

Abstract
The serotonin transporter (SERT) regulates extracellular levels of serotonin (5-hydroxytryptamine, 5HT) in the brain by transporting 5HT into neurons and glial cells. The human SERT (hSERT) is the primary target for drugs used in the treatment of emotional disorders, including depression. hSERT belongs to the solute carrier 6 family that includes a bacterial leucine transporter (LeuT), for which a high resolution crystal structure has become available. LeuT has proved to be an excellent model for human transporters and has advanced the understanding of solute carrier 6 transporter structure-function relationships. However, the precise structural mechanism by which antidepressants inhibit hSERT and the location of their binding pockets are still elusive. We have identified a residue (Ser-438) located within the 5HT-binding pocket in hSERT to be a critical determinant for the potency of several antidepressants, including the selective serotonin reuptake inhibitor citalopram and the tricyclic antidepressants imipramine, clomipramine, and amitriptyline. A conservative mutation of Ser-438 to threonine (S438T) selectively increased the Ki values for these antidepressants up to 175-fold. The effects of introducing a protein methyl group into the 5HT-binding pocket by S438T were absent or reduced for analogs of these antidepressants lacking a single methyl group. This suggests that these antidepressants interact directly with Ser-438 during binding to hSERT, implying an overlapping localization of substrate- and inhibitor-binding sites in hSERT suggesting that antidepressants function by a mechanism that involves direct occlusion of the 5HT-binding site.
Drugs that inhibit SERT,4 such as the tricyclic antidepressants (TCAs) and the selective serotonin reuptake inhibitors (SSRIs), are widely used to treat emotional disorders such as depression and anxiety (1). Despite the vast clinical significance of SERT as a drug target, structural aspects of drug recognition and inhibition are poorly understood. SERT belongs to the solute carrier 6 (SLC6) family that includes transporters for neurotransmitters such as γ-aminobutyric acid, norepinephrine, dopamine, and glycine. The structural mechanism underlying SLC6 transporter function was largely unknown until 2005, when a high resolution crystal structure of a bacterial homolog to mammalian SLC6 transporters, LeuT (2), provided the first structural insight into SLC6 transporter function. Since then, the LeuT structure has proven an excellent platform for constructing experimentally validated three-dimensional models of binding pockets for ions, substrate, and inhibitors in the human transporters (3-8).
Similar progress in understanding the structural mechanism of antidepressant inhibition of SERT has so far been absent. Prior to arrival of the LeuT structure, mutagenesis studies have identified several residues where mutations can perturb the potency or binding affinity of SSRIs and TCAs (Fig. 1). LeuT-based homology models of SERT show that several of these residues are localized within the 5HT-binding pocket, thus supporting a model in which antidepressants bind within or in close proximity to the substrate-binding pocket (9). A competitive mechanism is supported by pharmacological studies that show the affinity of TCAs and SSRIs at SERT is sensitive to 5HT concentration (10-12).
FIGURE 1.

Topology of SERT derived from LeuT. The substrate-binding pocket is formed by residues positioned in TM1, TM3, TM6, and TM8 (shown in gray). Residues elucidated as critical in recognition of SSRIs and TCAs are indicated as follows: Tyr-95 (29), Asp-98 (23), Ala-169 (31), Ile-172 (28), Met-180 (30), Ser-276 (40), Phe-513 (30), Ser-545 (41), and Phe-586 (42).
However, conclusive interpretation of mutagenesis studies has been hampered by the lack of structural information on SERT, and a consensus regarding the location of the binding sites for TCAs and SSRIs has therefore not been reached. Recently, this lack of consensus was further substantiated when two studies found LeuT to be inhibited by TCAs in a binding pocket outside the substrate-binding site (Fig. 2A) (13, 14). Based on mutations of ortholog residues in the human monoamine transporters, Zhou et al. (13) proposed that the primary effects of TCAs and possibly SSRIs are mediated by binding in this region in hSERT. Here we report that a subtle mutation in hSERT, S438T, dramatically affects the potency of antidepressants containing a dimethylaminopropyl chain. This effect is reduced or absent in the analogs of these antidepressants in which a single methyl is removed from the dimethylamino moiety, implicating that antidepressants and Ser-438 are in close proximity. Comparative modeling of hSERT suggests Ser-438 to be located within the 5HT-binding pocket. In contrast, mutations within the region in hSERT orthologous to the TCA-binding site found in LeuT did not decrease antidepressant potency significantly. Combined with previous data, our results could clarify the current disagreement regarding the position of the binding pocket for antidepressants in hSERT.
FIGURE 2.

Mutation of Ser-438 in the hSERT substrate-binding pocket affects the inhibitory potency of antidepressants. A, structure of LeuT in complex with leucine and desipramine (PDB code 2QJU). TM1 (blue), TM8 (green), and TM10 (pink) are highlighted. TM6 and TM11 are removed for clarity. Leucine and desipramine are shown as CPK representations in yellow and orange, respectively. Arg-30 and Asp-404 (shown as sticks) form the extracellular gate that separates leucine from the bound desipramine. B, left, substrate-binding site in LeuT (PDB code 2A65). Right, homology model of the substrate-binding site in hSERT. Conserved residues are shown in gray, and divergent residues are shown in dark red. TM1 (blue), TM6 (gray), and TM8 (green) are shown in both panels. C, graphical summary of the fold change (mean ± S.E.; n = 6-8) in potency of inhibitors at hSERT S438T compared with hSERT WT. Ki values and statistics are shown in Table 1 and supplemental Table S1. # indicates that fold change is less than 1. D, inhibition of 5HT uptake at hSERT WT (▪) and S438T (□) by citalopram and aminoethyl citalopram. The concentration-response curve as shown is a composite of six independent experiments. Error bars are S.E. and are shown when larger than symbol size.
EXPERIMENTAL PROCEDURES
Chemicals—Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, trypsin, and penicillin/streptomycin were purchased from Invitrogen. Cell culture flasks and 96-well plates were from NUNC (VWR International, West Chester, PA). [3H]5HT (20.3 Ci/mmol) and MicroScint-20 scintillation mixture were obtained from PerkinElmer Life Sciences. [3H]-2-(2-dimethylaminomethyl-phenylsulphanyl)-5-methyl-phenylamine ([3H]MADAM) (71.0 Ci/mmol) was from GE Healthcare (Buckinghamshire, England). Imipramine, desipramine, clomipramine, monomethyl-clomipramine, amitriptyline, nortriptyline, citalopram, monomethyl-citalopram, des-methyl-citalopram, (S)-citalopram, (S)-monomethyl-citalopram, venlafaxine, sertraline, duloxetine, fluoxetine, paroxetine, nisoxetine, MADAM, and aminoethyl-citalopram were kindly provided from H. Lundbeck A/S, Copenhagen, Denmark. RTI-55 was purchased from ABX (Radeberg, Germany).
Molecular Biology—The mammalian expression plasmid pcDNA3-hSERT containing human SERT cDNA has been described previously (15). Generation of point mutations (I179D, I179F, D400F, D400K, D400L, L406D, L406F, L406K, S438T, V489D, V489F, V489K, K490D, K490F, and K490T) in pcDNA3-hSERT was performed by site-directed mutagenesis using the QuickChange mutagenesis kit (Stratagene, Carlsbad, CA), followed by sequencing of the entire gene (MWG Biotec, Martinsried, Germany).
Cell Culturing and Expression of Human SERTs—COS7 cells (ATCC, Manassas, VA) were cultured in DMEM with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5% CO2 environment. Cells were transfected using TransIT DNA transfection reagent following the protocol supplied by the manufacturer. Prior to transfection, confluent cells growing in monolayer were resuspended in DMEM at a concentration of 1.3 × 106 cells/ml. Per 96-well plate, 6 μg of DNA and 18 μl of transfection reagent were mixed in 0.6 ml of DMEM and incubated at 20 °C for 20 min. Subsequently, this mixture was added to the cell suspension, and immediately afterward the cells were dispensed into white 96-well plates at 50% confluence.
Uptake Assays—Uptake assays were performed 40 h after transfection. Cells were washed twice with PBSCM buffer (in mm: NaCl, 137; KCl, 2.7; Na2HPO4, 4.3; KH2PO4, 1.4; CaCl2, 0.5; MgCl2, 0.5) prior to uptake experiments. In inhibition studies, cells were incubated with increasing concentrations of inhibitors in PBSCM and 50 or 150 nm [3H]5HT at 20 °C for 30 min. In saturation experiments, cells were incubated at 20 °C for 10 min in PBSCM containing increasing concentrations of [3H]5HT diluted 10-fold with unlabeled 5HT. Uptake was terminated by washing three times with PBSCM. The amount of accumulated [3H]5HT was determined by solubilizing cells in scintillant with counting of plates in a Packard TopCounter (PerkinElmer Life Sciences). Nonspecific uptake was determined as uptake in nontransfected cells. Assays were carried out in triplicate and repeated at least six times.
Cell Membrane Preparation and Radioligand-binding Displacement Assay—COS7 cells transiently expressing WT or S438T hSERT growing in 150-mm tissue culture Petri plates were washed with phosphate-buffered saline with 1 mm EDTA to detach from the plate. Cell suspension was centrifuged at low speed (700 × g) at room temperature for 5 min. Cell pellet was resuspended in cold H2O and frozen at -20 °C for 1 h. The suspension was thawed on ice and subjected to 10-15 passages through a 21-gauge needle to disrupt cells. Homogenate was transferred to cold 2-ml microcentrifuge tubes and centrifuged at 18,000 × g at 4 °C for 30 min. Supernatant was aspirated, and the pellet was resuspended in PBSCM. Protein concentration of the resulting membrane preparation was determined according to the BCA method using the Pierce BCA protein assay (ThermoFisher, Rockford, IL). Membranes were used directly for binding experiments or stored at -80 °C until use. For saturation binding studies, increasing concentrations of [3H]MADAM and 30-50 μg of total membrane protein per sample were combined in 96-well plates, and total volume was adjusted to 200 μl per sample. Binding was allowed to proceed for 2 h at room temperature with gentle rocking. Subsequently, membranes were transferred to 96-well glass fiber filter plates (Unifilter C, PerkinElmer Life Sciences) preincubated with 0.1% polyethyleneimine using a Packard Bell cell harvester (PerkinElmer Life Sciences) and washed four times with water. Nonspecific binding was determined in parallel at membranes from nontransfected COS7 cells. Filter plates were dried and soaked in scintillant followed by counting in a Packard Top-counter (PerkinElmer Life Sciences). Saturation binding assays were carried out in duplicate and repeated at least five times.
For competition binding assays, 30-50 μg of total membrane protein was incubated with a fixed concentration of [3H]MADAM (5 nm for WT and 25 nm for S438T) in the presence of increasing concentrations of inhibitor using the same protocol as for saturation binding experiments. Competition binding assays were carried out in duplicate and repeated at least three times.
Molecular Modeling—The crystal structure of LeuT (PDB accession code 2A65) was used to construct a model of hSERT using the MODELLER comparative modeling package (16) following the alignment of SERT and LeuT by Beuming et al. (17). The sodium ions were inserted manually in the same position as in PDB accession code 2A65. 5HT was docked into the hSERT model using the Glide docking program (18, 19) in Maestro (Schrödinger, LLC, version 8.5) with default settings applied. Because LeuT was crystallized with the small substrate leucine, the cavity was rather small for accommodating larger ligands. Therefore, we used a flexible docking approach using the induced fit docking procedure in Maestro for docking of (S)-citalopram and imipramine. Because the side chain of Phe-335 was placed on the extracellular side of the binding site as an aromatic lid, this side chain prevents access into the cavity. Therefore, the option in the induced fit docking workflow was used, in which this residue initially was mutated to an Ala residue and then added again later in the refinement.
Data Analysis—All data analysis was performed using Prism 4.0 software (GraphPad Inc., San Diego). For determination of IC50 values, dose-response data from [3H]5HT uptake inhibition assays were fitted by Equation 1,
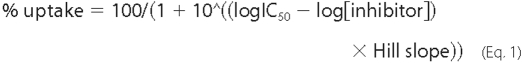
where IC50 is the concentration of inhibitor that produces a half-maximal inhibition of uptake. For determination of Km (the Michaelis-Menten constant) and Vmax (maximal uptake rate), the uptake rate was plotted as function of substrate concentration and fitted by Equation 2,
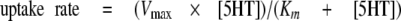 |
(Eq.2) |
IC50 values were converted to Ki values using the Cheng-Prusoff approximation (20), Equation 3,
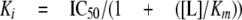 |
(Eq.3) |
where [L] is the concentration of [3H]5HT. Ki values were compared using Student's t test unless otherwise indicated.
For determination of Kd (the dissociation constant), data from saturation binding experiments were fitted to Equation 4,
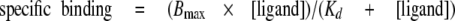 |
(Eq.4) |
where Bmax is the maximal binding, and Kd is the concentration of ligand required to reach half-maximal binding. For determination of IC50, dose-response data from competition binding experiments were fitted to Equation 5,
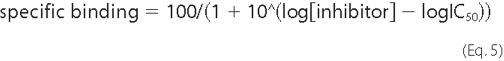
where IC50 is the concentration of inhibitor that produces a half-maximal binding of radioligand. IC50 values were converted to Ki values using the Cheng-Prusoff Equation 6,
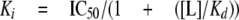 |
(Eq.6) |
where [L] is the concentration of [3H]MADAM.
RESULTS
Conservative Mutation S438T Selectively Affects hSERT Inhibitors—The importance of Ser-438 for antidepressant binding was initially discovered during a LeuT-guided mutational study of hSERT aimed at identifying residues forming the binding pocket for the SSRI citalopram (43). Our initial observation was that a conservative mutation of Ser-438 to threonine (S438T), thus introducing only a methyl group, increased the Ki value for citalopram inhibition of [3H]5HT uptake 175-fold (Fig. 2, C and D)(p < 0.0001; n = 6, paired t test). The Km value for 5HT transport was 6.9-fold decreased by this mutation (0.97 ± 0.09 μm, n = 9, for WT versus 0.14 ± 0.02 μm, n = 4, for S438T, p < 0.0001, t test), and Vmax was 5.5-fold decreased (6.6 ± 1.1 pmol/well/min, n = 9, for WT versus 1.2 ± 0.1 pmol/well/min, n = 4, for S438T; p < 0.0001, t test). Concomitant characterization of 11 prototypical SERT inhibitors showed a remarkably selective influence of S438T on inhibitor potency (Fig. 2C; Table 1; supplemental Fig. S1; supplemental Table S1). Specifically, seven inhibitors showed decreased Ki values at S438T compared with WT ranging from 7-fold for the TCA imipramine to 450-fold for the cocaine analog RTI-55. The remaining compounds displayed no change or less than a 2-fold change in Ki.
TABLE 1.
Impact of hSERT S438T on [3H]5HT uptake inhibition by SERT inhibitors
The Ki values for SERT inhibitors were determined at hSERT WT and hSERT S438T in a [3H]5HT uptake inhibition assay as described under “Experimental Procedures.” Data represent mean ± S.E. from at least six independent experiments (number of replicates are given in parentheses) each performed in triplicate. The number of methyl groups on the aminopropyl chain of the inhibitors is indicated.

** indicates p < 0.01.
*** indicates p < 0.001 (Student's t test).
Ser-438 Is Located in the 5HT-binding Pocket—Structural sequence alignment shows that Ser-438 in hSERT corresponds to Ser-355 in LeuT (Fig. 2B) (2, 17, 21). In LeuT, Ser-355 is located in the substrate-binding pocket interacting directly with the alkyl side chain of the substrate leucine and a Na+ ion. Previous experiments with hSERT have identified several residues important for substrate recognition. Most of these residues align well with residues in LeuT that form the substrate-binding pocket (21), substantiating that LeuT is a valid structural model of the 5HT-binding pocket in hSERT. In particular, evidence for a key role of Ser-438 in 5HT recognition has recently been shown in hSERT (8). In LeuT, Ser-355 is located 5 Å from Gly-24, which is replaced by an aspartate in hSERT (Fig. 2B; Asp-98 in hSERT) (2). This aspartate residue is conserved in all SLC6 transporters that transport monoamines (5HT, dopamine, and norepinephrine), and several lines of experimental data have established that the aspartate carboxylate group coordinates the primary amino group of monoamines (7, 8, 22-24). All TCAs and SSRIs contain an essential amino group that has been proposed to form similar interactions with Asp-98 in hSERT (23). Our LeuT-based homology model of hSERT shows that Ser-438 is located within 4 Å of Asp-98 (Fig. 2B). Considering this close proximity of Asp-98 to Ser-438 in hSERT, the S438T mutation might thus specifically perturb the ability of the substrate-binding pocket to accommodate the aminopropyl chain of citalopram.
Reciprocal Modification of Citalopram Modified the Effect of the S438T Mutation—We hypothesized that a citalopram analog with a shorter aminoalkyl chain would be less affected by the S438T mutation. We tested this idea by evaluating the effect of S438T on a citalopram analog containing a shorter aminoalkyl chain, and we found a 1.6-fold difference in Ki between WT and S438T hSERT (Fig. 2D; supplemental Table S1)(p = 0.11, n = 6; t test). This striking effect indicates that S438T may introduce a steric clash between the dimethylaminopropyl chain of citalopram and the γ-methyl group of the threonine (Fig. 3). To explore this further, we determined the effect of S438T on citalopram analogs where one or two methyl groups were removed from the aminopropyl chain, anticipating these subtle modifications to gradually decrease the effect of the S438T mutation on inhibitor potency. Indeed, Ki values for the monomethyl and des-methyl citalopram analogs (Fig. 4; Table 1) were remarkably less affected by the S438T mutation displaying only a 5.8- and 1.7-fold loss in potency, respectively, compared with the 175-fold decrease observed for citalopram (p < 0.0001, n = 6 (citalopram versus monomethyl-citalopram); p < 0.0001, n = 6 (monomethyl-citalopram versus des-methyl-citalopram); t test). This demonstrates that the addition of the protein methyl group at position 438 is compensated by removal of methyl groups on the aminopropyl chain of the ligand. We also characterized (S)-citalopram, which is the more potent enantiomer of the racemic citalopram, and its monomethyl analog (S)-monomethyl-citalopram at WT and S438T, and we observed a similar pattern with 320-fold loss of potency for (S)-citalopram compared with only a 12-fold loss of potency for (S)-monomethyl-citalopram (p < 0.001, n = 6-8 ((S)-citalopram versus (S)-monomethyl-citalopram); t test) (Table 1).
FIGURE 3.

Proposed working model for the impact of S438T mutation. Schematic showing hSERT interaction with the aminopropyl chain of TCAs and SSRIs. Asp-98 on TM1 (blue) coordinates the aminopropyl group. Introduction of a methyl group at Ser-438 on TM8 (green) by the S438T mutation leads to steric clash (indicated by the red dashed line) with a methyl group on the aminopropyl chain. The steric clash is removed with monomethyl analogs.
FIGURE 4.

Functional [3H]5HT uptake inhibition analysis at hSERT WT and hSERT S438T. A, upper, structure of inhibitors having one (R = H) or two (R = CH3) methyl groups at the aminopropyl chain. Lower, characterization of pairs of inhibitors having a single (blue squares, WT; blue triangles, S438T) or two (▪, WT; ▴, S438T) methyl groups at the aminopropyl chain. B, graphical summary of observed fold change in Ki between WT and S438T for each inhibitor pair (mean ± S.E.; n = 6-8). Ki values and statistics are shown in Table 1.
Differential Effect of Removing Methyl Groups on TCAs at WT and S438T hSERT—The ability of the S438T mutation to differentiate between one or two methyl groups on the aminopropyl chain of citalopram strongly suggests that Ser-438 is a direct contact site for citalopram. To investigate the generality of this phenomenon, we investigated the effect of S438T on another major class of antidepressants, the TCAs. The TCA imipramine contains the same dimethylaminopropyl chain as citalopram, and desipramine is the monomethyl analog of imipramine. This close analogy to citalopram and monomethyl-citalopram allowed us to test if the potency at hSERT of these TCAs also is differentially affected by the S438T mutation following similar patterns. We therefore determined the Ki value for imipramine and desipramine at WT and S438T (Fig. 4; Table 1). It is very well documented that a dimethylamine moiety in TCAs and SSRIs increases affinity and selectivity for SERT (whereas the corresponding monomethyl analogs are preferred by norepinephrine transporter) (25-27). Indeed, the determined Ki values at WT hSERT for imipramine and desipramine verify this preference as the dimethyl analog imipramine has lower Ki than the monomethyl analog desipramine (Table 1). However, when these two analogs were tested on S438T, we found imipramine to be more sensitive to this mutation compared with desipramine. Specifically, the S438T mutation induced a 7.1-fold loss of potency for imipramine in contrast to a much smaller 1.7-fold loss for desipramine (p < 0.0001, n = 6 (imipramine versus desipramine); t test). Thus, as observed for monomethyl-citalopram and citalopram, the S438T-induced increase in Ki is larger for the dimethyl analog (Fig. 4B). Thus, it appears that for the imipramine/desipramine pair of analogs, the monomethyl tolerates the added protein methyl much better than its dimethyl counterpart.
Next, we characterized clomipramine and its monomethyl analog, monomethyl-clomipramine, at WT and S438T and observed the same pattern (Fig. 4; Table 1); clomipramine had an 11.4-fold loss of potency, whereas monomethyl-clomipramine displayed only a 4.0-fold loss (p = 0.0084, n = 8 (clomipramine versus monomethyl-clomipramine); t test). Finally, we characterized the TCAs amitriptyline and nortriptyline, which differ in the same way as the other TCAs (Fig. 4; Table 1). Amitriptyline showed an 11.5-fold loss of potency, whereas nortriptyline had no change of potency on S438T compared with WT (p < 0.0001, n = 8 (amitriptyline versus nortriptyline); t test).
Competitive Binding Analysis of the S438T Mutation—Rather than directly perturbing ligand interaction with hSERT, the effects of the S438T mutation on the inhibitory potency of antidepressants could be caused via long range allosteric modification of the inhibitor-binding pocket. Therefore, we performed [3H]MADAM competitive binding assays to test if S438T-induced changes in inhibitory potency reflect a concomitant loss in binding affinity of the inhibitors (Table 2). [3H]MADAM was used to label SERT because the apparent Ki value of this radioligand was almost insensitive to the S438T mutation in contrast to other hSERT radioligands ([3H]imipramine, [3H]paroxetine, [3H]escitalopram, and [125I]RTI-55). Saturation binding analysis of [3H]MADAM showed that the Kd value was increased at hSERT S438T compared with hSERT WT (1.3 ± 0.3 nm, n = 6, for WT versus 21.6 ± 3.7 nm, n = 5, for S438T). Results from the competition binding assays corroborated the observations from the functional uptake assay. For all sets of mono- and dimethyl analog pairs, the dimethyl analog had the highest affinity for wild type hSERT (Table 2). Also, dimethyl analogs were in all cases more sensitive to the S438T mutation than their monomethyl counterpart, i.e. the affinity decrease caused by introduction of the protein methyl group in the S438T mutant was significantly smaller for all inhibitors containing a monomethylaminopropyl chain than the affinity decrease observed for their dimethyl analogs (Table 2). Notably, the observed Ki values obtained from the radioligand displacement assay are for all tested compounds on average an order of magnitude lower than the Ki values obtained from the functional uptake assay (Tables 1 and 2). Specifically, the concentration of compound required for displacing [3H]MADAM is lower than that required for inhibiting [3H]5HT transport. This assay-dependent difference is often observed for SERT inhibitors (15, 28-31) and is suggested to result from not well understood kinetic characteristics of transporter function in which an inhibitor can display a different Ki value for transport inhibition relative to its true dissociation constant determined from equilibrium binding experiments (32-34). However, the discrepancies between the Ki values obtained in either assay did not change the general rank order of inhibitor affinity or potency at either WT or S438T hSERT, and thus did not influence our hypothesis for the role of Ser-438 in inhibitor binding.
TABLE 2.
Impact of hSERT S438T on [3H]MADAM competition binding by SERT inhibitors
The Ki values for SERT inhibitors were determined at hSERT WT and hSERT S438T in a [3H]MADAM competition binding assay as described under “Experimental Procedures.” Data represent mean ± S.E. from at least three independent experiments (number of replicates are given in parentheses) each performed in duplicate. The number of methyl groups on the aminopropyl chain of the inhibitors is indicated.

** indicates p < 0.01 (Student's t test).
Induced Fit Docking of Citalopram and Imipramine—The inhibitory potency of SERT inhibitors containing a dimethylaminopropyl moiety are more sensitive to the S438T mutation compared with their monomethyl congeners. However, citalopram is remarkably more affected by the mutation compared with the three TCA molecules having a dimethylaminopropyl chain (imipramine, clomipramine, and amitriptyline). To better understand these differences, we performed induced fit docking of (S)-citalopram and the prototypical TCA, imipramine, into our homology model of hSERT (Fig. 5; supplemental Fig. S2). The dimethylamino group of both inhibitors is stabilized by the carboxylate group of Asp-98. However, the dimethylamino group of (S)-citalopram is located in closer proximity to Ser-438 compared with the same moiety of imipramine (4 versus 6 Å). Because the hydroxy group on the side chain of Ser-438 also coordinates one of the sodium ions, it cannot interact with the antidepressants through a hydrogen bond but is important in defining the cavity around the amino group. Given this difference in proximity of the inhibitors and Ser-438, this analysis provides a general explanation for the experimental results showing TCAs to be less affected by the introduction of a methyl group at Ser-438 compared with citalopram. Specifically, because the modeling shows imipramine to be located further away from Ser-438 compared with (S)-citalopram, the steric clash between a methyl group on the dimethylaminopropyl chain and the introduced protein methyl group at S438T would affect imipramine to a lesser extent than (S)-citalopram.
FIGURE 5.

Induced fit docking of (S)-citalopram (A) and imipramine (B). The highest scoring binding modes of both compounds are shown. TM1 (blue), TM3 and TM8 (green) are shown in both panels. Distances between the amino groups of the inhibitors and the carboxylate oxygen atom of Asp-98 and the C-β atom of Ser-438 are shown.
Role of hSERT Vestibule Residues for Antidepressant Binding—The ability of S438T to discern the presence of methyl groups on antidepressants suggests that this residue is a direct contact point for these ligands, implicating the 5HT-binding pocket as a likely binding site for these inhibitors. In contrast, the TCA-binding pocket recently found in LeuT is located in a region that forms a vestibule in the substrate permeation pathway that is probably shared by all SLC6 transporters (13, 14, 35, 36). The orthologous region in hSERT could presumably form a secondary binding pocket for antidepressants and contribute to the inhibitory mechanism by influencing the en route passage to the primary binding pocket or by allosteric modulating of binding at the primary pocket. We therefore examined the contribution of this region in hSERT to antidepressant function by introducing a range of nonconservative mutations (Fig. 6; supplemental Table S2). Specifically, we introduced considerably different side chains at five positions to perturb potential molecular contact to the antidepressants. Of the resulting 14 mutants, 9 had intact 5HT transport function, but none of these mutations increased Ki values significantly for (S)-citalopram, imipramine, or clomipramine. In contrast, a few mutants displayed slightly decreased Ki. Based on this lack of effect of vestibule mutations to perturb antidepressant potency, we suggest that this region in hSERT has minor influence on the inhibitory mechanism of TCAs and SSRIs.
FIGURE 6.

Impact of vestibule mutations in hSERT on inhibitor potency. A, left, structure of the TCA-binding site in LeuT with imipramine bound (PDB code 2Q72). Right, overlay of homology model of hSERT with LeuT structure (shown in left panel) to show the equivalent site in hSERT. TM1 (blue) and TM3, TM6, EL4, and TM10 (pink) are shown. B, graphical summary of fold change (mean ± S.E.; n = 6-10) in potency of (S)-citalopram, imipramine, and clomipramine at hSERT mutants compared with WT hSERT. For comparison, the fold changes in potency for (S)-citalopram, imipramine, and clomipramine at S438T compared with WT hSERT are shown. Ki values and statistics are shown in supplemental Table S2. * indicates a significant change (p < 0.05) in Ki values.
DISCUSSION
Fundamental requirements for understanding the structural mechanism underlying antidepressant inhibition of SERT include unambiguous identification of the location of the inhibitor-binding site and elucidation of specific protein-ligand contacts. The LeuT structure has great potential to guide functional studies aimed at providing this information, but to take full advantage of this structural template, it is critically important to establish similarities and discrepancies between the bacterial transporter and its mammalian relatives. Zhou et al. (13) suggests TCAs and possibly SSRIs inhibit human monoamine transporters by the same mechanism as observed in LeuT, implicating that the primary binding site is located in the transporter vestibule, separated from the substrate-binding pocket. This hypothesis seems in conflict with the fact that SSRIs and TCAs act as competitive inhibitors at SERT (10-12). However, accessibility of a distinct binding pocket in the vestibule is likely to vary during transport activity and could therefore be sensitive to increasing 5HT concentrations. Molecular dynamics simulations of LeuT have suggested that the vestibule in LeuT holds a second substrate-binding site (37). However, a recent study found that crystallization of LeuT in the presence of 30 mm leucine did not produce crystals where this suggested site was occupied by substrate (38). Still, although the existence of a second substrate-binding site in the LeuT vestibule remains unresolved, the vestibule in SERT might still have a second 5HT-binding site that is occupied during substrate permeation toward the central binding pocket.
However, comprehensive mutational studies have identified residues in the 5HT-binding pocket that are critical determinants for recognition of inhibitors (Fig. 1). In particular, Henry et al. (28) found that mutation of Ile-172 on TM3 decreased potency of the SSRIs citalopram and fluoxetine by up to 2 orders of magnitude. Ile-172 likely corresponds to Val-104 in LeuT, which is located in the leucine-binding pocket (2). Celik et al. (8) have recently verified that Ile-172 has an ortholog position in the substrate-binding site in SERT. TM3 is the anti-parallel homolog of TM8, and it is noteworthy that Ser-438 and Ile-172 are located on opposite sides of the 5HT-binding pocket. Together these data are best explained by an overlapping binding site for 5HT and inhibitors in which Ile-172 and Ser-438 form direct contact points for inhibitor binding. However, differentiation between direct and indirect effects in mutagenesis studies is inherently difficult. Effects from a mutation could arise from a long range allosteric effect that perturbs an inhibitor-binding site physically distinct from the 5HT pocket. A mutation could also induce a shift in equilibrium between the conformational states SERT assumes during substrate translocation. In both cases, the temporal accessibility of the inhibitor-binding site decreases. Indeed, it has been observed for imipramine that the dissociation rate is decreased in the presence of 5HT, which suggest cooperativity between two physically distinct binding sites (39). Therefore, the effects of mutation of Ile-172 and Ser-438 cannot be interpreted unambiguously without substantiating direct interactions between these residues and the inhibitor molecule. In this study, we address this problem by showing that analogs of TCAs and SSRIs with reciprocal chemical modifications become insensitive to the S438T mutation, thus substantiating a steric clash between the inhibitor aminopropyl moiety and the introduced protein methyl group as reason for the observed loss of potency. Similar approaches have previously been used to map specific interactions between 5HT-binding pocket residues and functional 5HT groups (8, 22, 23). This implies that the Ser-438 side chain is within 3 Å of the ligand and likely constitutes a direct contact point for the aminopropyl group of these inhibitors. In comparison, the distance between the equivalent residue in LeuT, Ser-355, and the TCA molecule within the structure of LeuT in complex with TCA is 18 Å (PDB code 2QJU), thus suggesting the antidepressant-binding pocket in LeuT and hSERT to be separate structural entities and supporting that 5HT- and antidepressant-binding pockets overlaps in hSERT.
Although the I172M and S438T mutants specifically indicate that the location of the hSERT high affinity binding pocket for citalopram and several TCAs is different from the LeuT TCA-binding pocket, the orthologous region in hSERT could still hold a secondary inhibitor-binding site. There is substantial evidence for the existence of a secondary low affinity allosteric site that can modulate dissociation rates of several SERT ligands from the high affinity binding site (39). Although further experiments are needed to fully address the existence of a functional relevant vestibule inhibitor-binding pocket, our observation that nonconservative mutations in this region in hSERT fail to alter inhibitor Ki indicate that, if present, such a binding site has little influence on the overall inhibitory mechanism.
The results presented imply Ser-438 as a direct contact for antidepressants bearing an aminopropyl group and provide further evidence for the location of a high affinity antidepressant pocket in hSERT. Along with other SERT mutants, S438T is potentially useful for future studies of the structure of this binding pocket and the mechanism of inhibition of antidepressants.
Supplementary Material
Acknowledgments
We thank Dr. Benny Bang-Andersen for providing compounds from the H. Lundbeck A/S compound collection and Prof. Flemming S. Jørgensen for fruitful discussions and assistance with computational studies. We thank Drs. Claus J. Løland and Kevin Erreger for critical comments on the manuscript.
This work was supported by the Carlsberg Foundation and the Danish Medical Research Council (to L. O.), the Lundbeck Foundation (to K. B. H.), the Danish Council for Strategic Research NABIIT Programme (to O. T.), and the Drug Research Academy (to J. A.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Tables S1 and S2.
Footnotes
The abbreviations used are: SERT, serotonin transporter; hSERT, human SERT; 5HT, 5-hydroxytryptamine; LeuT, leucine transporter; MADAM, 2-(2-dimethylaminomethyl-phenylsulphanyl)-5-methyl-phenylamine; SSRI, selective serotonin reuptake inhibitor; SLC6, solute carrier 6; TCA, tricyclic antidepressants; DMEM, Dulbecco's modified Eagle's medium; WT, wild type; PDB, Protein Data Bank; TM, transmembrane domain.
References
- 1.Wong, D. T., Perry, K. W., and Bymaster, F. P. (2005) Nat. Rev. Drug Discov. 4 764-774 [DOI] [PubMed] [Google Scholar]
- 2.Yamashita, A., Singh, S. K., Kawate, T., Jin, Y., and Gouaux, E. (2005) Nature 437 215-223 [DOI] [PubMed] [Google Scholar]
- 3.Forrest, L. R., Tavoulari, S., Zhang, Y. W., Rudnick, G., and Honig, B. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 12761-12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandenberg, R. J., Shaddick, K., and Ju, P. (2007) J. Biol. Chem. 282 14447-14453 [DOI] [PubMed] [Google Scholar]
- 5.Dodd, J. R., and Christie, D. L. (2007) J. Biol. Chem. 282 15528-15533 [DOI] [PubMed] [Google Scholar]
- 6.Zomot, E., Bendahan, A., Quick, M., Zhao, Y. F., Javitch, J. A., and Kanner, B. I. (2007) Nature 449 726-729 [DOI] [PubMed] [Google Scholar]
- 7.Beuming, T., Kniazeff, J., Bergmann, M. L., Shi, L., Gracia, L., Raniszewska, K., Newman, A. H., Javitch, J. A., Weinstein, H., Gether, U., and Loland, C. J. (2008) Nat. Neurosci. 11 780-789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celik, L., Sinning, S., Severinsen, K., Hansen, C. G., Møller, M. S., Bols, M., Wiborg, O., and Schiøtt, B. (2008) J. Am. Chem. Soc. 130 3853-3865 [DOI] [PubMed] [Google Scholar]
- 9.Henry, L. K., DeFelice, L. J., and Blakely, R. D. (2006) Neuron 49 791-796 [DOI] [PubMed] [Google Scholar]
- 10.Talvenheimo, J., Nelson, P. J., and Rudnick, G. (1979) J. Biol. Chem. 254 4631-4635 [PubMed] [Google Scholar]
- 11.Humphreys, C. J., Levin, J., and Rudnick, G. (1988) Mol. Pharmacol. 33 657-663 [PubMed] [Google Scholar]
- 12.Graham, D., Esnaud, H., Habert, E., and Langer, S. Z. (1989) Biochem. Pharmacol. 38 3819-3826 [DOI] [PubMed] [Google Scholar]
- 13.Zhou, Z., Zhen, J., Karpowich, N. K., Goetz, R. M., Law, C. J., Reith, M. E. A., and Wang, D. N. (2007) Science 317 1390-1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh, S. K., Yamashita, A., and Gouaux, E. (2007) Nature 448 952-956 [DOI] [PubMed] [Google Scholar]
- 15.Kristensen, A. S., Larsen, M. B., Johnsen, L. B., and Wiborg, O. (2004) Eur. J. Neurosci. 19 1513-1523 [DOI] [PubMed] [Google Scholar]
- 16.Fiser, A., and Sali, A. (2003) Methods Enzymol. 374 461-491 [DOI] [PubMed] [Google Scholar]
- 17.Beuming, T., Shi, L., Javitch, J. A., and Weinstein, H. (2006) Mol. Pharmacol. 70 1630-1642 [DOI] [PubMed] [Google Scholar]
- 18.Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., Repasky, M. P., Knoll, E. H., Shelley, M., Perry, J. K., Shaw, D. E., Francis, P., and Shenkin, P. S. (2004) J. Med. Chem. 47 1739-1749 [DOI] [PubMed] [Google Scholar]
- 19.Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., and Banks, J. L. (2004) J. Med. Chem. 47 1750-1759 [DOI] [PubMed] [Google Scholar]
- 20.Cheng, Y., and Prusoff, W. H. (1973) Biochem. Pharmacol. 22 3099-3108 [DOI] [PubMed] [Google Scholar]
- 21.Rudnick, G. (2006) J. Membr. Biol. 213 101-110 [DOI] [PubMed] [Google Scholar]
- 22.Adkins, E. M., Barker, E. L., and Blakely, R. D. (2001) Mol. Pharmacol. 59 514-523 [DOI] [PubMed] [Google Scholar]
- 23.Barker, E. L., Moore, K. R., Rakhshan, F., and Blakely, R. D. (1999) J. Neurosci. 19 4705-4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitayama, S., Shimada, S., Xu, H., Markham, L., Donovan, D. M., and Uhl, G. R. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 7782-7785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Owens, M. J., Morgan, W. N., Plott, S. J., and Nemeroff, C. B. (1997) J. Pharmacol. Exp. Ther. 283 1305-1322 [PubMed] [Google Scholar]
- 26.Tatsumi, M., Groshan, K., Blakely, R. D., and Richelson, E. (1997) Eur. J. Pharmacol. 340 249-258 [DOI] [PubMed] [Google Scholar]
- 27.Eshleman, A. J., Carmolli, M., Cumbay, M., Martens, C. R., Neve, K. A., and Janowsky, A. (1999) J. Pharmacol. Exp. Ther. 289 877-885 [PubMed] [Google Scholar]
- 28.Henry, L. K., Field, J. R., Adkins, E. M., Parnas, M. L., Vaughan, R. A., Zou, M. F., Newman, A. H., and Blakely, R. D. (2006) J. Biol. Chem. 281 2012-2023 [DOI] [PubMed] [Google Scholar]
- 29.Barker, E. L., Perlman, M. A., Adkins, E. M., Houlihan, W. J., Pristupa, Z. B., Niznik, H. B., and Blakely, R. D. (1998) J. Biol. Chem. 273 19459-19468 [DOI] [PubMed] [Google Scholar]
- 30.Mortensen, O. V., Kristensen, A. S., and Wiborg, O. (2001) J. Neurochem. 79 237-247 [DOI] [PubMed] [Google Scholar]
- 31.Larsen, M. B., Elfving, B., and Wiborg, O. (2004) J. Biol. Chem. 279 42147-42156 [DOI] [PubMed] [Google Scholar]
- 32.Rudnick, G., and Wall, S. C. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 1817-1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Talvenheimo, J., Fishkes, H., Nelson, P. J., and Rudnick, G. (1983) J. Biol. Chem. 258 6115-6119 [PubMed] [Google Scholar]
- 34.Marcusson, J. O., Backstrom, I. T., and Ross, S. B. (1986) Mol. Pharmacol. 30 121-128 [PubMed] [Google Scholar]
- 35.Rudnick, G. (2007) ACS Chem. Biol. 2 606-609 [DOI] [PubMed] [Google Scholar]
- 36.Henry, L. K., Meiler, J., and Blakely, R. D. (2007) Mol. Interv. 7 306-309 [DOI] [PubMed] [Google Scholar]
- 37.Shi, L., Quick, M., Zhao, Y., Weinstein, H., and Javitch, J. A. (2008) Mol. Cell 30 667-677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh, S. K., Piscitelli, C. L., Yamashita, A., and Gouaux, E. (2008) Science 322 1655-1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wennogle, L. P., and Meyerson, L. R. (1982) Eur. J. Pharmacol. 86 303-307 [DOI] [PubMed] [Google Scholar]
- 40.Zhang, Y. W., and Rudnick, G. (2005) J. Biol. Chem. 280 30807-30813 [DOI] [PubMed] [Google Scholar]
- 41.Sur, C., Betz, H., and Schloss, P. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 7639-7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barker, E. L., and Blakely, R. D. (1996) Mol. Pharmacol. 50 957-965 [PubMed] [Google Scholar]
- 43.Andersen, J., Taboureau, O., Olsen, L., Jørgensen, A. M., Hansen, K. B., Jensen, A. A., Egebjerg, J., Bang-Andersen, B., Jørgensen, F. S., Strømgaard, K., and Kristensen, A. S. (2007) Soc. Neurosci. Abstr. 248 15 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.