffolding proteins, they suggest that Kv4.2 remains bound to PSD-95 after forskolin-induced internalization.
Another possible mechanism through which PKA may influence Kv4.2 trafficking is through its effects on the Ca2+ permeability of NMDARs. PKA activation enhances the relative Ca2+ influx through NMDARs (Skeberdis et al., 2006), and could thereby increase Kv4.2 internalization, which requires NMDAR activation and intracellular Ca2+ (Kim et al., 2007). Interestingly, we have observed in a separate study (Jung et al., 2007) that Kv4.2 downregulation alters NMDAR subunit composition and enhances NMDAR currents similarly to that seen in (Skeberdis et al., 2006). Therefore, PKA may also indirectly alter NMDAR Ca2+ permeability through changes in NMDAR subunit composition after Kv4.2 internalization, further illustrating the complexity of postsynaptic molecular dynamics during synaptic transmission and plasticity.
In conclusion, this study clearly establishes that the activity-dependent internalization of Kv4.2 channels that occurs during neuronal plasticity (Kim et al., 2007; Kim and Hoffman, 2008) is mediated by PKA. Specifically, PKA phosphorylation of Kv4.2 at Ser552 is necessary for its activity-dependent internalization (Fig. 4; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This process also can involve ERK-specific signaling pathways, and requires a functional Kv4.2 C-terminal dileucine site (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These findings raise many exciting questions about the specific mechanisms of Kv4.2 activity-dependent internalization, and the role of targeted trafficking of Kv4.2 channels in synaptic plasticity.
Footnotes
We thank Dr. Arnold for providing the Kv4.2myc construct and Faith Kung for assistance with constructing the Kv4.2gS552A construct.
References
- Anderson et al., 2000.Anderson AE, Adams JP, Qian Y, Cook RG, Pfaffinger PJ, Sweatt JD. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J Biol Chem. 2000;275:5337–5346. doi: 10.1074/jbc.275.8.5337. [DOI] [PubMed] [Google Scholar]
- Bi and Poo, 1998.Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer et al., 1995.Blitzer RD, Wong T, Nouranifar R, Iyengar R, Landau EM. Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron. 1995;15:1403–1414. doi: 10.1016/0896-6273(95)90018-7. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2006.Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen et al., 1996.Cohen NA, Brenman JE, Snyder SH, Bredt DS. Binding of the inward rectifier K+, channel Kir 2.3 to PSD-95 is regulated by protein kinase A phosphorylation. Neuron. 1996;17:759–767. doi: 10.1016/s0896-6273(00)80207-x. [DOI] [PubMed] [Google Scholar]
- Esteban et al., 2003.Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- Hanyaloglu and von Zastrow, 2008.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- Hoffman and Johnston, 1998.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman et al., 1997.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Jerng et al., 2005.Jerng HH, Kunjilwar K, Pfaffinger PJ. Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J Physiol. 2005;568:767–788. doi: 10.1113/jphysiol.2005.087858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung et al., 2007.Jung S, Kim J, Hoffman DA. The A-type K+ channel subunit Kv4.2 regulates synaptic NMDAR subunit composition through an active CaMKII-dependent mechanism. Soc Neurosci Abstr. 2007;33 362.14. [Google Scholar]
- Kim and Hoffman, 2008.Kim J, Hoffman DA. Potassium channels: newly found players in synaptic plasticity. Neuroscientist. 2008;14:276–286. doi: 10.1177/1073858408315041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim et al., 2004.Kim J, Dittgen T, Nimmerjahn A, Waters J, Pawlak V, Helmchen F, Schlesinger S, Seeburg PH, Osten P. Sindbis vector SINrep(nsP2S726): a tool for rapid heterologous expression with attenuated cytotoxicity in neurons. J Neurosci Methods. 2004;133:81–90. doi: 10.1016/j.jneumeth.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Kim et al., 2005.Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim et al., 2007.Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin et al., 2008.Lin MT, Luján R, Watanabe M, Adelman JP, Maylie J. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci. 2008;11:170–177. doi: 10.1038/nn2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram et al., 1997.Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Nguyen and Woo, 2003.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401, 437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Otmakhova et al., 2000.Otmakhova NA, Ottmakhov N, Mortenson LH, Lisman JE. Inhibition of the cAMP pathway decreases early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 2000;20:4446–4451. doi: 10.1523/JNEUROSCI.20-12-04446.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera et al., 2003.Rivera JF, Ahmad S, Quick MW, Liman ER, Arnold DB. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat Neurosci. 2003;6:243–250. doi: 10.1038/nn1020. [DOI] [PubMed] [Google Scholar]
- Schrader et al., 2002.Schrader LA, Anderson AE, Mayne A, Pfaffinger PJ, Sweatt JD. PKA modulation of Kv4.2-encoded A-type potassium channels requires formation of a supramolecular complex. J Neurosci. 2002;22:10123–10133. doi: 10.1523/JNEUROSCI.22-23-10123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis et al., 2006.Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Sokolova et al., 2006.Sokolova IV, Lester HA, Davidson N. Postsynaptic mechanisms are essential for forskolin-induced potentiation of synaptic transmission. J Neurophysiol. 2006;95:2570–2579. doi: 10.1152/jn.00617.2005. [DOI] [PubMed] [Google Scholar]
- Varga et al., 2000.Varga AW, Anderson AE, Adams JP, Vogel H, Sweatt JD. Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn Mem. 2000;7:321–332. doi: 10.1101/lm.35300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe et al., 2002.Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan et al., 2002.Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]