

Abstract
Aminoacyl-tRNA synthetases are multidomain proteins that catalyze the covalent attachment of amino acids to their cognate transfer RNA. Various domains of an aminoacyl-tRNA synthetase perform their specific functions in a highly coordinated manner to maintain high accuracy in protein synthesis in cells. The coordination of their function, therefore, requires communication between domains. In this study we explored the relevance of enzyme motion in domain-domain communications. Specifically, we attempted to probe whether the communication between distantly located domains of a multidomain protein is accomplished through a coordinated movement of structural elements. We investigated the collective motion in Thermus thermophilus leucyl-tRNA synthetase by studying the low frequency normal modes. We identified the mode that best described the experimentally observed conformational changes of T. thermophilus leucyl-tRNA synthetase upon substrate binding and analyzed the correlated and anticorrelated motions between different domains. Furthermore, we used statistical coupling analysis to explore if the amino acid pairs and/or clusters whose motions are thermally coupled have also coevolved. Our study demonstrates that a small number of residues belong to the category whose coupled thermal motions correspond to evolutionary coupling as well. These residue clusters constitute a distinguished set of interacting networks that are sparsely distributed in the domain interface. Residues of these networking clusters are within van der Waals contact, and we suggest that they are critical in the propagation of long range mechanochemical motions in T. thermophilus leucyl-tRNA synthetase.
Domain-domain communication plays an important role in the function of aminoacyl-tRNA synthetases (ARSs)2 (1, 2). ARSs are present in all three domains of life and are responsible for catalyzing the aminoacylation of tRNA, which is a critical step in the translation of the genetic code. The core chemistry involves the covalent attachment of an amino acid onto the 3′-adenosine moiety of the cognate tRNA. ARSs consist of multiple domains (3) (Fig. 1). The central catalytic domain (CCD) is responsible for the activation of an amino acid in the presence of ATP to form an enzyme-bound aminoacyl-adenylate (AA-AMP; Equation 1) followed by transfer of that activated amino acid to the 3′-end of its cognate tRNA (Equation 2).
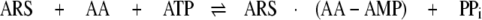 |
(Eq.1) |
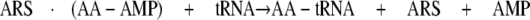 |
(Eq.2) |
The anticodon binding domain (Fig. 1) is often responsible for selecting the correct tRNA. To maintain high accuracy in the aminoacylation of tRNA, some ARSs have evolved amino acid editing mechanisms, which employed additional domains for proofreading (3-5).
FIGURE 1.

Schematic diagrams of the Tt LeuRS structure. a, various domains of the enzyme are labeled and rotations of the editing and the leucine-specific domains are shown. b, overlapped structures of the tRNA-bound (purple) and unbound (gray) Tt LeuRS displaying the linear displacement of various structural elements.
Aminoacylation of tRNA involves a series of events that includes selection of the correct amino acid and its activation in the presence of ATP, binding of the cognate tRNA, transfer of the activated amino acid onto the cognate tRNA, release of the aminoacylated tRNA from the enzyme active site, and in some cases, proofreading (4, 5). Moreover, proofreading may require translocation of the misactivated amino acid and/or the mischarged tRNA from the aminoacylation active site to the editing active site. Therefore, to maintain high precision in the aminoacylation of tRNAs, these events need to be synchronized, requiring a high degree of coordination between the various ARS domains. Indeed, a number of structural and mutational studies have provided strong evidence for domain-domain communication in ARSs (1, 2, 6). Although molecular mechanism of signal propagation from one domain to another domain has remained poorly understood, recent studies suggest that domain dynamics might play an important role in long range communication (7, 8).
Structural studies demonstrated large domain displacements in Thermus thermophilus LeuRS (Tt LeuRS) during various steps of catalysis (9, 10). Tt LeuRS possesses a complex modular architecture (9, 11). It has three distinct flexible domains: the editing domain (ED; residues 224-417), zinc-1 binding domain (ZBD; residues 154-189), and leucine-specific domain (LSD; residues 577-634). These three domains are linked with the CCD by β-ribbon structures (Fig. 1) (11). The crystal structure of the Tt LeuRS-tRNALeu complex (in post-transfer editing conformation) demonstrated that the ED undergoes a 35° rotation compared with the tRNA unbound form (Fig. 1). The LSD, which is critical for aminoacylation, undergoes a rigid-body rotation of 19° (9, 12). These conformational changes of various structural elements suggest that large scale flexibility and domain motions play a critical role in the aminoacylation and editing reactions catalyzed by LeuRS. However, these structural studies failed to provide insight into the nature of these motions (correlated or anticorrelated) among various domains and functional sites. Moreover, from the existing structural and biochemical studies, little insight was obtained about the effect of mutation/deletion of distant structural elements on the function of LeuRS. Several questions remained unanswered regarding the interplay between domain dynamics and enzyme function.
In a multidomain enzyme like an ARS, one possible way to accomplish these long range communications is through coupled domain dynamics. These coordinated domain movements are essential for the protein functions and are cooperative in nature. These cooperative motions mediating long range communications are believed to play a major role in enzyme functions including chemical catalysis (13-17). In fact, exploring the coupling between mechanical and chemical events in multidomain proteins, especially at the molecular level, has remained one of the challenging areas of contemporary biochemical research (7, 18).
In this work we have investigated the motions of various domains of Tt LeuRS and examined the role of conserved and co-evolved residues on protein dynamics using bioinformatics and computational means. The collective motions were studied by computing their normal modes. The evolutionary basis for retaining specific residue pairs was examined by using statistical coupling analysis. By combining the results of these studies, we were able to extract clusters of residues that are coupled not only by thermal motions but also through evolution. We suggest that these clusters of residues, which are at the core of cooperative domain dynamics, are involved in mediating domain-domain communication in Tt LeuRS.
EXPERIMENTAL PROCEDURES
General Strategy—To study the collective nature of these domain motions, we first performed normal mode analysis (NMA) (15, 18-28) and identified the low frequency normal mode that adequately describes the domain displacements as observed in the tRNA-bound and unbound conformations of Tt LeuRS. After identifying the correct mode, we explored the collective dynamics among various domains and characterized them by analyzing their correlated and anticorrelated patterns. In parallel, a statistical coupling analysis (SCA) (29, 30) was performed to identify residues/clusters whose mutations are evolutionarily correlated. This analysis provided the coupling of two sites that may even be separated by large distances in a protein (30). Finally, to understand how cooperative dynamics between two distant domains are accomplished, residues identified by combining these two studies were mapped onto protein surfaces and analyzed through visualizations using Visual Molecular Dynamics software (31). The NMA and SCA plots were generated by using MATLAB R2006b (The MathWorks Inc., Natick, MA).
Starting Structure—Two crystal structures of Tt LeuRS were used in this study. The crystal structure of Tt LeuRS complexed with a leucyl-adenylate analogue (PDB code 1H3N; residues 1-814) represents the tRNA unbound enzyme conformation. In contrast, the post-transfer editing conformation of the enzyme was taken from the tRNALeu bound structure (PDB code 2BYT; residues 1-876). The various conformational changes of LeuRS structural elements are shown in Fig. 1.
Normal Mode Calculations—Functionally important protein motions are often characterized by slow (low frequency) and large amplitude motions (19, 25, 32-34). These motions in biomolecules can be studied by normal mode analysis, which is a fast and powerful method especially useful for studying the dynamics of large proteins like ARSs. The key premise of NMA is that the slower lowest frequency (large amplitude) normal modes describe the largest movements in a protein that are functionally relevant (35). NMA has been successful in describing the domain motions and their collective nature in diverse set of proteins including hexokinase (33), lysozyme (35), citrate synthetase (20), adenylate kinase (22), RNA polymerase (23), icosahedral virus capsid (26), and the Escherichia. coli 70 S ribosome (36). Therefore, in an enzyme like ARS, where cooperative domain dynamics play a key role in maintaining high accuracy in the aminoacylation of the cognate tRNA, NMA is expected to provide significant information about these domain dynamics.
Elastic Network Model—The normal mode calculation is based on the harmonic approximation of the potential energy function around a minimum energy conformation. The small displacements ri(t) of atomic coordinate i, close to a stationary point of the potential energy surface, is given by
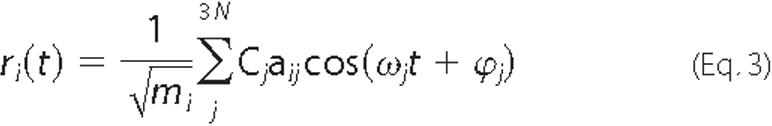
where mi is the mass of the corresponding atom, and aij is the ith coordinate of the normal mode j. The corresponding frequency vj equals to ωj/2π, where ω 2j is obtained as the jth eigenvalue of the 3N × 3N mass-weighted second derivatives of the potential energy matrix. Cj and ϕj are the amplitude and the phase of mode j. In this work NMA was carried out using the coarse-grained elastic network model (21, 24, 27). In particular, we used the anisotropic network model (ANM), where the protein is simplified to a string of beads (37). Each bead represents aCα atom. In ANM, the fluctuations are anisotropic, and the overall potential of the system is expressed as the sum of harmonic potentials between the interacting Cα atoms.

In Equation 4, γ represents the uniform spring constant,
 and rpq are the original
and instantaneous distance vectors between residues p and q,
and Γpq is the pqth element of the binary
connection matrix of inter-residue contacts. Based on an interaction cut-off
distance of rc, Γpq is
equal to 1 if
and rpq are the original
and instantaneous distance vectors between residues p and q,
and Γpq is the pqth element of the binary
connection matrix of inter-residue contacts. Based on an interaction cut-off
distance of rc, Γpq is
equal to 1 if 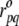 and zero otherwise
(21). The online Anisotropic
Network Model web server was used to obtain the simulated functional motion
from the Tt LeuRS structure
(24,
27). The optimal cut-off
distance (rc) between Cα atoms was kept
at 15 Å in this study
(24). The correlated or
anticorrelated motions between residue pairs of distant structural elements
were analyzed from the cross-correlations of residue pairs (see below).
and zero otherwise
(21). The online Anisotropic
Network Model web server was used to obtain the simulated functional motion
from the Tt LeuRS structure
(24,
27). The optimal cut-off
distance (rc) between Cα atoms was kept
at 15 Å in this study
(24). The correlated or
anticorrelated motions between residue pairs of distant structural elements
were analyzed from the cross-correlations of residue pairs (see below).
Comparison with Experimentally Observed Conformational Transition—Results of the NMA are compared with the experimentally observed conformational changes. The direction of motions and their magnitude as well as the collectivity were compared in identifying the mode most involved.
Overlap—The overlap, Oj, between the conformational change Δri observed by crystallographers and the jth normal mode was calculated according to Marques and Sanejouand (20),
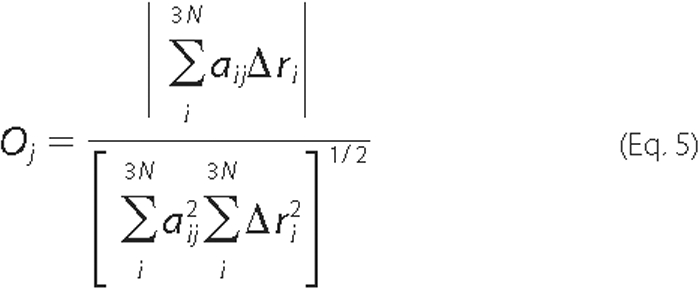
where ΔrI =
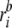 -
- 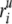 ,
,
 and
and 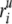 are the
ith atomic coordinates for the substrate bound and the unbound
structures of the protein, respectively. aij is the same
as defined in Equation 3. A value of 1.0 for the overlap means that the
direction given by normal mode j is identical with
Δri.
are the
ith atomic coordinates for the substrate bound and the unbound
structures of the protein, respectively. aij is the same
as defined in Equation 3. A value of 1.0 for the overlap means that the
direction given by normal mode j is identical with
Δri.
Correlation Coefficient—To measure the similarity in the relative magnitude of the atomic displacements observed experimentally and in mode j, the correlation coefficient (Cj) was calculated by
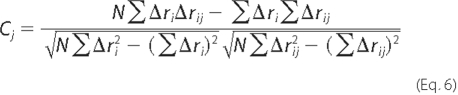
where Δri is the observed displacement of the ith atom in the crystal structure (as discussed Equation 5), Δrij is the observed displacement of the same atom i but in the normal mode j, and N represents the number of residues in the protein. A correlation coefficient of 1.0 means that that the relative magnitudes of atomic displacements are identical.
Collectivity of a Conformational Change—The collectivity of the movement of a protein in a given mode describes the extent by which various structural elements undergo a conformational change together. The higher degree of collectivity implies that a larger fraction of residues is affected by a given mode. In the case of least collective (localized) motion, normal mode movements involve only a few atoms.
To quantitatively measure the extent of collectivity of the protein motion, a collectivity index, as proposed by Bruschweiler (38), was calculated for all three modes. Accordingly, the collectivity (κ) is computed by
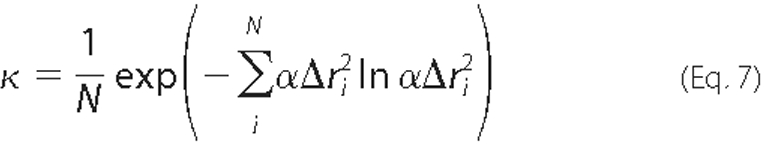
where α is a chosen normalization factor, and Δri is defined previously, so that

The value of κ is confined between N-1 and 1; κ = 1 indicates the conformational change is maximally collective.
Correlated and Anticorrelated Motions—The nature of the inter-residue motions, correlated or anticorrelated, in the protein were identified by computing the cross-correlations between residue fluctuations from the simulated motion trajectory. The cross-correlation coefficient between fluctuations of residues p and q (Cpq) was calculated by

where the atomic (Cα) displacements of residues p and q are represented by rp and rq, respectively, and the angular brackets represent an ensemble average calculated using the structures for a particular normal mode. The cross-correlation coefficients were obtained as a matrix and are represented as a two-dimensional cross-correlation map. A negative correlation value indicates anticorrelated motion, whereas a positive value identifies correlated pattern of dynamics between two compared residues.
Statistical Coupling Analysis—SCA is based upon the assumption that “coupling of two sites in a protein, whether for structural or functional reasons, should cause those two sites to coevolve” (8, 29, 30). This method takes a MSA of a protein and calculates the change in the amino acid distribution at one position with respect to a perturbation at another position.
Energetic Coupling of Amino Acid Positions in LeuRS—LeuRS
sequences from multiple species were collected from PSI-BLAST
(39), and a multiple sequence
alignment (MSA) was constructed using ClustalW
(40). To measure the extent of
coupling between two sites i and j of a protein sequence,
calculation of statistical coupling energy
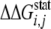 was carried out
as described in the literature
(29). In this method a large
scale perturbation experiment on the MSA of the LeuRS family was performed by
introducing a change in the amino acid distribution at one position and
examining the effect at another site
(29,
30). Briefly, the method
consists of two steps. First, the parameter that represents the overall
evolutionary conservation at a position i in the sequence of the
chosen protein family (expressed as a statistical energy
was carried out
as described in the literature
(29). In this method a large
scale perturbation experiment on the MSA of the LeuRS family was performed by
introducing a change in the amino acid distribution at one position and
examining the effect at another site
(29,
30). Briefly, the method
consists of two steps. First, the parameter that represents the overall
evolutionary conservation at a position i in the sequence of the
chosen protein family (expressed as a statistical energy
 ) is calculated using the
principles of Boltzmann statistics as follows:
) is calculated using the
principles of Boltzmann statistics as follows:

where kT* is an arbitrary energy unit,
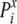 is the probability of observing amino acid
x at site i, and PxMSA is the
probability of observing amino acid x in the overall MSA
(29).
is the probability of observing amino acid
x at site i, and PxMSA is the
probability of observing amino acid x in the overall MSA
(29).
In the second step a perturbation experiment was carried out on the MSA following the method described in Suel et al. (30). Briefly, a perturbation at a specific position of a MSA refers to the creation of a subalignment, which is sufficiently large and diverse similar to the parent MSA. However, the probability of occurrence of a residue at a given position has changed considerably. For two coevolving positions on the sequence, the perturbation process, therefore, introduces a change in the amino acid distribution at one position and measures the change of the same at the other.
The statistical coupling energy between sites i and j is calculated and expressed as

where  is the
probability of a particular amino acid x at site i dependent
on a perturbation at site j. The sum over x indicates the
combined effect of the perturbation on all amino acids at site i due
to a perturbation at site j. Therefore, a large
is the
probability of a particular amino acid x at site i dependent
on a perturbation at site j. The sum over x indicates the
combined effect of the perturbation on all amino acids at site i due
to a perturbation at site j. Therefore, a large
 value represents a highly
conserved residue at ith site in the MSA
(29), whereas a large
statistical coupling value,
value represents a highly
conserved residue at ith site in the MSA
(29), whereas a large
statistical coupling value,
 , indicates strong
energetic coupling between the residues at the two sites, i and
j (29,
30).
, indicates strong
energetic coupling between the residues at the two sites, i and
j (29,
30).
Hierarchical clustering was carried out using the statistical toolbox
utility of MATLAB. Using the clustering algorithm, the raw unclustered data
were grouped into several clusters of residues that are strongly coupled. In
the subsequent steps, these data are analyzed, truncated, and re-clustered
into smaller datasets bearing strong coupling value
(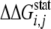 ).
).
Mapping and Visualization of the Evolutionarily and Thermally Coupled Residues on Three-dimensional Structures of the Enzyme—The thermal as well as statistical coupling of residues across the Tt LeuRS domains was further explored by visualization. The coevolved residue pairs were first mapped onto the three-dimensional structure of LeuRS and analyzed with the aid of the molecular surface generation tool of the Visual Molecular Dynamics program (31). Based on van der Waals contacts, these residues were grouped into five clusters, each of which represents a network of contiguous residues. Contribution of these coevolving residue clusters to the cooperative domain dynamics was further investigated by quantifying the amount of dynamic coupling between them from the NMA cross-correlation matrix.
RESULTS
Analysis of the Domain Dynamics of Tt LeuRS—In the first step of this study, NMA was performed to characterize domain motions of Tt LeuRS. The computed low frequency normal modes were then evaluated by comparing them against structural transitions, observed in the x-ray crystal structures. These comparative studies were based on the direction, magnitude, and the collectivity of conformational changes.
Conformational Flexibility—To explore protein flexibility, we used the tRNA unbound structure of Tt LeuRS (“open” form; PDB code 1H3N) (11). The NMA for open-form structure 1H3N yielded a number of low frequency normal modes. Among the 20 lowest frequency modes, the first three modes depicted the interdomain motions, whereas the relatively higher frequency modes represented the subdomain motions.
Earlier NMA studies on 20 different proteins in open and “closed” forms demonstrated that the normal mode(s) obtained from the open form compares better with experimentally observed conformational changes than that obtained from the closed form (22). To verify if the same motions are predicted starting from the closed conformation, we computed the normal modes using the tRNA-bound Tt LeuRS structure. No significant change in the simulated motions was observed, which clearly demonstrated that the protein moves to acquire similar domain motions from either end of the conformational continuum.
Direction of Motions—We analyzed the directions of motions for the first three low frequency modes (Fig. 2). The mode that best describes the experimentally observed direction of the conformational changes in Tt LeuRS was judged by the overlap values calculated by Equation 5. The overlap value (Oj) of >0.7 was obtained for both modes 1 and 3 (Table 1), suggesting the possible involvement of either of these two modes in the observed conformational transition in Tt LeuRS upon tRNA binding.
FIGURE 2.

The three lowest frequency normal modes (modes 1-3 from left to right), obtained for the Tt LeuRS structure (PDB code 1H3N) using ANM (27). Only the backbone Cα atoms are shown for clarity. Atoms red in color indicate large fluctuations, whereas blue colors correspond to small fluctuations. The magnitude and direction of the displacement are represented by purple arrows.
TABLE 1.
Computed overlaps (Oj), correlation coefficients (Cj), and collectivities (κ) for the three individual lowest frequency modes when compared to the experimentally observed conformational changes of Tt LeuRS upon substrate binding
Equations 5-7 were used in computing these quantities.
| Properties | Mode 1 | Mode 2 | Mode 3 |
|---|---|---|---|
| Overlap (Oj) | 0.72 | 0.60 | 0.71 |
| Correlation (Cj) | 0.51 | 0.56 | 0.31 |
| Collectivity (κ) | 0.37 | 0.20 | 0.42 |
Magnitude of Motions—The similarity between the magnitude of the atomic displacements, determined experimentally or given by the mode most involved in the conformational change, was analyzed by comparing the correlation coefficient. The correlation coefficient (Cj) for each mode was calculated using Equation 6 and is given in Table 1. Among the three lowest frequency modes, mode 3 had the lowest value of correlation coefficient (0.31), whereas modes 1 and 2 exhibited higher correlation of motion (Cj ≥ 0.5).
Collectivity of Motions—It has been reported that NMA effectively simulates the highly collective motions (22). The collectivity of motion was analyzed for Tt LeuRS and a value of κ = 0.27 was obtained using Equation 7 (Table 1). This higher value of κ indicates that the experimentally observed conformational changes in Tt LeuRS can be adequately described by a single mode (22). The degrees of collectivity for individual modes were also computed and are compiled in Table 1. The degrees of collectivity obtained for modes 1 and 3 were comparable (κ = ∼0.4), suggesting that the observed open/closed conformational change in Tt LeuRS can be described adequately by one of these two modes. Comparing the overlap, correlation coefficients, and collectivity for the first three lowest frequency modes (Table 1), mode 1 appeared to illustrate the collective domain motions of Tt LeuRS in the best way and was chosen for further analysis.
Correlated and Anticorrelated Motions between Various Structural Elements—Results of NMA provided correlations and anticorrelations in fluctuations of various Tt LeuRS domains. The cross-correlation coefficients between the residue fluctuations of the protein were calculated using Equation 9 for mode 1 and are given as an 814 × 814 matrix (Fig. 3). Correlated residues are colored in red, anticorrelated residues are in blue, and the relative magnitude of these correlated and anti-correlated fluctuations are indicated in a normalized (+1 to -1) scale as shown in the color bar. This analysis was further extended to identify key Tt LeuRS structural entities that are engaged in either correlated or anticorrelated motions. Correlation coefficients of selective coupled segments of the protein were extracted from the parent matrix and are illustrated in Fig. 4.
FIGURE 3.

NMA cross-correlation matrix for the 814 residues of Tt LeuRS. The cross-correlations between residue fluctuations (Cpq), calculated using Equation 9, are normalized and expressed with a color-coded scale. A value of +1.0 was set for the most strongly correlated motion and is colored red, whereas -1.0 was used for the most strongly anticorrelated motion and is colored blue.
FIGURE 4.

Cooperativity of dynamics between various segments of Tt LeuRS, as observed in the normal mode calculations. a-d represents the submatrices of cross-correlations between residue fluctuations of various structural elements (e). These submatrices are extracted from the parent 814 × 814 cross-correlation matrix using MATLAB R2006b. The cross-correlations are expressed by the same color-coded scale as described in Fig. 3. Relevant structural elements are shown in different colors: tan, 49HMGH; orange, LSD; cyan, 637VMSKS; blue, residues 538-546; yellow, ZBD; red, 420RDW; magenta, residues 290-300; green, residues 330-340.
Analysis of the two-dimensional cross-correlation matrix revealed that the motion of the LSD was predominately anti-correlated with respect to the ED (Fig. 4, a and e). However, the LSD exhibited correlated motion with respect to a helical region (residues 290-300) and a polypeptide segment (residues 330-340) of the ED. Moreover, the LSD was engaged in highly correlated motion with the catalytically important 49HMGH and 637VMSKS loops (Fig. 4, and e). Additionally, these loops were also engaged in correlated motions with respect to other structural elements in the CCD including the polypeptide 537GGVEHAVLH, a protein segment that is critical for leucyl-adenylate binding (Fig. 4, c and e). The 537GGVEHAVLH polypeptide exhibited strong correlation with the ZBD (Fig. 4, d and e), which in turn was engaged in strong correlated motion with the helix (residues 290-300) of the ED.
The correlated/anticorrelated motions among various functional sites demonstrated a clear link between the coordination of Tt LeuRS functions and its domain dynamics. This dynamics-functional interdependence is described below.
Anticorrelated Motion between the ED and the LSD—As mentioned above, the NMA study of Tt LeuRS showed that the relative motions between the ED and the LSD are predominantly anticorrelated (Fig. 4, a and e). This is consistent with the structural studies which revealed that the ED of Tt LeuRS, when complexed with tRNALeu in the post-transfer-editing conformation, undergoes a rotation of 35° (Fig. 1) relative to the tRNA unbound form (9). Similar rotation of the ED (47° relative to the main body) has also been observed in the case of another class 1 enzyme, isoleucyl-tRNA synthetase (41). It has been proposed that this change in conformation of the editing domain opens up a deep channel (41) between the aminoacylating (synthetic) and editing active sites. The opening of this channel facilitates the shuttling of the 3′ terminus of mischarged tRNALeu from the synthetic active site to the editing active site (10, 41). It is now evident from our study that the anticorrelated motion between the ED and the LSD (Fig. 2) actually assists the collective atomic displacements that are needed to open up a deep cleft between these two domains. The periodic compression and expansion of the cleft existing between the ED and the LSD through an anticorrelated motion is likely to be a key regulatory element to the movement of the 3′-end of tRNALeu from the CCD to the ED during the translocation process. Therefore, the present NMA result is in excellent agreement with the predicted “pseudoreversible” dynamics (42) of tRNA translocation based on biochemical and structural studies (9, 10, 12, 41).
Correlated Motion between the ZBD and the Glu-292-containing Helix—One of the intriguing observations of the present study is the existence of strong correlations between the dynamics of a thin segment of the ED (Glu-292-containing helix) and the ZBD (Fig. 4, d and e). Structural studies revealed that the ZBD (residues 154-189) is highly mobile and attains two conformations: open and closed (9, 43). The closed form of the ZBD is responsible for protecting the leucyl adenylate from water hydrolysis. In the open form the ZBD is believed to undergo a large rotation away from the catalytic center, which makes the adenylate intermediate accessible to the cognate tRNA (43). Therefore, the oscillatory dynamics of the ZBD has an important role in the aminoacylation of tRNALeu. From biochemical studies it was revealed that any disruption of the secondary structure of the Glu-292-containing helix has significant impact on the aminoacylation (44-46). There was no clear explanation of why a change in this distant structural element could have such a significant impact on the aminoacylation reaction. The very existence of the strongly coupled motion between the Glu-292-containing helix and the ZBD, found in this study, demonstrates that the aminoacylation process is fundamentally dependent upon the cooperative dynamics between these two structural elements, located in separate domains.
The correlated motion between the ZBD and the Glu-292-containing helix also supports the unmasking of the pretransfer editing mechanism in a post-transfer-defective mutant and the ED (also called connective polypeptide 1 (CP1) domain) deletion mutant of E. coli LeuRS (47, 48). The existence of a correlated motion between the Glu-292-containing helix and the ZBD suggests that a point mutation in the Glu-292-containing helix (47) or a complete deletion of CP1 domain (48) might disrupt the coordinated movement of these peptide segments, thereby preventing the ZBD to acquire a conformation essential for the adenylate binding and its protection against water hydrolysis. Although the NMA study alone could not shed any light on the exact mechanism of the pretransfer editing reaction (for example, if the hydrolysis of noncognate adenylate is enzyme-catalyzed-selective hydrolysis or it is the selective release of noncognate adenylate from the enzyme active site followed by rapid nonenzymatic hydrolysis (49)), the present study provides a mechanistic insight into the role of coupled domain motion in the unmasking of pretransfer editing reaction of the LeuRS.
Correlated Motion among the Structural Elements of the LSD, the CCD, and the Glu-292-containing Helix—Another interesting finding of the present NMA study is the trajectory of a correlated motion existing between residue clusters of the functionally relevant structural elements of the LSD, the CCD, and the Glu-292-containing helix of the ED (Fig. 4, b-e). Previous structural studies predicted the need for a concerted movement of the LSD and the class 1-specific 637VMSKS and 49HMGH loops in amino acid selection and binding (11). The present study of the simulated domain dynamics is in complete agreement with the anticipated motion from structural studies. As described previously, the evidence of the correlated dynamics was found among the functionally relevant 637VMSKS, 49HMGH, and 537GGVEHAVLH polypeptides of the CCD. Additionally, these structural elements are also engaged in correlated motion with the LSD and the ZBD (Fig. 4). Therefore, the present study not only supports structural observations (11) but also provides insight into how the cooperative dynamics among various structural elements play a role in enzyme functions, namely, the substrate binding and aminoacylation (11, 12, 50). Taken together, cooperative domain dynamics take center stage in this protein function.
SCA of the LeuRS Family—SCA was performed on an alignment of 484 protein sequences of the LeuRS family by systematically perturbing each position where a specific amino acid was present in at least 57% of the sequences in the alignment.
Highly Conserved Tryptophan Residues—The statistical
analysis revealed a significant number of strongly conserved tryptophan
residues in the central catalytic domain. The degree of conservation of any
residue, expressed as  is
calculated by using Equation 10
(29). By choosing an arbitrary
cut-off value of
is
calculated by using Equation 10
(29). By choosing an arbitrary
cut-off value of 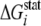 (
(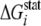 ≥ 3.50
kT*), we identified 25 residues that are extremely
conserved in the LeuRS family (Table
2). About 50% of these residues are tryptophan, belonging to the
less dynamic central core of the enzyme (supplemental Fig. S1). The presence
of such a high number of tryptophan residues indicates the requirement of
strong hydrophobic groups in maintaining the structure and function. It seems
that these tryptophan residues are involved in van der Waals packing and
thereby remained evolutionarily conserved. A list of highly conserved but
non-catalytic residues, their type, location on the protein structure, and the
computed
≥ 3.50
kT*), we identified 25 residues that are extremely
conserved in the LeuRS family (Table
2). About 50% of these residues are tryptophan, belonging to the
less dynamic central core of the enzyme (supplemental Fig. S1). The presence
of such a high number of tryptophan residues indicates the requirement of
strong hydrophobic groups in maintaining the structure and function. It seems
that these tryptophan residues are involved in van der Waals packing and
thereby remained evolutionarily conserved. A list of highly conserved but
non-catalytic residues, their type, location on the protein structure, and the
computed 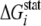 values (Equation
10) in the LeuRS family are given in Table
2.
values (Equation
10) in the LeuRS family are given in Table
2.
TABLE 2.
Highly conserved residues of the LeuRS family
Conserved residues are identified based on a threshold value of ΔGistat of 3.5 kT*, where kT* is an arbitrary energy unit (29). Computation of ΔGistat is performed using Equation 10.
| Residue number | Computed ΔGistat | Residue type | Location of the residue |
|---|---|---|---|
| kT* | |||
| Trp-17 | 4.37 | Hydrophobic | CCD |
| Met-40 | 3.75 | Hydrophobic | CCD-LSD interface |
| His-49 | 3.76 | Polar | CCD-LSD interface |
| His-52 | 3.76 | Polar | CCD |
| Trp-79 | 3.87 | Hydrophobic | CCD |
| Trp-121 | 4.35 | Hydrophobic | CCD |
| Trp-158 | 4.37 | Hydrophobic | ZBD |
| Trp-212 | 3.70 | Hydrophobic | CCD |
| Met-219 | 4.37 | Hydrophobic | CCD |
| Trp-223 | 3.42 | Hydrophobic | CCD-ZBD interface |
| Tyr-332 | 3.56 | Polar | CCD |
| His-343 | 4.37 | Polar | CCD |
| Trp-422 | 4.37 | Hydrophobic | CCD |
| Trp-430 | 4.35 | Hydrophobic | CCD-ZBD interface |
| Trp-506 | 3.42 | Hydrophobic | CCD-ZBD interface |
| Trp-529 | 4.03 | Hydrophobic | CCD |
| Tyr-535 | 3.42 | Hydrophobic | CCD-ZBD interface |
| His-541 | 3.76 | Polar | CCD-LSD interface |
| His-545 | 3.76 | Polar | CCD-ZBD interface |
| Tyr-548 | 3.42 | Hydrophobic | CCD-ZBD interface |
| Met-576 | 3.53 | Hydrophobic | CCD-LSD interface |
| Trp-676 | 4.32 | Hydrophobic | CCD-LSD interface |
| His-785 | 3.70 | Polar | CCD |
| Trp-791 | 3.94 | Hydrophobic | CCD |
| Trp-804 | 3.80 | Hydrophobic | CCD |
Coevolved Residue Clusters—The degree of coupling between
residue pairs was calculated using Equation 11 and expressed as statistical
coupling energy  for residue pair at sites i and j. Coupling between various
residue pairs were represented as a two-dimensional coupling matrix as shown
in Fig. 5. The SCA was carried
out in three stages. In stage 1, the initial round of clustering resulted in a
matrix of 876 (residue number) × 218 (perturbation site) matrix elements
representing the coupling between residues.
Fig. 5, a and
b, represent the unclustered and the first-round of
clustered matrices, respectively. As it is apparent from
Fig. 5b, a large part
of the SCA plot (blue squares) shows no coupling at all (0
kT*), whereas strong coupling was observed in the
left and right bottom-most corners (red squares,
> 0.8 kT*) as indicated. In stage 2 we performed a
two-dimensional iterative clustering and obtained three subclusters
(supplemental Fig. S2, a-c) following the methods described in the
literature (29). This process
led to the identification of several residues that have coevolved. In stage 3
we mapped all these coevolved residues (stage 2 clusters) and the 25 conserved
residues (discussed in the previous section) onto the LeuRS three-dimensional
structure. Finally, subclusters of residues that are within van der Waals
contact were identified through visualization
(Table 3). These residue
clusters, conveniently grouped into five, are located across the three
domains, namely, the LSD, the CCD, and the ED as well as at their interfaces.
The statistical couplings among intra- and intercluster residues are
represented in four separate SCA plots
(Fig. 6).
for residue pair at sites i and j. Coupling between various
residue pairs were represented as a two-dimensional coupling matrix as shown
in Fig. 5. The SCA was carried
out in three stages. In stage 1, the initial round of clustering resulted in a
matrix of 876 (residue number) × 218 (perturbation site) matrix elements
representing the coupling between residues.
Fig. 5, a and
b, represent the unclustered and the first-round of
clustered matrices, respectively. As it is apparent from
Fig. 5b, a large part
of the SCA plot (blue squares) shows no coupling at all (0
kT*), whereas strong coupling was observed in the
left and right bottom-most corners (red squares,
> 0.8 kT*) as indicated. In stage 2 we performed a
two-dimensional iterative clustering and obtained three subclusters
(supplemental Fig. S2, a-c) following the methods described in the
literature (29). This process
led to the identification of several residues that have coevolved. In stage 3
we mapped all these coevolved residues (stage 2 clusters) and the 25 conserved
residues (discussed in the previous section) onto the LeuRS three-dimensional
structure. Finally, subclusters of residues that are within van der Waals
contact were identified through visualization
(Table 3). These residue
clusters, conveniently grouped into five, are located across the three
domains, namely, the LSD, the CCD, and the ED as well as at their interfaces.
The statistical couplings among intra- and intercluster residues are
represented in four separate SCA plots
(Fig. 6).
FIGURE 5.

Statistical coupling analysis of the LeuRS family. Statistical
coupling matrix: a, no clustering; b, one-dimensional
hierarchical clustering showing the co-evolving residues. The color gradient,
as indicated in the color bar, is as follows: blue squares represent
the lowest (0 kT*), and red squares represent the
highest (1 kT*) statistical coupling energies,
 (calculated using
Equation 11).
(calculated using
Equation 11).
TABLE 3.
| Clusters | Residues |
|---|---|
| Cluster A | Met-576 Val-577 Val-637 Met-638 Lys-640 Ser-641 Lys-642 Gly-643 Asn-644 Gly-645 Met-647 Val-648 Thr-663 Ala-667 Met-674 Val-675 Trp-676 Val-681 |
| Cluster B | Val-37 Val-39 Met-40 Met-50 L53 Lys-54 Asn-55 Met-58 Val-61 His-75 Gln-534 Tyr-535 I536 Phe-572 Ala-738 |
| Cluster C | C162 Gln-170 Trp-177 Trp-212 Val-216 Met-219 Trp-223 Trp-422 Leu-423 Met-498 Asp-499 Thr-500 Phe-501 Trp-506 Tyr-507 Pro-520 Val-539 His-541 Val-543 His-545 Tyr-548 Phe-552 Leu-556 His-557 |
| Cluster D | Val-156 Asn-157 Trp-158 Glu-188 Leu-189 Thr-268 Ala-273 Tyr-283 Ala-287 Glu-292 Arg-295 Glu-298 Gly-299 Arg-300 |
| Cluster E | Trp-79 Ala-89 Phe-F93 Trp-100 Tyr-102 Gln-107 Ala-108 Trp-121 Thr-126 Cys-128 Trp-430 Ser-639 Lys-640 |
FIGURE 6.

Trajectory of correlated motions observed through clusters of coevolving residues. The motion path is shown with broken-lined arrows. The five clusters (A-E) are mapped on to the Tt LeuRS structure and are displayed at the center. The four SCA plots showing both intra- and intercouplings between A with four other clusters are displayed at corners. Residue numbers shown in italics belong to Cluster A.
Combined NMA-SCA Study—Results from the NMA provided a discrete pattern of cooperative motion in Tt LeuRS that is spread across various domains, whereas the evolutionarily coupled residues were identified by using the SCA. To explore the thermal coupling between these SCA-generated clusters, we repeated the thermal motion analysis and extracted cross-correlations between residue fluctuations in these clusters (Fig. 7).
FIGURE 7.

Coupling of thermal motions among residues in the coevolved clusters. Cross-correlations between various clusters are shown. a, clusters A-B. b, clusters B-C. c, clusters C-D. d, clusters C-E. e, clusters A-E. The cross-correlations are expressed by the same color-coded scale as used in Fig. 3.
Thermally and Evolutionarily Coupled Residues—Through this combined NMA-SCA study, we were able to identify residues that are both evolutionarily and thermally coupled. Analysis of dynamics (from the computed normal mode simulation data) of the SCA-generated clusters (Fig. 6) demonstrated strong cooperative motions among them. These cross-correlation matrices are shown in Fig. 7, a-e. From the LSD end, strong correlated dynamics were observed between clusters A (coevolved residues at the interface of the LSD and the CCD) and B (coevolved residues mostly belonging to the CCD) (Fig. 7a) and between clusters B and C (coevolved residues mostly belonging to the CCD) (Fig. 7b). These correlated dynamics are spread up to the Glu-292-containing helix of the ED through cluster C and a part of cluster D (coevolved residues at the interface of the ZBD and the ED) (Fig. 7c). Interestingly, a part of cluster D (residues 268, 273, 283, and 287) shows a strong anticorrelated fluctuation with cluster C. It appears that a network of coupled motions exists between the two distantly located structural elements, the LSD and the Glu-292-containing helix of the ED. Therefore, it is quite possible that signals originating at one domain could propagate through a path of correlated thermal motions between neighboring clusters (as shown by the broken lined arrows in Fig. 6) to finally reach the other domain.
Existing Mutational Results—Several biochemical results indicate strong impact on enzyme functions upon mutations of residues that belong to the network of interacting clusters identified by the combined NMA-SCA study. For example, a point mutation at position 5783 (E. coli Leu-570 and Lys-600 in human mitochondrial LeuRS) located at the interface of the LSD and the CCD has been reported to have a significant effect on amino acid discrimination and tRNA aminoacylation (50). The crystal structure data (11) suggest that the residue at this position is not in direct contact with the amino acid moiety of the bound adenylate substrate. Thus, it was not clear how the mutation at the position 578 can alter amino acid specificity for this enzyme. In our study we found that Leu-578 is in van der Waals contact with Met-576, which is highly conserved (Table 2), and Val-577, which exhibits a strong evolutionary coupling with residues of functionally important 49HMGH and 637VMSKS loops (Fig. 4e and Fig. 6). The analysis of dynamics also revealed that Val-577 belongs to the interaction network and is engaged in strong correlated motion with residues of 49HMGH and 637VMSKS motifs (Fig. 4, d and e). Thus, mutation of residue at position 578 of LeuRS is likely to influence the motion of the 637VMSKS loop, which in turn could fail to modulate the active site conformation necessary for substrate recognition.
Another case of experimental evidence of the critical role of the network on function was revealed when the entire LSD was deleted. Removal of the LSD has severe impact on the aminoacylation reaction, the molecular basis of which remained unclear (12). The present study indicates strong coupling of cluster A (comprising residues at the interface of the LSD and CCD including 637VMSKS loop) and cluster B (comprised of 537GGVEHAVLH546 and 49HMGH segments) in terms of both coevolution (Figs. 4e and 6) and motion (Fig. 7). The existence of such correlated dynamics explains the role of LSD in the aminoacylation reaction.
In the present study we observed a significant coupling of motion between ZBD (154-189) and the 290-300 helix of the ED. Visualization of residues 290-300 confirms that this protein segment is in van der Waals contact with residues at positions 187 and 188. The SCA studies demonstrate that residues in these two functional sites have indeed coevolved (Fig. 6) and exhibited strong correlated motion. Therefore, mutations at these sites could have an effect on enzyme functions. In fact, mutations of residues at positions 294 and 295 (Glu-292 and Ala-293 of E. coli LeuRS) have significant impact on the editing and the aminoacylation function of LeuRS (44-46).
In addition to the existing mutational data involving residues at the
domain interfaces, the effect of mutation of a residue that is located at the
center of the networking cluster was also observed. Mutation of the highly
conserved residue at position 422 of the RDW peptide has significant impact
(30-fold decrease in kcat) on the aminoacylation reaction
(51). Our study suggests that
this position corresponds to an absolutely conserved tryptophan residue
(Trp-422 of Tt LeuRS and Trp-445 of yeast mitochondrial LeuRS
(52)) which possesses a very
high 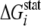 value (4.37
kT*). Also, Trp-422 is in van der Waals contact with
Leu-423, which shows significant correlation of dynamics with the ZBD and
Glu-292-containing helix (Fig.
7c). Therefore, any perturbation within the RDW peptide
is expected to have significant impact on the enzyme function.
value (4.37
kT*). Also, Trp-422 is in van der Waals contact with
Leu-423, which shows significant correlation of dynamics with the ZBD and
Glu-292-containing helix (Fig.
7c). Therefore, any perturbation within the RDW peptide
is expected to have significant impact on the enzyme function.
DISCUSSION
The polypeptide backbone and side chains in a protein exist in constant motion. These internal local motions are quite fast (in the range of femto- and picosecond time scales) (53). However, in some cases specific enzymatic function requires a slower conformational transition involving medium to large scale motions (nano- to microseconds) (54, 55). These slower motions encompass displacement of much larger structural elements such as a whole domain. It is well known that multidomain proteins utilize these collective motions to accomplish specific functions, and these functionally important protein motions can then be ascribed as “cooperative.”
Exploring Cooperative Domain Dynamics by Molecular Simulations—One way to study these slower domain motions in proteins is to perform normal mode analysis (24, 25, 56). A domain is a compactly folded region of a protein that exhibits considerable independence in terms of both stability and motion. Therefore, a protein can be conveniently described as an assembly of these compact regions (domains) joined by flexible connectors (loops). Low frequency normal modes calculated for such an assembly accurately illustrate the slower motions of domains. The coarse-grain model and the harmonic approximation of the atomic fluctuations used in the analysis (24, 56) do not seem to affect the accuracy of the results. More importantly, this method significantly reduces the computational cost so that the collective motion in a large protein like an ARS can be analyzed in great detail.
The information about the slower protein dynamics can also be obtained using a very long (in the nanosecond timescale) molecular dynamics simulation. However, the use of such expensive computational scheme is not expected to provide any additional information about these collective slower motions. On the other hand, molecular dynamics (57, 58) simulations are extremely beneficial to explore chemical events that are influenced by faster motions such as vibration effects (quantum mechanical tunneling) on enzyme kinetics (54, 55, 59-61), the role of protein electrostatics and solvents on redox transitions (62, 63), and the role of specific mutations and conformational flexibility on substrate binding and inhibition (64-68). In the present context we have rather focused on the slower displacements of functional domains that are involved in long range communications in Tt LeuRS. Hence, the use of a normal mode approximation is justified for exploring these functionally relevant domain motions.
Regulations of Cooperative Domain Dynamics in LeuRS—As illustrated in the results section, the NMA study of the domain dynamics in Tt LeuRS revealed a significant amount of cooperativity among various structural elements. Closer scrutiny of the experimental results further demonstrated a clear correspondence between protein function and the cooperative dynamics of these domains. But perhaps the bigger question that has remained unanswered is the mechanism by which this cooperativity of dynamics among various domains is regulated. Does a multidomain enzyme like LeuRS need a set of non-catalytic residues for promoting domain and subdomain dynamics? If so, how are they arranged across domains? An insight into the molecular basis of the cooperativity between such distantly located domains could possibly address these questions.
One way to explore this communicating pathway among domains is to use a simple hypothesis of the evolutionary basis for the conservation and/or coevolution of residues that are relevant for both structure and function and in the present context, the dynamics itself. Through a systematic SCA, it is possible to determine energetically coupled residues involved in allosteric communication in a protein. This method was successfully applied for a number of protein families with different structural and functional variations (8, 29, 30, 69, 70). In some of these cases, SCA identified the functional coupling of specific residue pairs that are located distantly and were not evident from the three-dimensional structure of protein alone (8, 30). So far, SCA was performed to identify the evolutionarily and energetically coupled residues within a domain of a protein. Because the underlying assumption of coevolution of two sites (for functional/structural reasons) is still valid, in this work we extended this approach to a multidomain protein family.
Coevolution of Residues under the Constraints of Cooperative Domain Dynamics—The SCA method is based on the hypothesis that the structural and functional constraints on a protein sequence allow two distant functional sites to evolve cooperatively. Therefore, in that evolutionary perspective, to maintain the structure, function, and dynamics, residues at these two sites will be either completely retained, or a mutation at one site will be suitably counterbalanced by another. Then these two residue pairs (or sites) can be considered to be evolutionarily constrained.
The present SCA study resulted in the identification of five clusters in which both intra- and intercluster evolutionary coupling of residues were observed (see above). Analysis of cross-correlations between residue fluctuations (combined SCA-NMA study) demonstrated that these coevolving residues are distributed across domains that are involved in cooperative domain dynamics. In particular, it revealed the existence of a well connected network of residues from the LSD to the ED through the CCD. Within a cluster, the van der Waal surfaces of these residues are in contact with each other. When visualized together, these clusters appear to constitute a contiguous network of discrete clusters of residues spanning from the LSD to the ED (Fig. 6, Table 3). Taken together, it is the coupling of motions that connect the two distantly located domains (the LSD and the Glu-292-containing helix of the ED) of Tt LeuRS. Therefore, we suggest that Tt LeuRS is likely to utilize this network of coupled motions in the propagation of long range signals for functional reasons such as substrate binding/release and catalysis.
Conclusions—The study of collective motions demonstrates that cooperative domain dynamics are inherent to the Tt LeuRS structure and functions. Additionally, through the structure-guided analysis of the effect of evolutionary constraints on the LeuRS sequence, the coevolved network of residues was successfully identified that is sparsely distributed across all the functional domains. Further analysis of the dynamics of these core networking groups demonstrated significant thermal coupling. Taken together, these clusters represent a distinguished set of residues that are not only coupled by motion but also retained by the evolution. It has been reported earlier that evolution preferentially conserves those contacts (residues) that are critical to the functional dynamics of an enzyme (71). Results of our study show that not only the conserved residues but also the coevolving residue pairs are critical for functional dynamics, which is cooperative in nature. Mutational studies of some key networking residues are under way.
Supplementary Material
Acknowledgments
We thank Dr. Ranganathan (University of Texas, Southwestern Medical Center, Dallas) for the MATLAB scripts of the SCA. We thank Prof. K. Musier-Forsyth and Dr. Anjali Mascarenhas for helpful discussions. We are grateful to Prof. Karen Havholm and Prof. Warren Gallagher for critical reading of the manuscript. We also thank three anonymous reviewers for constructive comments.
This work was supported by the Cottrell College Science Award, Research Corp. (Tucson, AZ) (to S. H.) and the Office of Research and Sponsored Programs, University of Wisconsin, Eau Claire, WI.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: ARS, aminoacyl-tRNA synthetase; ANM, anisotropic network model; NMA, normal mode analysis; SCA, statistical coupling analysis; MSA, multiple sequence alignment; CCD, central catalytic domain; ED, editing domain; LSD, leucine-specific domain; ZBD, zinc-1 binding domain; Tt LeuRS, T. thermophilus Leucyl-tRNA synthetase.
In the present context, a position is defined as the residue number corresponding to the query protein sequence (Tt LeuRS) used in the PSI-BLAST search.
References
- 1.Alexander, R. W., and Schimmel, P. (2001) Prog. Nucleic Acid Res. Mol. Biol. 69 317-349 [DOI] [PubMed] [Google Scholar]
- 2.Zhang, C. M., and Hou, Y. M. (2005) Biochemistry 44 7240-7249 [DOI] [PubMed] [Google Scholar]
- 3.Ibba, M., and Söll, D. (2000) Annu. Rev. Biochem. 69 617-650 [DOI] [PubMed] [Google Scholar]
- 4.Jakubowski, H., and Goldman, E. (1992) Microbiol. Rev. 56 412-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mascarenhas, A., Martinis, S. A., An, S., Rosen, A. E., and Musier-Forsyth, K. (2009) in Protein Engineering (RajBhandary, U. L., and Koehrer, C. eds) pp. 153-200, Springer-Verlag New York Inc., New York
- 6.Uter, N. T., and Perona, J. J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 14396-14401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budiman, M. E., Knaggs, M. H., Fetrow, J. S., and Alexander, R. W. (2007) Proteins 68 670-689 [DOI] [PubMed] [Google Scholar]
- 8.Estabrook, R. A., Luo, J., Purdy, M. M., Sharma, V., Weakliem, P., Bruice, T. C., and Reich, N. O. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 994-999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tukalo, M., Yaremchuk, A., Fukunaga, R., Yokoyama, S., and Cusack, S. (2005) Nat. Struct. Mol. Biol. 12 923-930 [DOI] [PubMed] [Google Scholar]
- 10.Mascarenhas, A. P., and Martinis, S. A. (2008) Biochemistry 47 4808-4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cusack, S., Yaremchuk, A., and Tukalo, M. (2000) EMBO J. 19 2351-2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vu, M. T., and Martinis, S. A. (2007) Biochemistry 46 5170-5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harte, W. E., Jr., Swaminathan, S., Mansuri, M. M., Martin, J. C., Rosenberg, I. E., and Beveridge, D. L. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 8864-8868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benkovic, S. J., and Hammes-Schiffer, S. (2003) Science 301 1196-1202 [DOI] [PubMed] [Google Scholar]
- 15.Bu, Z., Biehl, R., Monkenbusch, M., Richter, D., and Callaway, D. J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 17646-17651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benkovic, S. J., and Hammes-Schiffer, S. (2006) Science 312 208-209 [DOI] [PubMed] [Google Scholar]
- 17.Hawkins, R. J., and McLeish, T. C. (2006) Biophys. J. 91 2055-2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu, H., Ma, L., Yang, Y., and Cui, Q. (2007) PLOS Comput. Biol. 3 214-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks, B., and Karplus, M. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 6571-6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marques, O., and Sanejouand, Y. H. (1995) Proteins 23 557-560 [DOI] [PubMed] [Google Scholar]
- 21.Bahar, I., Atilgan, A. R., and Erman, B. (1997) Fold. Des. 2 173-181 [DOI] [PubMed] [Google Scholar]
- 22.Tama, F., and Sanejouand, Y. H. (2001) Protein Eng. 14 1-6 [DOI] [PubMed] [Google Scholar]
- 23.Yildirim, Y., and Doruker, P. (2004) J. Biomol. Struct. Dyn. 22 267-280 [DOI] [PubMed] [Google Scholar]
- 24.Bahar, I., and Rader, A. J. (2005) Curr. Opin. Struct. Biol. 15 586-592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma, J. (2005) Structure 13 373-380 [DOI] [PubMed] [Google Scholar]
- 26.Tama, F., and Brooks, C. L., III (2005) J. Mol. Biol. 345 299-314 [DOI] [PubMed] [Google Scholar]
- 27.Eyal, E., Yang, L. W., and Bahar, I. (2006) Bioinformatics 22 2619-2627 [DOI] [PubMed] [Google Scholar]
- 28.Kong, Y., Ma, J., Karplus, M., and Lipscomb, W. N. (2006) J. Mol. Biol. 356 237-247 [DOI] [PubMed] [Google Scholar]
- 29.Lockless, S. W., and Ranganathan, R. (1999) Science 286 295-299 [DOI] [PubMed] [Google Scholar]
- 30.Suel, G. M., Lockless, S. W., Wall, M. A., and Ranganathan, R. (2003) Nat. Struct. Biol. 10 59-69 [DOI] [PubMed] [Google Scholar]
- 31.Humphrey, W., Dalke, A., and Schulten, K. (1996) J. Mol. Graph. 14 33-38 [DOI] [PubMed] [Google Scholar]
- 32.Go, N., Noguti, T., and Nishikawa, T. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 3696-3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrison, R. W. (1984) Biopolymers 23 2943-2949 [DOI] [PubMed] [Google Scholar]
- 34.Kitao, A., and Go, N. (1999) Curr. Opin. Struct. Biol. 9 164-169 [DOI] [PubMed] [Google Scholar]
- 35.Brooks, B., and Karplus, M. (1985) Proc. Natl. Acad. Sci. U. S. A. 82 4995-4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank, J., and Agrawal, R. K. (2000) Nature 406 318-322 [DOI] [PubMed] [Google Scholar]
- 37.Atilgan, A. R., Durell, S. R., Jernigan, R. L., Demirel, M. C., Keskin, O., and Bahar, I. (2001) Biophys. J. 80 505-515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruschweiler, R. (1995) J. Chem. Phys. 102 3396-3403 [Google Scholar]
- 39.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic Acids Res. 25 3389-3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silvian, L. F., Wang, J., and Steitz, T. A. (1999) Science 285 1074-1077 [PubMed] [Google Scholar]
- 42.Rock, F. L., Mao, W., Yaremchuk, A., Tukalo, M., Crepin, T., Zhou, H., Zhang, Y. K., Hernandez, V., Akama, T., Baker, S. J., Plattner, J. J., Shapiro, L., Martinis, S. A., Benkovic, S. J., Cusack, S., and Alley, M. R. (2007) Science 316 1759-1761 [DOI] [PubMed] [Google Scholar]
- 43.Fukunaga, R., and Yokoyama, S. (2005) Nat. Struct. Mol. Biol. 12 915-922 [DOI] [PubMed] [Google Scholar]
- 44.Li, T., Guo, N., Xia, X., Wang, E. D., and Wang, Y. L. (1999) Biochemistry 38 13063-13069 [DOI] [PubMed] [Google Scholar]
- 45.Chen, J. F., Li, T., Wang, E. D., and Wang, Y. L. (2001) Biochemistry 40 1144-1149 [DOI] [PubMed] [Google Scholar]
- 46.Du, X., and Wang, E. D. (2002) Biochemistry 41 10623-10628 [DOI] [PubMed] [Google Scholar]
- 47.Williams, A. M., and Martinis, S. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 3586-3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boniecki, M. T., Vu, M. T., Betha, A. K., and Martinis, S. A. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 19223-19228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hati, S., Ziervogel, B., Sternjohn, J., Wong, F. C., Nagan, M. C., Rosen, A. E., Siliciano, P. G., Chihade, J. W., and Musier-Forsyth, K. (2006) J. Biol. Chem. 281 27862-27872 [DOI] [PubMed] [Google Scholar]
- 50.Lue, S. W., and Kelley, S. O. (2007) Biochemistry 46 4466-4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nawaz, M. H., Pang, Y. L., and Martinis, S. A. (2007) J. Mol. Biol. 367 384-394 [DOI] [PubMed] [Google Scholar]
- 52.Rho, S. B., Lincecum, T. L., Jr., and Martinis, S. A. (2002) EMBO J. 21 6874-6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tousignant, A., and Pelletier, J. N. (2004) Chem. Biol. 11 1037-1042 [DOI] [PubMed] [Google Scholar]
- 54.Hammes-Schiffer, S. (2002) Biochemistry 41 13335-13343 [DOI] [PubMed] [Google Scholar]
- 55.Hammes-Schiffer, S., and Benkovic, S. J. (2006) Annu. Rev. Biochem. 75 519-541 [DOI] [PubMed] [Google Scholar]
- 56.Ma, J., and Karplus, M. (1997) J. Mol. Biol. 274 114-131 [DOI] [PubMed] [Google Scholar]
- 57.Hansson, T., Oostenbrink, C., and van Gunsteren, W. (2002) Curr. Opin. Struct. Biol. 12 190-196 [DOI] [PubMed] [Google Scholar]
- 58.Karplus, M. (2002) Acc. Chem. Res. 35 321-323 [DOI] [PubMed] [Google Scholar]
- 59.Truhlar, D. G., Gao, J., Alhambra, C., Garcia-Viloca, M., Corchado, J., Sanchez, M. L., and Villa, J. (2002) Acc. Chem. Res. 35 341-349 [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Viloca, M., Gao, J., Karplus, M., and Truhlar, D. G. (2004) Science 303 186-195 [DOI] [PubMed] [Google Scholar]
- 61.Truhlar, D. G., Gao, J., Garcia-Viloca, M., Alhambra, C., Corchado, J., Sanchez, M. L., and Poulsen, T. D. (2004) Int. J. Quantum Chem. 100 1136-1152 [Google Scholar]
- 62.Bhattacharyya, S., Ma, S., Stankovich, M. T., Truhlar, D. G., and Gao, J. (2005) Biochemistry 44 16549-16562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhattacharyya, S., Stankovich, M. T., Truhlar, D. G., and Gao, J. (2007) J. Phys. Chem. A 111 5729-5742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soares, T. A., Lins, R. D., Straatsma, T. P., and Briggs, J. M. (2002) Biopolymers 65 313-323 [DOI] [PubMed] [Google Scholar]
- 65.Mustata, G. I., Soares, T. A., and Briggs, J. M. (2003) Biopolymers 70 186-200 [DOI] [PubMed] [Google Scholar]
- 66.Barreca, M. L., Lee, K. W., Chimirri, A., and Briggs, J. M. (2003) Biophys. J. 84 1450-1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mursinna, R. S., Lee, K. W., Briggs, J. M., and Martinis, S. A. (2004) Biochemistry 43 155-165 [DOI] [PubMed] [Google Scholar]
- 68.Brigo, A., Lee, K. W., Fogolari, F., Mustata, G. I., and Briggs, J. M. (2005) Proteins 59 723-741 [DOI] [PubMed] [Google Scholar]
- 69.Bartlett, G. J., and Taylor, W. R. (2008) Proteins 71 950-959 [DOI] [PubMed] [Google Scholar]
- 70.Nascimento, A. S., Krauchenco, S., Golubev, A. M., Gustchina, A., Wlodawer, A., and Polikarpov, I. (2008) J. Mol. Biol. 382 763-778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng, W., Brooks, B. R., and Thirumalai, D. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 7664-7669 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.