

Abstract
Aldo-ketoreductase 1C (AKR1C) enzymes catalyze the NADPH-dependent reduction of ketosteroids to hydroxysteroids. They are Phase I metabolizing enzymes for natural and synthetic steroid hormones. They convert 5α-dihydrotestosterone (Dht, potent androgen) to 3α/β-androstanediols (inactive androgens) and the prodrug tibolone (Tib) to estrogenic 3α/β-hydroxytibolones. Herein we demonstrate for the first time that human AKR1C enzymes (AKR1C1-4) are able to reduce conjugated steroids such as Dht-17β-glucuronide (DhtG), Dht-17β-sulfate (DhtS), and Tib-17β-sulfate (TibS). Product identities were characterized by liquid chromatography-mass spectrometry, and kinetic parameters of the reactions were determined. The product profile of the reduction of each steroid conjugate by the individual AKR1C isoform was similar to that of the corresponding free steroid except for the reduction of DhtG catalyzed by AKR1C2, where a complete inversion in stereochemical preference to 3β-reduction (with DhtG) from 3α-reduction (with Dht and DhtS) was observed. The catalytic efficiency of 3-keto reduction was modestly affected by the presence of a 17-sulfate group but severely impaired by the presence of a 17-glucuronide group for AKR1C1-3 isoforms. AKR1C4, however, showed superior catalytic efficiencies versus the other isoforms, and those were unaffected by steroid conjugation. Our findings provide evidence for alternative pathways of steroid metabolism where the phase I reaction (reduction) occurs after the phase II reaction (conjugation). Specifically, it is indicated that Dht is metabolized to its metabolite 3α-androstanediol-17-glucuronide via the previously unrecognized “conjugation pathway” involving the sequential reactions of UGT2B17 and AKR1C4 in liver but via the conventional “reduction pathway” involving the sequential reactions of AKR1C2 and UGT2B15/17 in prostate.
Aldo-ketoreductase (AKR)2 1C enzymes are cytosolic hydroxysteroid dehydrogenases (HSDs) that catalyze the NADPH-dependent reduction of ketosteroids to hydroxysteroids (1). Four isoforms are known to exist in humans: AKR1C1 (20α[3α]-HSD), AKR1C2 (human 3α-HSD type 3; also known as bile acid-binding protein), AKR1C3 (human 3α-HSD type 2; also known as human 17β-HSD type 5), and AKR1C4 (human 3α-HSD type 1). Despite their high sequence identity (>86%), each individual human AKR1C enzyme displays distinct positional and stereochemical substrate preference and tissue distribution patterns. These enzymes are able to reduce 3-, 17-, and 20-ketosteroids and accept a broad spectrum of natural or synthetic steroids as substrates. They are implicated in diverse physiological functions by regulating the metabolism of androgens, estrogens, and progestins (1-3). Interestingly, distantly related human aldose reductase AKR1B1 shows superior turnover of glutathione conjugated lipid aldehydes than the unconjugated aldehydes (4). However, the ability of AKR1C isoforms to turnover steroid conjugates has not been previously examined.
AKR1C isoforms are involved in the metabolism of 5α-dihydrotestosterone (Dht). Dht is a potent natural androgen that controls the development and growth of the human prostate. AKR1C1-4 reduce Dht at the 3-position to form inactive androgens 5α-androstane-3α,17β-diol (3α-Diol) and 5α-androstane-3β,17β-diol (3β-Diol) (Scheme 1). Individual AKR1C enzymes display different catalytic efficiencies and product stereochemistry in the reduction of Dht. AKR1C1 is a reductive 3β-HSD and yields 3β-Diol as the main product, AKR1C2 and AKR1C4 catalyze the highly efficient formation of 3α-Diol, and AKR1C3 catalyzes the slow formation of a mixture of 3α- and 3β-Diol (5). Although the liver-specific location of AKR1C4 limits this enzyme to the clearance of Dht in that organ, AKR1C1 and AKR1C2 are believed to be responsible for the formation of 3β-Diol (a pro-apoptotic ligand of ERβ) and 3α-Diol (an inactive androgen), respectively, in the androgen target tissues, such as the human prostate.
SCHEME 1.

Examples of steroid transformation reactions catalyzed by AKR1C isoforms: inactivation of Dht and activation of Tib.
AKR1C enzymes can also act on prodrugs such as the synthetic steroid hormone tibolone (Tib) by converting it into biologically active 3α/β-hydroxy metabolites (Scheme 1) (6, 7). Tib is used to treat menopausal symptoms without the undesirable stimulation of the breast and endometrium (8). The tissue-specific actions of Tib rely on its metabolism to estrogenic metabolites 3α/β-hydroxytibolones (3α/β-OH-Tibs). It has been shown that AKR1C enzymes are responsible for the activation of Tib (6, 7). AKR1C1 and AKR1C2 selectively reduce Tib to 3β-OH-Tib, but not to 3α-OH-Tib, and are responsible for the activation of Tib to its 3β-OH-Tib metabolite in target tissues.
AKR1C enzymes are recognized as Phase I metabolizing enzymes. Their reactions introduce a hydroxyl group into the product, which is then available for sulfonation and glucuronidation by phase II enzymes. It is a generally accepted dogma that the phase I reaction occurs prior to the phase II reaction. Therefore, for steroids such as Dht and Tib, which possess both the keto- and the hydroxygroups, the accepted metabolic pathway follows the sequence of 3-keto reduction first then conjugation at either the 3- or 17-position. However, alternative pathways are possible where the steroid might be conjugated first at the 17-position then reduced at the 3-position. This alternative pathway requires that the AKR1C enzymes accept conjugated steroids as substrates. There have been no previous reports to support such a proposal. Although the efficiency and specificity with which AKR1C enzymes recognize free steroids have been extensively studied, it is unknown whether these enzymes are able to turnover conjugated steroids. Herein, we investigated the ability of human AKR1C enzymes to reduce glucuronide or sulfate-conjugated steroids by using Dht-17-glucuronide (DhtG), Dht-17-sulfate (DhtS), and Tib-17-sulfate (TibS) as substrates. We demonstrate for the first time that AKR1C1-4 catalyze the reduction of conjugated steroids with activities comparable to those seen with the free steroids, thus providing experimental evidence for alternative pathways in steroid hormone metabolism.
EXPERIMENTAL PROCEDURES
Materials—Dht, DhtG, DhtS, 3α-Diol-17-G, and 3β-Diol-17-S were obtained from Steraloids (Wilton, NH), of which 3β-Diol-17-S was custom synthesized. Pyridine nucleotides were purchased from Roche Applied Science. Steroids of the Tib series, including Tib, TibS, 3α- and 3β-OH-Tib, and 3α- and 3β-OH-TibS, were provided by NV Organon (Oss, The Netherlands). All other reagents were purchased from Sigma-Aldrich and were of ACS (American Chemical Society) grade or better. Recombinant AKR1C enzymes were overexpressed and purified to homogeneity as previously described (9, 10). The specific activities of AKR1C1-3 were standardized with 1-acenaphthenol as the substrate using a modified assay. Reactions were performed in 1-ml systems containing 100 mm potassium phosphate buffer (pH 7.0), 0.02% bovine serum albumin, 2.3 mm NAD+, and 200 μm 1-acenaphthenol with 4% acetonitrile as a cosolvent. Initial rates were calculated using ε = 11,209 m-1 cm-1, which took into account the absorbance of both NADH and 1-acenaphthenone at 340 nm. The specific activities were determined to be 2.1 μmol/min/mg for AKR1C1, 2.5 μmol/min/mg for AKR1C2, and 2.8 μmol/min/mg for AKR1C3. The specific activity of AKR1C4 was 0.21 μmol/min/mg (for 75 μm androsterone oxidation) under standard assay conditions (9).
Product Identification by LC/MS—Products formed during the reduction of conjugated steroids catalyzed by AKR1C enzymes were prepared for LC/MS analyses as follows. Reaction mixtures contained 100 mm potassium phosphate buffer (pH 7.0), steroid (36 μm DhtG, 45 μm DhtS, or 45 μm TibS), 0.5 mm NADPH, and 4% methanol in 450 μl of total volume. The reaction was initiated by the addition of purified enzyme (buffer for no-enzyme control, 15 μg of AKR1C1, 19 μg of AKR1C2, 33 μg of AKR1C3, or 13 μg of AKR1C4) and incubated at 37 °C. Incubation times were 90 min for no-enzyme control, 60 min for reactions with AKR1C1 and AKR1C2, 90 min for AKR1C3, and 20 and 60 min for AKR1C4. Reaction mixtures were extracted twice with 1.5 ml of water-saturated ethyl acetate. The pooled organic extracts were vacuum-dried, and the residues were re-dissolved in 200 μl of 50% methanol.
LC was performed using a Waters Alliance 2690 HPLC system (Waters Corp., Milford, MA) coupled to the mass spectrometer. For DhtG and DhtS, a SunFire C8 column (4.6 × 150 mm, Waters) was employed. Solvent A was 5 mm aqueous ammonium acetate in water, and solvent B was 5 mm ammonium acetate in acetonitrile. The linear gradient used was as follows: 30% solvent B at 0 and 3 min, 50% solvent B at 13 min, 80% solvent B at 14 and 19 min, and 30% solvent B at 20 and 30 min with a flow rate of 0.3 ml/min. For TibS, a Jupiter C18 column (2.0 × 150 mm, Phenomenex, Torrance, CA) was employed. Solvent A was 5 mm aqueous ammonium acetate, and solvent B was 5 mm ammonium acetate in acetonitrile. The linear gradient was as follows: 20% B at 0 and 2 min, 30% B at 8 and 12 min, and 20% B at 14 min with a flow rate of 0.5 ml/min. All separations were performed at ambient temperature.
MS was conducted using an LCQ ion-trap mass spectrometer (Thermo Fisher, San Jose, CA) equipped with an electrospray ionization source. The mass spectrometer was operated in the negative ion mode with a potential of 4.5 kV applied to the electrospray ionization needle. Operating conditions for DhtG and DhtS were as follows: heated capillary temperature 220 °C, capillary voltage -4 V, tube lens offset 10 V, nitrogen was used for the sheath gas at 80 p.s.i., and for the auxiliary at 10 (arbitrary units). Operating conditions for TibS were as follows: heated capillary temperature 230 °C, capillary voltage -23 V, tube lens offset -25 V, nitrogen was used for the sheath gas at 50 p.s.i., and for the auxiliary gas at 30 (arbitrary units). Full scanning analyses were performed in the range of m/z 100 to m/z 600. Products of the reactions were identified based on their LC retention times, and mass spectra relative to those observed with the authentic standards. The fragment ions monitored for the steroid conjugates were as follows: DhtG (M-H-, m/z = 465); 3α- and 3β-Diol-17-G (M-H-, m/z = 467); DhtS (M-H-, m/z = 369); 3α- and 3β-Diol-17-S (M-H-, m/z = 371); TibS (M-H-, m/z = 391.5); and 3α- and 3β-OH-TibS (M-H-, m/z = 393.5).
Determination of Steady-state Kinetic Parameters—Initial rates of the NADPH-dependent reduction of ketosteroids and their conjugates catalyzed by AKR1C were measured using a Hitachi F-2500 fluorescence spectrophotometer (Hitachi America, Ltd., New York, NY) by monitoring the change in fluorescence emission of NADPH. Excitation and emission wavelengths were set at 340 and 450 nm, respectively. Changes in fluorescence units were converted to nanomoles of cofactor by using standard curves of fluorescence emission versus known NADPH concentrations. Typical reaction samples contained enzyme (2.9 μg of AKR1C1, 2.4 μg of AKR1C2, 4.3 μg of AKR1C3, or 1.7 μg of AKR1C4), NADPH at saturating concentration (12 μm to 25 μm) and steroid at varied concentrations (0.1 μm to 50 μm) in a total volume of 1 ml. Data were analyzed by nonlinear least-squares fitting to the equation,
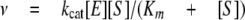 |
(Eq.1) |
where ν is the initial velocity, [E] and [S] are the total molar concentrations of the enzyme and steroid substrate, respectively, kcat (s-1) is the turnover number, and Km (μm) is the apparent Michaelis-Menten constant for the steroid substrate. When decreases in rates were noticed at higher concentrations of substrate, a modified equation that took into consideration substrate inhibition was used,
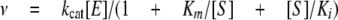 |
(Eq.2) |
where Ki (μm) is the apparent dissociation constant for the presumed inhibitory enzyme-substrate complex (11).
Molecular Modeling—Models of Dht and Tib bound to AKR1C2 previously found using the AUTODOCK program (12) were used as templates to model the binding of steroid conjugates in AKR1C. Specifically, DhtS was modeled into the active site of AKR1C2 (PDB entry 1IHI) (13) based on the docked model of Dht in the productive conformation, which could account for the formation of 3α-Diol. A model of the sulfate group (CH3OSO3) was generated using Chem3D, which was built onto the template model of Dht by overlaying the C-O bond of the conjugate group with the C17-O bond of Dht. The SO3 group was allowed to rotate freely around the C-O bond to avoid clashing with surrounding residues. It appeared that the extra SO3 group could be accommodated by the enzyme in multiple positions without changing the position of the steroid scaffold. Similarly, TibS were modeled into the active site of AKR1C2.
Docking of DhtG was performed as follows. A model of DhtG was built with Chem3D using Dht and the glucuronide group (CO(C5H8O4CO2)) retrieved from the estradiol-3β-d-glucuronide (PDB entry 1BFV). The conjugate was subjected to energy minimization with the steroid scaffold fixed. The resulting DhtG model was then placed into the binding pocket of AKR1C2 by overlaying the steroid moiety onto the Dht template in the enzyme.
Crystal structures of AKR1C1 (PDB entry 1MRQ) (14), AKR1C3 (PDB entry 1XF0) (15), and AKR1C4 (PDB entry 2FVL) were superimposed onto the structure of AKR1C2 (13) to allow direct comparison of the steroid binding pockets and inspection of the accommodation of steroid conjugates in these enzymes.
RESULTS
DhtG, DhtS, and TibS were used as example substrates for AKR1C1-4 to assess the ability of AKR1C isoforms to transform conjugated steroids (Scheme 2). To compare the reaction of steroid conjugates with those of the corresponding free steroids, the product profile of each reaction was characterized, and kinetic constants were determined. Previous characterization of the reduction of the free steroids Dht and Tib catalyzed by the AKR1C1-4 enzymes showed that individual isoforms displayed distinct stereochemical preference and kinetic characteristics (5, 6). Thus, these reactions represent unique reaction systems to examine the effect of a bulky conjugate group at the 17-position on the stereochemistry and the catalytic efficiency of 3-ketosteroid reduction.
SCHEME 2.

Structures of the steroid conjugates studied.
Product Profiles of Reduction of DhtG Catalyzed by AKR1C1-4—Enzymatic activities were observed by LC/MS analysis when AKR1C enzymes were incubated with DhtG. This was evident by the disappearance of the substrate peak, and the appearance of product peaks in LC/MS chromatograms of samples containing enzymes compared with the no enzyme controls (Fig. 1).
FIGURE 1.

LC/MS analysis of the reduction of DhtG catalyzed by human AKR1C isoforms. A, the ion chromatogram (m/z 450-500) of a mixture of authentic standards of DhtG and 3α-Diol-17-G; B-F, corresponding ion chromatograms of reaction samples containing no enzyme (B) and AKR1C1-4 (C-F). Samples were prepared as described under “Experimental Procedures.”
Two product peaks were found in the incubations of DhtG with AKR1C isoforms. The product peak with an LC retention time of 12.6 min was identified as 3α-Diol-17-G by reference to the authentic standard. The peak at 7.4 min had the same MS spectrum as 3α-Diol-17-G (see supplemental Fig. S1) and was assigned as 3β-Diol-17-G. Peak areas of the ions on the chromatograms provided an estimated ratio of the two metabolites generated. In the reactions catalyzed by AKR1C1, AKR1C2, and AKR1C3, the formation of 3β-Diol-17-G was 6.2-, 4.6-, and 2.3-fold higher than that of 3α-Diol-17-G, respectively. Thus 70-80% of the total product formed by the three isoforms was 3β-Diol-17-G (Table 1). In contrast, 3α-Diol-17-G was the preferred product from the reduction of DhtG catalyzed by AKR1C4, and accounted for 89% of the total product in 20-min incubations. The ratio between 3α- and 3β-reduced products formed by AKR1C4 decreased from 7.9 to 4.7 when the incubation time was extended from 20 to 60 min. This was consistent with the epimerase activity of AKR1C4 that converts the 3α-reduced product formed initially to the 3β-reduced product. This epimerization reaction was previously observed with Dht as the substrate (5).
TABLE 1.
Product profiles for the reduction of Dht and Tib and their conjugates catalyzed by AKR1C isoforms
Values for Dht were calculated based on the specific activity values for 3α- and 3β-reduction measured by radiometric assay (5). Values for other steroids are ratios of peak areas of the ions of 3α and 3β products on LC-MS chromatograms.
|
3α:3β
|
||||
|---|---|---|---|---|
| AKR1C1 | AKR1C2 | AKR1C3 | AKR1C4 | |
| Dht | 1:2.9 | 20:1 | 1.6:1 | 3.6:1 |
| DhtG | 1:6.2 | 1:4.6 | 1:2.3 | 7.9:1 |
| DhtS | 1:2.9 | 5.5:1 | 1:3.0 | 1:1.1 |
| Tib | 3β only | 3β only | 1:5.4 | 4.7:1 |
| TibS | 1:120 | 1:40 | 1:53 | 1.4:1 |
The product profiles for the reduction of DhtG and Dht catalyzed by AKR1C enzymes were compared (Table 1). It was found that AKR1C1 and AKR1C4 showed unchanged stereochemical preference between the conjugated and the free steroids. In contrast, a complete inversion in stereo-preference of AKR1C2 from forming 3α-Diol (3α to 3β ratio 20:1) with Dht to forming 3β-Diol-17-G (3α to 3β ratio 1:4.6) with DhtG was observed. A shift to 3β-reduction with DhtG versus Dht was also seen with AKR1C3; however, this may not be a true reflection of a change in stereoselectivity. Longer incubation times were used for the reactions catalyzed by AKR1C3 due to its weak 3-ketosteroid reductase activity (5) such that its epimerase activity (data not shown) may have affected the product distribution.
Product Profiles of Reduction of DhtS Catalyzed by AKR1C1-4—Two product peaks were also found when DhtS was used as substrate for AKR1C1-4 (Fig. 2). The first product peak (at 13.8 min) could be positively identified as the 3β-Diol-17-S based on the available authentic standard. The second product peak (at 18.0 min) that eluted after the substrate DhtS was assigned as 3α-Diol-17-S, based on its MS properties that were identical to 3β-Diol-17-S (see supplemental Fig. S2). Note that the elution order of the sulfates were the same as the glucuronides, i.e. the 3β-reduced conjugates (3β-Diol-17-G and 3β-Diol-17-S) eluted well before the corresponding Dht conjugates (DhtG and DhtS, respectively), although the 3α-reduced conjugates (3α-Diol-17-G and 3α-Diol-17-S) eluted shortly after the corresponding Dht conjugates (DhtG and DhtS, respectively).
FIGURE 2.

LC/MS analysis of the reduction of DhtS catalyzed by human AKR1C isoforms. A, the ion chromatogram (m/z 350-400) of a mixture of authentic standards of DhtS and 3β-Diol-17-S; B-F, corresponding ion chromatograms of reaction samples containing no enzyme (B) and AKR1C1-4 (C-F). Samples were prepared as described under “Experimental Procedures.”
In the reactions of DhtS catalyzed by AKR1C1 and AKR1C3, the formation of 3β-Diol-17-S was 2.9- and 3.0-fold higher than that of 3α-Diol-17-S, respectively. In contrast, the formation of 3α-Diol-17-S was 5.5-fold higher than that of 3β-Diol-17-S for the reaction catalyzed by AKR1C2. In the reaction catalyzed by AKR1C4, approximately equal amounts of 3α- and 3β-reduced products were generated from the 20-min incubation. The 3α to 3β product ratio decreased from 0.99 to 0.56 for the 60-min incubation of AKR1C4, indicating epimerase activity of the enzyme with this substrate.
The product profiles for the reaction of DhtS catalyzed by AKR1C1 and AKR1C2 were largely unchanged from those observed with Dht (Table 1). As such, AKR1C1 acted predominantly as a reductive 3β-HSD and AKR1C2 acted predominantly as a reductive 3α-HSD with DhtS and Dht. A shift to more 3β-reduced product was seen in the reaction of DhtS versus that of Dht with AKR1C3 and AKR1C4, which was attributed to the epimerase activity of both enzymes.
It thus seems that AKR1C1-4 catalyzed the reductions of Dht and DhtS with generally unchanged stereochemical outcome, whereas this is not the case for Dht and DhtG, because the stereochemical outcome of AKR1C2 was inverted with DhtG versus Dht.
Product Profiles of Reduction of TibS Catalyzed by AKR1C1-4—LC/MS analyses of the products from the reduction of TibS catalyzed by the AKR1C isoforms are shown in Fig. 3. The identification of the products was accomplished by reference to the authentic standards of the two prospective products 3α- and 3β-OH-TibS. A major metabolite was observed with an LC retention time of 9.8 min (which corresponded to 3β-OH-TibS) in incubations of TibS with AKR1C1-3 (Fig. 3, C and D). A very minor product was observed at 7.0 min, which corresponded to 3α-OH-TibS. In contrast, the two metabolites were formed in comparable amounts in incubations of TibS with AKR1C4 (Fig. 3E).
FIGURE 3.

LC/MS analysis of the reduction of TibS catalyzed by human AKR1C isoforms. A, the ion chromatogram (m/z 200-400) of a mixture of authentic standards of TibS, 3α-OH-TibS, and 3β-OH-TibS; B-F, corresponding ion chromatograms of reaction samples containing no enzyme (B) and AKR1C1-4 (C-F). Samples were prepared as described under “Experimental Procedures.”
The product profiles were very similar for Tib and TibS with each individual AKR1C isoform (Table 1). The reduction of Tib catalyzed by AKR1C1-3 was rather stereospecific in comparison to the reduction of Dht by AKR1C isoforms. The 3β-reduced product was the sole product for Tib reduction with AKR1C1 and AKR1C2, and the 3β-reduced product was formed 5.4 times more than the 3α-reduced product with AKR1C3, whereas with AKR1C4 the ratio between the 3α- and the 3β-products was 4.7. The above pattern was largely maintained for TibS, the 3β-reduced product accounted for >98% of the total products for AKR1C1-3 catalyzed reactions, whereas with AKR1C4 the ratio between the 3α- and 3β-reduced products was 1.4.
Kinetic Parameters—The kcat and Km values for the reduction of the steroids catalyzed by AKR1C isoforms were determined by continuous spectrofluorimetric assays and are listed in Table 2 (see supplemental Fig. S3 for representative data). The depletion of NADPH was monitored during the assay, thus for reactions where a mixture of products were formed the activity measured represented the total activity.
TABLE 2.
Kinetic parameters for the reduction of Dht, DhtG, and DhtS catalyzed by AKR1C isoforms
Steady-state parameters were determined by fluorometric assay. Values are given as means ± S.D. (n > 2). Values for AKR1C3-catalyzed reactions were not determined due to low turnover.
| AKR1C1 | AKR1C2 | AKR1C4 | |
|---|---|---|---|
| Dht | |||
| kcat (min−1) | 0.74 ± 0.03 | 1.98 ± 0.12 | 3.08 ± 0.15 |
| Km (μm) | 4.2 ± 0.5 | 2.9 ± 0.2 | <0.2 |
| kcat/Km (min−1m−1) | 1.8 × 105 | 6.8 × 105 | >1.5 × 107 |
| DhtG | |||
| kcat (min−1) | 0.53 ± 0.03 | 0.24 ± 0.03 | 3.28 ± 0.08 |
| Km (μm) | 16.1 ± 2.4 | 4.1 ± 1.2 | <0.2 |
| kcat/Km (min−1m−1) | 3.3 × 104 | 5.9 × 104 | >1.6 × 107 |
| DhtS | |||
| kcat (min−1) | 0.75 ± 0.05 | 1.0 ± 0.1 | 3.07 ± 0.07 |
| Km (μm) | 5.2 ± 0.8 | 4.2 ± 0.5 | <0.2 |
| kcat/Km (min−1m−1) | 1.4 × 105 | 2.4 × 105 | >1.5 × 107 |
AKR1C1 turned over Dht and DhtS with similar kcat and Km values, whereas its reactivity was reduced with DhtG. Similarly, AKR1C2 is a relatively efficient enzyme for both Dht and DhtS reduction, whereas its reactivity was significantly impaired in the reduction of DhtG. Thus, AKR1C1 and AKR1C2 displayed a 5.5-fold and a 9.3-fold decrease in catalytic efficiency, respectively, for the reactions of DhtG versus Dht, whereas the two enzymes displayed only 1.3-fold and 2.8-fold decreases in catalytic efficiency for DhtS versus Dht.
AKR1C3 was a very poor enzyme for the turnover of all the steroids in the Dht series and gave estimates of kcat of <0.2 min-1. Due to the low turnover, it was not possible to accurately determine kcat and Km values of these reactions. In contrast, AKR1C4 displayed superior kcat values with all the steroids in the Dht series. This enzyme also displayed low Km values in these reactions. The v/[S] curves were essentially zero order for the steroid concentrations used, indicating a Km that is lower than 0.2 μm. Thus, AKR1C4 has high catalytic efficiency for the turnover of the free and conjugated Dht.
In the Tib series, comparable kinetics were observed for the reduction of the free and conjugated steroids (Table 3). AKR1C1 showed a 2-fold higher kcat and a 3-fold higher Km with TibS than Tib. AKR1C2 exhibited the highest catalytic efficiency (kcat/Km) among the human enzymes with both TibS and Tib as substrates. Previously a strong substrate inhibition was observed in the reduction of Tib catalyzed by AKR1C2 (6). This unique kinetic feature was retained in the reduction of TibS catalyzed by the same enzyme. AKR1C3 was a slow enzyme with TibS and gave an estimated kcat lower than 0.2 min-1. Similar kinetic parameters were also observed for the reduction of Tib and TibS catalyzed by AKR1C4. Interestingly, a decrease in Km to 0.5 μm suggested a slight preference for the conjugated steroid than the free steroid by AKR1C4.
TABLE 3.
Kinetic parameters for the reduction of Tib and TibS catalyzed by AKR1C isoforms
Steady-state parameters were determined by fluorometric assay. Values are given as means ± S.D. (n > 2). Data for Tib were from Ref. 6. Values for AKR1C3-catalyzed reactions were not determined due to low turnover.
| AKR1C1 | AKR1C2 | AKR1C4 | |
|---|---|---|---|
| Tib | |||
| kcat (min−1) | 0.88 ± 0.06 | 12.7 ± 3.7 | 1.83 ± 0.06 |
| Km (μm) | 0.76 ± 0.13 | 0.87 ± 0.34 | 1.02 ± 0.07 |
| kcat/Km (min−1m−1) | 1.2 × 106 | 1.4 × 107 | 1.8 × 106 |
| Ki (μm) | 0.7 ± 0.29 | ||
| TibS | |||
| kcat (min−1) | 1.9 ± 0.3 | 12.1 ± 2.7 | 1.20 ± 0.03 |
| Km (μm) | 2.5 ± 0.28 | 1.2 ± 0.5 | 0.5 ± 0.1 |
| kcat/Km (min−1m−1) | 7.6 × 105 | 1.0 × 107 | 2.4 × 106 |
| Ki (μm) | 2.1 ± 0.6 |
Molecular Modeling of Steroid Conjugate Binding in AKR1C—The observed reactivity of AKR1C enzymes on steroid conjugates was rationalized by molecular docking of steroid conjugates into the binding sites of AKR1C. Crystal structures of AKR1C1-4 confirmed that these enzymes all share the structural motif of the family, i.e. an (α/β)8 barrel structure with three large associated loops and an elongated steroid binding site formed by residues from several loops (Fig. 4) (13-17). Previously, we have conducted molecular docking studies of Dht and Tib into the active sites of AKR1C1 and AKR1C2 and shown “productive” docked positions of steroid substrates that could account for reactivity (12). In addition, the binding channels of AKR1C1 and AKR1C2 were found to be wide near the entry point such that steroids were also docked in the opening roughly perpendicular to the channel in a “non-productive” manner.
FIGURE 4.

The steroid binding site of AKR1C enzymes can accommodate conjugated steroids. A binding model of DhtG in AKR1C2 is shown. The details of the model building are given under “Experimental Procedures.” The (α/β)8-barrel structure of AKR1C2 is shown in ribbon representation with α-helices in purple and β-strands in yellow. NADP+, shown in stick representation in orange, binds in a highly conserved position. DhtG, shown in sphere representation with carbon in green and oxygen in red, binds in the steroid binding site that is formed by residues from several loops (in blue). The 3-keto group of the steroid is in close proximity with the nicotinamide ring of the cofactor for 3-ketosteroid reduction. The conjugate group of the steroid is accommodated in the wide opening at the entry point of the steroid binding pocket. The closest neighboring residues (in light blue) to the glucuronide group are shown in stick and dot representations. The figure was prepared using PyMOL.
Using the productive docked model of Dht in AKR1C2 as the template ligand, we first examined whether DhtS could be accommodated. Indeed, the sulfate group could be added onto the docked Dht without causing any steric clashing between the steroid and the surrounding residues. The sulfate group was housed in the opening of the substrate channel, and several rotamers of the sulfate were allowed. Docking of DhtG into AKR1C2 was performed slightly differently. A model of DhtG was built and energy-minimized prior to docking. The resulting DhtG model was then docked into the binding site of AKR1C2 by overlaying its steroid moiety onto the Dht template in the enzyme. Surprisingly, DhtG too can be accommodated in the binding pocket of AKR1C2 (Fig. 4). Docking studies revealed that residues 127-129 are most close to the conjugate group, which were >4 Å away from the sulfate group and >3 Å from the glucuronide group.
Steroid conjugates were also successfully docked to the binding site of AKR1C1, which was expected, because the steroid binding pockets of AKR1C1 and AKR1C2 differ only by one residue. Inspection of the crystal structures of all four enzymes revealed that the relative broad opening of the steroid binding channel is a common feature, consistent with the high sequence identity (>86%) and their shared ability to convert conjugated steroids.
The molecular modeling of sulfated steroids in AKR1C isoforms showed that the accommodation of the 17-sulfate group of a steroid substrate by these enzymes may occur with little effects on the position of the 3-keto group. This explains the similarity in kinetic behavior and stereochemistry observed for free and sulfated steroids as substrates for AKR1C enzymes. In contrast, when the reduction of DhtG is compared with Dht, severely impaired catalytic efficiencies were observed with AKR1C1 and AKR1C2, and a complete inversion of stereochemistry was seen with AKR1C2. These experimental observations suggest that the accommodation of the 17-glucuronide group of a steroid substrate by these enzymes significantly affects the binding of the 3-keto group. Compared with a sulfate group, a glucuronide group is bulkier, which places it closer to residues 127-129 when docked. A glucuronide group is also more flexible in that, in addition to the freely rotatable glycosidic bond, its monosaccharide can assume multiple conformations. It is thus possible that interactions between the glucuronide group of DhtG and residues 127-129 force the steroid to bind in a conformation different from the one observed in our modeling studies. As a result, the 3-keto group is in a position not optimal for the reaction, and changes in stereochemistry may occur. Our docking results suggest that residue 127, which is Glu in AKR1C1-3, but Thr in AKR1C4, may play an important role in the reduction of catalytic efficiency observed with AKR1C1 and AKR1C2, but not with AKR1C4.
The ability of AKR1C4 to accept DhtG just as well as the free steroid is a very interesting finding, which suggests that the binding site of AKR1C4 may be wider for this enzyme compared with the other isoforms. Unfortunately, less is known about the mature structure of the ligand binding site of AKR1C4, because no ternary structure is available. Because it is known that the binding sites of AKR1C enzymes undergo significant conformational change when the binary complex is converted to the ternary complex (18, 19), docking of the steroid conjugates into AKR1C4 was not further pursued.
DISCUSSION
The activities of human AKR1C enzymes on steroid hormone substrates have been thoroughly studied to show that in vivo these enzymes can regulate the metabolism of androgens, estrogens, and progestins via their 3-, 17-, and 20-ketosteroid reductase activities (2, 5). In the present study, we have extended our knowledge of the substrate specificity of AKR1C enzymes. Using DhtG, DhtS, and TibS as example substrates, we now demonstrate that AKR1C enzymes transform not only the free steroids but also glucuronide and sulfate conjugated steroids as well. This finding significantly expands the substrate spectrum for these enzymes and more importantly, suggests alternative pathways in steroid metabolism where the important role of AKR1C in pre-receptor regulation may also be mediated through steroid conjugate metabolism.
Free versus Conjugated Steroids as Substrates for AKR1C Isoforms—Individual AKR1C isoforms have been shown to display distinct positional and stereochemical preference with ketosteroid substrates (2, 5). It is evident from the reduction of Dht and Tib catalyzed by AKR1C1-4 that the catalytic efficiency and stereochemical outcome of the reaction of these free steroids are isoform and steroid-dependent (Table 1). AKR1C1 is a reductive 3β-HSD with both these 3-ketosteroids and yields a3β-hydroxysteroid as the major product. AKR1C2, however, selectively reduces Dht to 3α-Diol and reverses its stereochemistry preference to form only the 3β-reduced product with Tib. AKR1C2 is a highly efficient enzyme for the reduction of Tib but also displayed strong substrate inhibition. AKR1C3 generates a mixture of 3α- and 3β-reduced products and reduces both Dht and Tib slowly. This is consistent with its weak 3-ketosteroid reductase activity and high 17-ketosteroid reductase activity. In contrast, AKR1C4 is very efficient at 3-keto reduction and acts mainly as a reductive 3α-HSD with both steroids. The kinetic and stereochemical characteristics of these reactions represent unique reaction systems to examine the effect of a bulky conjugate group at the 17-position of a steroid substrate on the kinetics and the stereochemical outcome of the reaction at the 3-position.
We find that conjugation of the 17β-hydroxy group did not abolish the reactivity of AKR1C1-4 on the 3-ketone group of Dht and Tib. In the case of AKR1C1, its preference for 3β-reduction remained unchanged by the conjugation at the 17-position, because almost the same stereochemical product profiles were observed with free steroids (Dht and Tib) and the corresponding steroid conjugates (DhtG/S and TibS). Conjugation reduced the catalytic efficiency of AKR1C1 on DhtG and DhtS compared with the free steroid Dht, whereas sulfonation increased the apparent kcat and Km of AKR1C1 for the reduction of TibS versus the free steroid. Interestingly, a complete inversion in stereoselectivity was observed with AKR1C2 in that the enzyme generated mainly 3α-hydroxy products from Dht and DhtS but gave predominantly the 3β-hydroxy steroid product with DhtG at a much depressed reaction rate. In contrast, AKR1C2 displayed the same stereochemical and kinetic behavior with Tib and TibS, generating selectively the 3β-hydroxy steroid product with high kcat values and strong substrate inhibition. AKR1C3 was a weak enzyme producing mixed 3α- and 3β-hydroxy products for either the conjugated or the free steroids in the Dht series, but forming mainly the 3β-hydroxy products with the steroids in the Tib series. Most strikingly, AKR1C4 retained its superior catalytic efficiency seen with Dht with both DhtG and DhtS. Its preference for 3α-reduction was also unchanged. AKR1C4 also displayed very similar stereochemistry and kinetic parameters with Tib and TibS. Therefore, with the exception of DhtG and AKR1C2, the stereochemical outcomes of the reactions of steroid conjugates by AKR1C1-4 were largely unchanged from those seen with the respective free steroid. Kinetically, AKR1C1 and AKR1C2 appeared to have decreased catalytic ability with steroid conjugates, whereas AKR1C4 was a slightly more efficient enzyme with the steroid conjugates.
Accommodating Nature of the AKR1C Substrate Binding Site—Available crystal structures and molecular modeling studies have suggested that the binding of the steroid substrate in AKR1C enzymes is achieved by (i) the anchoring of one end of the steroid (the keto-group) in the “oxyanion” hole by the catalytic residues and (ii) the positioning of the steroid nucleus by surrounding hydrophobic residues (12-17). The other end of the steroid is often untethered and located in a wide opening where steroids were found to bind non-productively.
When the binding of steroid conjugates to AKR1C enzymes was modeled, the present study showed that it is in this opening where the conjugate group is accommodated. The presence of a sulfate group at 17-position did not affect the position of the 3-ketone group in the active site, which is consistent with the largely unchanged kinetic constants and stereochemistry of all four AKR1C enzymes between the free and sulfated steroids. However, the severely impaired turnover rate and the inversion of stereochemistry for the reduction of DhtG catalyzed by AKR1C2 compared with that of Dht suggest that the bulky and flexible glucuronide group may have a more pronounced effect on the position of the keto-group. In contrast, the similar kinetic and stereochemical properties observed for the turnover of the free and conjugated steroids by AKR1C4 point to a more accommodating active site.
Considering the high sequence identity shared by AKR1 family members, it is conceivable that their ability to convert conjugated substrates may also extend to other enzymes in the family, e.g. AKR1D1 (steroid 5β-reductase). Indeed, AKR1B enzymes are able to reduce lipid aldehyde glutathione conjugates more efficiently than the free aldehyde (20). A glutathione binding site in AKR1B was identified to account for the reduced Km values observed for the conjugated substrate versus the free aldehyde. However, a lack of a specific trend in changes in kinetic parameters of AKR1C enzymes with the steroid conjugates versus the free steroids argue against a specific site in AKR1C for the binding of the conjugate group. This is supported by the modeling, because the conjugate group of a steroid substrate is largely outside of the steroid pocket of AKR1C enzymes, and no specific interaction between the conjugate group and the pocket residues could be identified.
Alternative Pathways in Steroid Metabolism—In steroid hormone metabolism, AKR1C isoforms act as phase I enzymes and are thought to precede the phase II conjugating enzymes. However, the finding that AKR1C isoforms act on steroid conjugates raises the possibility that for some steroids conjugation (phase II reaction) may occur before reduction (phase I reaction). Therefore, in addition to the generally accepted metabolic pathway where Dht is metabolized to Diol then to DiolG/S (“reduction pathway”), Dht may be converted to DhtG/S and then reduced to DiolG/S (“conjugation pathway”). Similarly, Tib can be metabolized following the sequence of either Tib → OH-Tib → OH-TibS (reduction pathway) or Tib → TibS → OH-TibS (conjugation pathway). Alternative pathways for androgen metabolism had been proposed earlier (21) but were mostly disregarded, because there has been a lack of experimental evidence to support such a proposal. Hochberg and colleagues found that the 17β-HSD in human red blood cells catalyzes the oxidation of both estradiol and its 3-sulfate and that side-chain cleavage enzymes were able to act on both cholesterol and cholesterol sulfate, supporting similar alternative pathways for these steroids (22, 23). The current report demonstrates for the first time that both the free and the conjugated steroid may serve as substrates for AKR1C enzymes. The novel activity of AKR1C isoforms on steroid conjugates prompted us to re-evaluate their role in steroid hormone metabolism.
Role of AKR1C Enzymes in Hepatic Metabolism of Dht—In the liver, testosterone from the gonad or from exogenously administrated testosterone esters is reduced by type 1 5α-reductase to form Dht (24). Dht is then metabolized to 3α-Diol glucuronides. 3α-Diol-17-G is the predominant circulating metabolite of Dht, which accounts for >80% of the total 3α-Diol glucuronides formed (25, 26). Levels of enzymatic activity and expression of UDP-glucuronosyltransferases (UGTs) and AKR1C isoforms indicate that the conjugation pathway involving the sequential actions UGT2B15/17 and AKR1C4 enzymes is an important pathway for the hepatic metabolism of Dht (Scheme 3).
SCHEME 3.

Conjugation pathway to 3α-Diol-17-G predominates in liver.
Liver is the principal site of glucuronidation, with UGT2B7 having the highest expression followed by UGT2B4, UGT2B15, and UGT2B17, where the relative expressions are 90%, 70%, and 50%, respectively, of UGT2B7 expression (27, 28). Of these enzymes, UGT2B4 has low activity toward androgens and is thus unlikely to play a major role in Dht metabolism. UGT2B7 favors conjugation at the 3-position, whereas UGT2B15 and UGT2B17 are efficient enzymes for 17β-glucuronidation. UGT2B17 was found to be responsible for forming DhtG locally in the liver, which can compete with the free Dht to act as an endogenous substrate for AKR1C enzymes. Expression of AKR1C isoforms in fresh liver biopsies were found to follow the rank order of AKR1C4 > AKR1C1 ≫ AKR1C2 > AKR1C3 (7). Thus, being the most abundant and catalytically efficient enzyme with both free and conjugated Dht, AKR1C4 is set to be the most important 3-keto reductase for Dht metabolism. The comparable kinetic parameters of Dht and DhtG suggest that both can be biologically relevant substrates for this enzyme. However, if Dht were mostly metabolized by the reduction pathway, formation of high levels of 3α-Diol-3-G by the most abundantly expressed UGT2B7 would be expected, but this is not supported by the metabolite profile of Dht. Because 3α-Diol-17-G is the major serum Dht metabolite, we conclude that, contrary to the general belief, the conjugation pathway is the major pathway for hepatic Dht metabolism.
No significant circulating levels of DhtS or Diol sulfates have been found, although DhtS is known to be formed by sulfotransferase (SULT) 2A1 in the liver (29, 30). AKR1C enzymes have similar stereochemistry and catalytic efficiencies with DhtS versus Dht as substrate, thus Diol sulfates may be formed via both the reduction pathway and the conjugation pathway. SULTs are low capacity enzymes in comparison to UGT. As a result, low levels of DhtS and Diol sulfates are formed in the liver. This is consistent with the Dht → DhtG → 3α-Dht-17-G pathway as the major metabolic pathway for Dht in liver.
Role of AKR1C Enzymes in Prostate Metabolism of Dht—In androgen target tissues such as the prostate, Dht regulates the growth and maintenance of the mature gland, and it is eliminated by its metabolism to 3α/β-Diol and Diol glucuronides. Although 3α-Diol is an inactive androgen, it can be converted back to Dht by oxidative HSDs. The RoDH-like 3α-HSD has recently been identified to be responsible for this back reaction (31). 3β-Diol, on the other hand, is a proapoptotic ligand for estrogen receptor β, the activation of which is an important growth constraint in the gland (32). Diol glucuronides have no known biological effects but can be converted back to the free steroids by active glucuronidases that are present in all tissues of the body (33). However, less is known about their expression levels, activities, and substrate specificity. Transcript profiling in the prostate showed that AKR1C1-AKR1C3 but not AKR1C4 are expressed in epithelial and stromal cells of the prostate (34, 35), and only UGT2B15 and UGT2B17 were found in the prostate (27, 28, 36). Taken together, the level of the potent androgen Dht in this organ is regulated by enzymes in the pathways shown in Scheme 4. Contribution from AKR1C3 is most likely to be small due to its low activity with Dht and Dht conjugates. Because AKR1C2 inverts its stereochemistry to 3β-reduction with DhtG from 3α-reduction with Dht, the only possible pathway for Dht to 3α-Diol-17-G is via the reduction pathway involving the sequential reaction of AKR1C2 and UGT2B15/17. This pathway would account for the formation of 3α-Diol-17-G in the gland. The conversion of Dht to 3β-Diol-17-G, on the other hand, can follow either the reduction pathway (involving the sequential reactions of AKR1C1 and UGT2B15/17) or the conjugation pathway (involving the sequential reactions of UGT2B17 and AKR1C1/2). However, the low catalytic efficiencies of UGT2B15/17 for 3β-Diol (37) and AKR1C1/2 for DhtG explains why 3β-Diol-17-G is not a major metabolite. Thus the action of AKR1C1 in the reduction pathway would be to provide a tissue reservoir of the free 3β-Diol that would remain unconjugated.
SCHEME 4.

Reduction pathway to 3α-Diol-17-G predominates in prostate. RL-HSD, RoDH Like 3α-HSD.
DhtS may be formed by SULT2B1 in prostate (30). However, SULT2B1 has no detectable activity toward 3α-Diol, and its activity on 3β-Diol has not been reported. SULT2B1 was found to specifically catalyze the sulfonation of 3β-hydroxysteroids such as dehydroepitestosterone with high catalytic efficiency, suggesting formation of 3β-Diol-3-S is possible. Formation of 3α/β-Diol-17-S may also occur via the conjugation pathway, but these metabolites have not been observed (20).
Role of AKR1C Enzymes in Tib Metabolism—The beneficial effects of Tib are due to its estrogenic metabolites 3α- and 3β-OH-Tib (8). Although all human AKR1C enzymes are able to convert Tib to 3α- and/or 3β-OH-Tib in vitro (6), only AKR1C1 and AKR1C2 were implicated for the metabolism of Tib to 3β-OH-Tib in liver and target tissues, and AKR1C4 was found not to contribute significantly in 3α-OH-Tib formation in liver (7). Sulfonation at the 17β-hydroxy position of Tib is an important phase II metabolic step (38, 39), suggesting that TibS could be a possible source of substrate for AKR1C1 and AKR1C2 and that contribution from a conjugation pathway where phase II SULTs precede AKR1C isoforms cannot be ruled out. In addition, oxidative 3β-HSD that converts 3β-OH-Tib back to Tib and sulfatases that catalyze the desulfonation of the 3-sulfonate group may be involved in Tib metabolism in target tissues. Taken together, metabolism of Tib in liver and target tissues may involve enzymes and pathways shown in Scheme 5.
SCHEME 5.

Role of human AKR1C isoforms in the metabolism of Tib.
SULT2A1 in liver is responsible for forming the majority of the sulfates of Tib and metabolites in plasma, and it is also the only isoform capable of catalyzing bis-sulfonation (40). 3β,17β-TibS2, the second major metabolite in human (41, 42), may be formed either via the conventional reduction pathway by the sequential reactions of AKR1C1/2 and SULT2A1 or vice versa via the conjugation pathway. SULTs, however, are low capacity and high affinity enzymes. The slower formation of TibS compared with that of 3β-OH-Tib suggests that the reduction pathway predominates.
In target tissues, Tib may be converted to 3β-S-Tib by the sequential reactions of AKR1C1/2 and SULT1E1 (reduction pathway) and to 3β-OH-Tib-17-S by the sequential reactions of SULT1E1 and AKR1C1/2 (conjugation pathway). Although AKR1C1 and AKR1C2 have comparable catalytic efficiency with Tib and TibS, the preference of SULT1E1 for sulfonation at 3-hydroxyl over the 17-hydroxyl group dictates that the reduction pathway dominates for Tib metabolism. It is important to note that the steps in the reduction pathway are potentially reversible. Thus, the levels of the steroids Tib (androgenic), 3β-OH-Tib (estrogenic), and 3β-S-Tib (inactive) are regulated by enzymes AKR1C1/2, SULT1E1, 3β-HSD, and sulfatase. In different tissues, these metabolizing enzymes display specific expression patterns that will regulate the availability of the active metabolite 3β-OH-Tib, contributing to tissue-specific actions of Tib.
In summary, we have demonstrated that AKR1C enzymes are able to turnover conjugated steroids that are normally considered the end point of steroid metabolism before secretion. This finding thus provides the evidence for alternative pathways of steroid metabolism where the phase I reaction (reduction) occurs after the phase II reaction (conjugation). It would assist in the identification of the major metabolic pathway of steroid hormones (and the enzymes involved) in different organs, e.g. Dht is metabolized to its metabolite 3α-Diol-17-G via the previously unrecognized conjugation pathway by the sequential reactions of UGT2B17 and AKR1C4 in the liver, but via the conventional reduction pathway by the sequential reactions of AKR1C2 and UGT2B15/17 in the prostate. Furthermore, the finding of the novel activity of AKR1C enzymes on steroid conjugates may also impact our understanding of the metabolism of many other xenobiotics. In addition to steroids, AKR1C enzymes are known to have a broad substrate spectrum that also includes but is not limited to prostaglandins, drugs, and polycyclic aromatic hydrocarbons (43). The current study extends the list of substrates for these enzymes to conjugates. When AKR1C enzymes are implicated in the metabolism of xenobiotics, possible activities toward the conjugate metabolites of the parent compound should be considered.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK47015, R01-CA90744, and P30 ES015857 (to T. M. P.). This work was also supported by a pilot project fund from NIH Grant P30 ES015857 (to Y. J.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S3.
Footnotes
The abbreviations used are: AKR, aldo-ketoreductase; HSD, hydroxysteroid dehydrogenase; AKR1C1, human 20α(3β)-HSD; AKR1C2, human type 3 3α-HSD; AKR1C3, human type 2 3α-HSD, human type 5 17β-HSD; AKR1C4, human type 1 3α-HSD; Dht, 5α-dihydrotestosterone; DhtG, 5α-dihydrotestosterone-17β-glucuronide; DhtS, 5α-dihydrotestosterone-17β-sulfate; 3α/β-Diol, 5α-androstane-3α/β,17β-diol; 3α/β-Diol-17-G, 5α-androstan-3α/β-ol-17β-glucuronide; 3α/β-Diol-3-G, 5α-androstan-17β-ol-3α/β-glucuronide; 3α/β-Diol-17-S, 5α-androstan-3α/β-ol-17β-sulfate; LC, liquid chromatography; MS, mass spectrometry; Tib, tibolone (Livial, [7α,17α]-17-hydroxy-7-methyl-19-norpregn-5(10)-en-20-yn-3-one); 3α/β-OH-Tib, 3α/β-hydroxytibolone; TibS, tibolone-17β-sulfate; 3α/β-OH-TibS, 3α/β-hydroxytibolone-17β-sulfate; 3β-S-Tib, tibolone-3β-sulfate; 3β,17-TibS2, 3β,17β-tibolone bis-sulfate; RL-HSD, RoDH like 3α-HSD; SULT, sulfotransferase; UGT, UDP-glucuronosyltransferase.
References
- 1.Penning, T. M. (2003) Hum. Reprod. Update 9 193-205 [DOI] [PubMed] [Google Scholar]
- 2.Penning, T. M., Burczynski, M. E., Jez, J. M., Hung, C. F., Lin, H. K., Ma, H., Moore, M., Palackal, N., and Ratnam, K. (2000) Biochem. J. 351 67-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizner, T. L., Lin, H. K., Peehl, D. M., Steckelbroeck, S., Bauman, D. R., and Penning, T. M. (2003) Endocrinology 144 2922-2932 [DOI] [PubMed] [Google Scholar]
- 4.Srivastava, S., Chandra, A., Wang, L. F., Seifert, W. E., Jr., DaGue, B. B., Ansari, N. H., Srivastava, S. K., and Bhatnagar, A. (1998) J. Biol. Chem. 273 10893-10900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steckelbroeck, S., Jin, Y., Gopishetty, S., Oyesanmi, B., and Penning, T. M. (2004) J. Biol. Chem. 279 10784-10795 [DOI] [PubMed] [Google Scholar]
- 6.Steckelbroeck, S., Jin, Y., Oyesanmi, B., Kloosterboer, H. J., and Penning, T. M. (2004) Mol. Pharmacol. 66 1702-1711 [DOI] [PubMed] [Google Scholar]
- 7.Steckelbroeck, S., Oyesanmi, B., Jin, Y., Lee, S. H., Kloosterboer, H. J., and Penning, T. M. (2006) J. Pharmacol. Exp. Ther. 316 1300-1309 [DOI] [PubMed] [Google Scholar]
- 8.Kloosterboer, H. J. (2001) J. Steroid Biochem. Mol. Biol. 76 231-238 [DOI] [PubMed] [Google Scholar]
- 9.Burczynski, M. E., Harvey, R. G., and Penning, T. M. (1999) Biochemistry 38 10626. [DOI] [PubMed] [Google Scholar]
- 10.Ratnam, K., Ma, H., and Penning, T. M. (1999) Biochemistry 38 7856-7864 [DOI] [PubMed] [Google Scholar]
- 11.Shen, P., and Larter, R. (1994) Biophys. J. 67 1414-1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin, Y., and Penning, T. M. (2006) Steroids 71 380-391 [DOI] [PubMed] [Google Scholar]
- 13.Jin, Y., Stayrook, S. E., Albert, R. H., Palackal, N. T., Penning, T. M., and Lewis, M. (2001) Biochemistry 40 10161-10168 [DOI] [PubMed] [Google Scholar]
- 14.Couture, J. F., Legrand, P., Cantin, L., Luu-The, V., Labrie, F., and Breton, R. (2003) J. Mol. Biol. 331 593-604 [DOI] [PubMed] [Google Scholar]
- 15.Qiu, W., Zhou, M., Labrie, F., and Lin, S. X. (2004) Mol. Endocrinol. 18 1798-1807 [DOI] [PubMed] [Google Scholar]
- 16.Komoto, J., Yamada, T., Watanabe, K., and Takusagawa, F. (2004) Biochemistry 43 2188-2198 [DOI] [PubMed] [Google Scholar]
- 17.Lovering, A. L., Ride, J. P., Bunce, C. M., Desmond, J. C., Cummings, S. M., and White, S. A. (2004) Cancer Res. 64 1802-1810 [DOI] [PubMed] [Google Scholar]
- 18.Bennett, M. J., Albert, R. H., Jez, J. M., Ma, H., Penning, T. M., and Lewis, M. (1997) Structure 5 799-812 [DOI] [PubMed] [Google Scholar]
- 19.Cooper, W. C., Jin, Y., and Penning, T. M. (2007) J. Biol. Chem. 282 33484-33493 [DOI] [PubMed] [Google Scholar]
- 20.Dixit, B. L., Balendiran, G. K., Watowich, S. J., Srivastava, S., Ramana, K. V., Petrash, J. M., Bhatnagar, A., and Srivastava, S. K. (2000) J. Biol. Chem. 275 21587-21595 [DOI] [PubMed] [Google Scholar]
- 21.Rittmaster, R. S. (1993) Endocr. Rev. 14 121-132 [DOI] [PubMed] [Google Scholar]
- 22.Hochberg, R. B., Ladany, S., Welch, M., and Lieberman, S. (1974) Biochemistry 13 1938-1945 [DOI] [PubMed] [Google Scholar]
- 23.Jacobsohn, G. M., and Hochberg, R. B. (1968) J. Biol. Chem. 243 2985-2994 [PubMed] [Google Scholar]
- 24.Russell, D. W., and Wilson, J. D. (1994) Annu. Rev. Biochem. 63 25-61 [DOI] [PubMed] [Google Scholar]
- 25.Guillemette, C., Hum, D. W., and Belanger, A. (1996) Am. J. Physiol. 271 E348-E353 [DOI] [PubMed] [Google Scholar]
- 26.Rittmaster, R. S., Thompson, D. L., Listwak, S., and Loriaux, D. L. (1988) J. Clin. Endocrinol. Metab. 66 212-216 [DOI] [PubMed] [Google Scholar]
- 27.Turgeon, D., Carrier, J. S., Levesque, E., Hum, D. W., and Belanger, A. (2001) Endocrinology 142 778-787 [DOI] [PubMed] [Google Scholar]
- 28.Chouinard, S., Yueh, M. F., Tukey, R. H., Giton, F., Fiet, J., Pelletier, G., Barbier, O., and Belanger, A. (2008) J. Steroid Biochem. Mol. Biol. 109 247-253 [DOI] [PubMed] [Google Scholar]
- 29.Chatterjee, B., Echchgadda, I., and Song, C. S. (2005) Methods Enzymol. 400 165-191 [DOI] [PubMed] [Google Scholar]
- 30.Geese, W. J., and Raftogianis, R. B. (2001) Biochem. Biophys. Res. Commun. 288 280-289 [DOI] [PubMed] [Google Scholar]
- 31.Bauman, D. R., Steckelbroeck, S., Williams, M. V., Peehl, D. M., and Penning, T. M. (2006) Mol. Endocrinol. 20 444-458 [DOI] [PubMed] [Google Scholar]
- 32.Weihua, Z., Lathe, R., Warner, M., and Gustafsson, J. A. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 13589-13594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulder, G. J. (1992) Annu. Rev. Pharmacol. Toxicol. 32 25-49 [DOI] [PubMed] [Google Scholar]
- 34.Bauman, D. R., Steckelbroeck, S., Peehl, D. M., and Penning, T. M. (2006) Endocrinology 147 5806-5816 [DOI] [PubMed] [Google Scholar]
- 35.Lin, H. K., Jez, J. M., Schlegel, B. P., Peehl, D. M., Pachter, J. A., and Penning, T. M. (1997) Mol. Endocrinol. 11 1971-1984 [DOI] [PubMed] [Google Scholar]
- 36.Belanger, A., Pelletier, G., Labrie, F., Barbier, O., and Chouinard, S. (2003) Trends Endocrinol. Metab. 14 473-479 [DOI] [PubMed] [Google Scholar]
- 37.Guillemette C, Hum, D. W., and Belanger A. (1995) J. Steroid Biochem. Mol. Biol. 55 355-362 [DOI] [PubMed] [Google Scholar]
- 38.Verheul, H. A., Timmer, C. J., van Iersel, M. L., Delbressine, L. P., and Kloosterboer, H. J. (2007) Drug Metab. Dispos. 35 1112-1118 [DOI] [PubMed] [Google Scholar]
- 39.Vos, R. M., Krebbers, S. F., Verhoeven, C. H., and Delbressine, L. P. (2002) Drug Metab. Dispos. 30 106-112 [DOI] [PubMed] [Google Scholar]
- 40.Falany, J. L., Macrina, N., and Falany, C. N. (2004) J. Steroid Biochem. Mol. Biol. 88 383-391 [DOI] [PubMed] [Google Scholar]
- 41.Verheul, H. A., Blok, L. J., Burger, C. W., Hanifi-Moghaddam, P., and Kloosterboer, H. J. (2007) Reprod. Sci. 14 160-168 [DOI] [PubMed] [Google Scholar]
- 42.Verheul, H. A., Timmer, C. J., and Kloosterboer, H. J. (2007) Clin. Pharmacol. Ther. 81 573-579 [DOI] [PubMed] [Google Scholar]
- 43.Jin, Y., and Penning, T. M. (2007) Annu. Rev. Pharmacol. Toxicol. 47 263-292 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.