

Abstract
Bacteria decode the isoleucine codon AUA using a tRNA species that is posttranscriptionally modified at the wobble position of the anticodon with a lysine-containing cytidine derivative called lysidine. The lysidine modification of tRNAIle2 is an essential identity determinant for proper aminoacylation by isoleucyl tRNA synthetase (IleRS) and codon recognition on the ribosome. The ATP- and lysine-dependent formation of lysidine is catalyzed by tRNAIle-lysidine synthetase. Using the purified recombinant enzyme from Escherichia coli and an in vitro transcribed tRNA substrate, we have confirmed that lysidine modification is both necessary and sufficient to convert tRNAIle2 into a substrate for IleRS. A series of lysine analogs were tested as potential inhibitors during the mechanistic characterization of tRNAIle-lysidine synthetase. Gel electrophoresis revealed that many of these analogs, including some simple alkyl amines, were alternative substrates. Incorporation of these amines into alternative tRNA products was confirmed by mass spectrometry. The availability of tRNAIle2 with differential modifications enabled an exploration of the structural requirements of the anticodon for aminoacylation by methionyl tRNA synthetase and IleRS. All of the modifications were effective at creating negative determinants for methionyl tRNA synthetase and positive determinants for IleRS, although the tolerance of IleRS differed between the enzymes from E. coli and Bacillus subtilis.
The fidelity of protein synthesis requires precise decoding of the genetic code. It is essential that aminoacyl tRNA synthetases match the appropriate amino acid-tRNA pairs and that the anticodons of charged tRNAs interact with the correct mRNA codons. Posttranscriptional modification of the first, or wobble, position of the tRNA anticodon contributes to the accuracy of the latter process (1). One example in eubacteria is the single tRNA for decoding the rare isoleucine codon AUA. This species, tRNAIle2, contains a lysine-containing cytidine derivative called lysidine at the anticodon wobble position (2). The lysidine modification of tRNAIle2 converts the original codon recognition specificity from AUG to AUA and the amino acid specificity for synthetase activation from methionine to isoleucine (3). The prevalence of tRNAIle2 in Escherichia coli is very low (4), and its availability may constitute a mechanism for the regulation of protein synthesis.
The single gene responsible for lysidine formation in both Bacillus subtilis and E. coli has been identified and named tilS (for tRNAIle-lysidine synthetase) (5). The tilS gene (formerly yacA) is essential for viability in B. subtilis (6), and homologs are nearly universally present in eubacteria, including human pathogens. Partial inactivation of the tilS gene in E. coli leads to an AUA codon-dependent translational defect (5). Loss or inhibition of TilS2 protein function is expected to disrupt bacterial protein synthesis as depletion of the pool of isoleucine-chargeable tRNAIle2 would leave ribosomes unable to translate past AUA codons. There is no mammalian counterpart of TilS, as eukaryotes use tRNAIle species with either inosine or a modified uridine at the wobble position of the anticodon to recognize all three isoleucine codons (7). Several high resolution protein structures of bacterial TilS are available (8, 9), and a straightforward reaction mechanism has been proposed (5, 10). Together these attributes contribute to the potential attractiveness of TilS as a novel, albeit unproven target for a broad-spectrum antibacterial agent.
Using a simplified assay format, we have begun mechanistic studies of this enzyme and confirmed that lysidine modification of tRNAIle2 is both necessary and sufficient to convert this tRNA into a substrate for isoleucyl tRNA synthetase (IleRS). During a systematic study of substrate requirements, we found that a number of lysine analogs and smaller primary amine-containing compounds can act as alternative substrates for the TilS-mediated modification of tRNAIle2. The availability of these differentially modified tRNAs has in turn enabled an exploration of the structural requirements of the tRNA anticodon for aminoacylation by IleRS.
EXPERIMENTAL PROCEDURES
Materials—l-[4,5-3H]Lysine monohydrochloride, l-[4,5-3H]isoleucine, l-[methyl-3H]methionine, and wheat germ agglutinin polyethyleneimine-treated type A polyvinyltoluene scintillation proximity assay (SPA) beads were obtained from GE Healthcare. Lysine analogs, α,β-methyleneadenosine-5′-triphosphate, and β,γ-methyleneadenosine-5′-triphosphate were obtained from Sigma-Aldrich. Other ATP analogs were obtained from TriLink BioTechnologies (San Diego, CA). TCEP was obtained from Pierce.
Preparation of Recombinant E. coli TilS—The tilS gene in accession number Z50870 was obtained by PCR amplification from E. coli K12 genomic DNA (American Type Culture Collection, Manassas, VA) using the following primers: 5′-GGCATATGACACTCACGCTCAATAGA-3′ (forward primer) and 5′-GGGAAGCTTACTAAGCGTTTTCTGCCA-3′ (reverse primer). The PCR product was ligated into the expression vector pET26B+ (Novagen, Madison, WI), and the clones were sequenced on an ABI 377 DNA sequencer using BigDye Terminator Version 3.1 chemistry. The C-terminal hexahistidine-tagged TilS fusion protein was expressed in E. coli BL21 (DE3) cells with 1 mm isopropyl 1-thio-β-d-galactopyranoside induction and purified using nickel-nitrilotriacetic acid resin (Qiagen, Valencia, CA) following the manufacturer's instructions. The pooled protein was further purified and buffer-exchanged on a Superdex 200 column (GE Healthcare). The mass of the recombinant TilS was verified by electrospray ionization mass spectrometry performed at Novatia, LLC (Monmouth Junction, NJ).
Preparation of Recombinant B. subtilis IleRS—The IleRS gene in accession number Q45477 was obtained from genomic DNA prepared from B. subtilis strain 168 (obtained from the Bacillus Genetic Stock Center). PCR amplification of the gene was conducted with the following primers: 5′-CACATTAATATGGATTTTAAAGACACGCTCTTAATG-3′ (forward primer) and 5′-GTCGCTGCAGTTATTTTTGATAGTATTTTTCAACGATTTC-3′ (reverse primer). The PCR product was ligated into the expression vector pET3D-FLAG (Novagen) and confirmed by DNA sequencing. The N-terminal FLAG-tagged fusion protein was expressed in E. coli BL21 (DE3) cells with 1 mm isopropyl 1-thio-β-d-galactopyranoside induction and purified using M2-FLAG resin (Sigma-Aldrich) following the manufacturer's instructions. The pooled protein peak was further purified and buffer-exchanged on a Superdex 200 column. The mass of the recombinant IleRS protein was verified by electrospray ionization mass spectrometry.
Preparation of E. coli S100 Extract—E. coli BL21 (DE3) cells, 3 g, were suspended in 7 ml of 50 mm HEPES (pH 7.5), 100 mm NaCl, 10 mm MgCl2, 10% glycerol and lysed in a French pressure cell at 18,000–20,000 p.s.i. After centrifugation at 100,000 × g for 60 min, the soluble proteins in the supernatant were loaded onto a Superdex 200 column. The peak fractions were pooled, and 0.1 volume of 10% streptomycin sulfate was added. The precipitate was removed by centrifugation at 16,000 × g for 10 min at 4 °C, and solid ammonium sulfate was added to the supernatant to 65% saturation. After collection of the precipitate by centrifugation at 16,000 × g for 10 min at 4 °C, the pellet was resuspended in 0.5 ml of 50 mm HEPES (pH 7.6), 10 mm MgCl2 and dialyzed against the same buffer at 4 °C overnight in a Slide-A-Lyzer-10K (Pierce). The protein preparation was frozen after the addition of glycerol to 10%.
Preparation of tRNAIle2—A template for in vitro transcription was built by putting the tRNA gene behind a T7 promoter (11). Overlapping oligonucleotides were prepared on an ABI 394 DNA synthesizer to construct a cDNA flanking the E. coli tRNAIle2 sequence with a 5′ T7 RNA polymerase promoter (5′-AATTCCTCTAATACGACTCACTATAGGCCCCTTAGCTCAGTGGTTAGAGCAGGCGACTCATAATCGCTTGGTCGC-3′) and a 3′ BstNI restriction site (5′-AATTCCTGGTGGCCCCTGCTGGACTTGAACCAGCGACCAAGCGATTATGAGTCGCCTGCTCTAACCACTGAGC-3′). Vector pLDR24 was generated via mutagenesis of pLDR21 (ATCC #87206) to add EcoRI and PstI restriction sites downstream of the tac promoter. The cDNA was cloned into pLDR24, and the sequence was verified. The resulting pLDR24(Ile2) clone was cut with BstNI to generate the transcription template and used without further purification at 50 μg/ml in the Ribomax Large Scale RNA Production System T7 kit (Promega, Madison, WI) following the manufacturer's instructions. After incubation, the synthesis reaction was extracted with phenol:chloroform: isoamyl alcohol (25:24:1) and purified on a Superdex 75 column (GE Healthcare). Purity and mass were verified by polyacrylamide gel electrophoresis on Tris borate-EDTA urea gels (Invitrogen) and electrospray ionization mass spectrometry.
Preparation of C34G-tRNAIle2—Mutagenesis on clone pLDR24(Ile2) was performed via PCR amplification with the Vent and Vent exo– polymerases (New England Biolabs, Ipswich, MA) using the following oligonucleotides 5′-GTGGTTAGAGCAGGCGACTGATAATCGCTTGGTCGCT-3′ (forward primer) and 5′-CCAGCGACCAAGCGATTATCAGTCGCCTGCTCTAACCAC-3′ (reverse primer). The sequence of the pLDR24(G34Ile2) clone was verified, and the vector was digested with BstNI for the preparation of tRNA as above.
Assay of TilS Enzymatic Activity—The standard TilS enzymatic assay was performed in a 96-well white polystyrene microplate with 30 μl of a reaction mix comprised of 50 mm TAPS buffer (pH 8.5), 3 mm MgCl2, 0.01% Tween 20, 0.5 mm TCEP, 0.1 mg/ml bovine serum albumin, 20 nm E. coli tRNAIle2, 5 μm ATP, 110 nm [3H]lysine (91 Ci/mmol), and 10 nm E. coli TilS enzyme. Samples were incubated for 2 h at room temperature before terminating the reaction by the addition of 30 μl of 2.7 mg/ml SPA beads suspended in 175 mm sodium citrate buffer (pH 2.0) with 100 mm NaCl. After 20 min at room temperature to allow tRNA capture, the plate was centrifuged at 400 × g for 5 min. Radioactivity was measured in a Packard Topcount NXT microplate scintillation counter. Inhibition curves were plotted, and IC50 values were calculated with a 2 parameter fit using KaleidaGraph (Synergy Software).
Preparation of tRNA with Lysine Analogs—Reactions were performed in a 75-μl reaction mix consisting of 50 mm TAPS (pH 8.5), 3 mm MgCl2, 0.5 mm TCEP, 0.1 mg/ml bovine serum albumin, 20 μm E. coli tRNAIle2, 100 μm ATP, 500 nm E. coli TilS enzyme, and 100 μm lysine or specified analog concentration for 4 h at 37°C and then frozen without further work up until analysis. The amount of unmodified tRNA was assessed by diluting an aliquot of the reaction mixture 1000-fold into a 30-μl reaction mix comprised of 50 mm TAPS buffer (pH 8.5), 3 mm MgCl2, 0.01% Tween 20, 0.5 mm TCEP, 0.1 mg/ml bovine serum albumin, 50 μm ATP, 110 nm [3H]lysine (91 Ci/mmol), and 20 nm E. coli TilS enzyme. Samples were incubated for 2 h at room temperature before terminating the reaction by the addition of SPA beads as in the standard assay above. The reactions with lysine analogs were also analyzed by gel electrophoresis using a 15% polyacrylamide Tris borate-EDTA, 8 m urea gel run at 120 V for 3 h and stained with ethidium bromide.
Assay of tRNA Synthetases—Assays for IleRS and MetRS activities were performed in 96-well white polystyrene microplates with 30 μl of a reaction mix comprised of 50 mm HEPES buffer (pH 7.3), 10 mm Mg(OAc)2, 100 mm KCl, 0.5 mm TCEP, 0.1 mg/ml bovine serum albumin, 100 nm TilS-modified tRNAIle2, 100 μm ATP, 100 nm [3H]amino acid (93 Ci/mmol for isoleucine; 76 Ci/mmol for methionine), and 40 μg/ml partially purified S-100 fraction of E. coli extract or 0.1 nm recombinant B. subtilis IleRS. The enzyme amounts chosen were within the linear response range of the assay to ensure that the measured rates did not reach a plateau. Samples were incubated for 2 h at room temperature before terminating the reaction by the addition of 30 μl of 8.3 mg/ml SPA beads suspended in 175 mm sodium citrate buffer (pH 2.0) with 100 mm NaCl. After 10 min at room temperature to allow tRNA capture, the plate was centrifuged at 400 × g for 5 min. Radioactivity was measured in a Packard Topcount NXT microplate scintillation counter.
RESULTS
The reaction catalyzed by TilS has been assayed with a radio-labeled lysine substrate by following the incorporation of acid precipitable radioactivity into tRNA (5, 10). To further simplify the procedure and make the TilS reaction amenable to high throughput screening, we applied SPA technology. This approach has been successfully used for aminoacyl tRNA synthetases, which also perform the ATP-dependent coupling of an amino acid to a tRNA (12). We found polyethyleneimine-coated wheat germ agglutinin type I polyvinyltoluene beads to perform best for the nonspecific capture of total tRNA. The tRNA substrate was prepared by run-off transcription; previous work indicates that the synthetic polynucleotide is nearly equivalent to the native tRNA substrate (10). The use of high specific activity tritiated lysine enabled good detection sensitivity using only nm levels of enzyme and tRNA.
TilS was found to have an alkaline pH optimum in the range of 8.5–9.5; pH 8.5 was adopted for the standard assay. Kinetic analysis established that the substrate levels were not saturating for the standard assay described under “Experimental Procedures.” Our examination of the enzymatic reactivity of TilS began with analogs of the three normal substrates as potential competitive inhibitors or alternative substrates. Because cytidine-34 is the site of modification in the tRNA substrate, both cytidine and CMP were tested for inhibition. No inhibition was detected up to 1 mm, however, which is not surprising as previous work had shown that binding determinants are located throughout the tRNA substrate, particularly in the anticodon stem-loop and the acceptor stem (10). In the cited work a C34G-mutated tRNAIle2 was tested as a TilS substrate and found to be inactive. We prepared the C34G-tRNAIle2 and evaluated it as an inhibitor instead of as a substrate; the mutated tRNAIle2 was in fact found to be a potent inhibitor with an IC50 of 85 nm. This result confirms the importance of the structure and sequence beyond cytidine 34 for enzymatic recognition.
A series of 12 commercially available analogs of ATP were also examined for inhibition in the standard TilS assay. As summarized in Table 1, five analogs, three with changes in the triphosphate moiety and two with alterations to the purine base, were found to be modest to weak inhibitors. The analogs were also tested as alternate substrates in a variant of the standard assay lacking ATP. Three analogs modified in the purine base were found to support the TilS reaction (Table 2), although their catalytic efficiencies, as determined by Vmax/Km, were at least 10-fold lower than ATP. Four other analogs (N1-methyladenosine-5′-triphosphate, 2-aminoadenosine-5′-triphosphate, 2-amino-6-chloropurineriboside-5′-triphosphate, and 6-chloropurineriboside-5′-triphosphate) were neither substrates nor inhibitors.
TABLE 1.
Inhibition constants for ATP analogs in the TilS reaction
| ATP analog | IC50 |
|---|---|
| μm | |
| Benzimidazoleriboside-5′-triphosphate | 105 ± 8 |
| 8-Azaadenosine-5′-triphosphate | 255 ± 60 |
| Adenosine-5′-O-(1-thiotriphosphate) | 16 ± 1 |
| α,β-Methyleneadenosine-5′-triphosphate | 35 ± 3 |
| β,γ-Methyleneadenosine-5′-triphosphate | 82 ± 6 |
TABLE 2.
Kinetic constants for ATP and analogs in the TilS reaction
| Nucleotide substrate | Km | Relative Vmax/Km |
|---|---|---|
| μm | ||
| ATP | 1.6 ± 0.5 | (100) |
| 8-Azidoadenosine-5′-triphosphate | 10.0 ± 2.4 | 2.3 |
| 7-Deazaadenosine-5′-triphosphate | 1.8 ± 0.5 | 8.6 |
| N6-Methyladenosine-5′-triphosphate | 21.7 ± 2.7 | 4.7 |
A series of commercially available lysine analogs were examined for inhibition in the standard TilS assay. The structures of these compounds and their inhibition constants are listed in Fig. 1 and Table 3. Lysine, 1, itself appears as an inhibitor because of the isotopic dilution of the tritiated lysine in the assay; the IC50 in this case approximates Km and provides a useful reference point for the binding affinity of the other compounds. Alteration of the amino or carboxyl functionalities on the α carbon of lysine resulted in only very weak inhibitors (compounds 5–8). Likewise, shortening the side chain, deleting the ε amino group, or substituting a guanidino moiety were also detrimental (compounds 9, 11, and 12). Modifications within the lysine side chain, however, were much better tolerated. Although the hydroxylysine 4 had reduced potency, dehydrolysine 10 and aminoethylserine 3 were very similar to lysine itself. Surprisingly, aminoethylcysteine 2 appeared to have significantly improved binding affinity relative to lysine. The relatively relaxed requirements of the lysine side chain prompted examination of some primary alkyl amines as potential side chain mimics. Although the simple amino acids 16 and 17 were not inhibitors of the TilS reaction, three other amines containing neutral functional groups instead of carboxylates (13–15) retained detectable binding affinity. The potency of N-acetylethylenediamine (13) is surprisingly good considering it completely lacks the binding determinants of the lysine α carbon.
FIGURE 1.

Structures of lysine and its analogs studied in this report.
TABLE 3.
Inhibition constants for lysine and the analogs in Fig. 1 in the TilS reaction
| No. | Compound | IC50 | R1 | R2 | X | R30 | R4 |
|---|---|---|---|---|---|---|---|
| μm | |||||||
| 1 | Lysinea | 0.68 ± 0.04 | 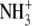 |
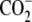 |
CH2 | H | 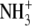 |
| 2 | Aminoethylcysteine | 0.045 ± 0.003 | 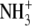 |
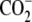 |
S | H | 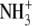 |
| 3 | Aminoethylserine | 0.40 ± 0.02 | 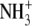 |
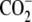 |
O | H | 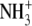 |
| 4 | (5R)-5-Hydroxylysine | 17.6 ± 0.8 | 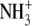 |
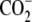 |
CH2 | OH | 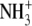 |
| 5 | Lysinamide | 620 ± 70 | 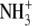 |
CONH2 | CH2 | H | 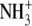 |
| 6 | Cadaverine | 910 ± 40 |  |
H | CH2 | H | 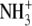 |
| 7 | N-α-methyl-lysineb | 1300 ± 120 | 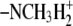 |
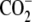 |
CH2 | H | 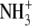 |
| 8 | 6-Aminohexanoic acid | >1000 | -H | 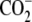 |
CH2 | H | 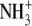 |
| 9 | Norleucine | >1000 | 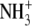 |
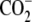 |
CH2 | H | H |
| 10 | 4,5-trans-Dehydrolysine | 2.5 ± 0.1 | |||||
| 11 | Ornithineb | 700 ± 150 | |||||
| 12 | Arginine | >1000 | |||||
| 13 | N-Acetylethylenediamine | 6.8 ± 0.9 | |||||
| 14 | Histamine | 330 ± 20 | |||||
| 15 | Ethanolamine | 1770 ± 150 | |||||
| 16 | Glycine | >1000 | |||||
| 17 | β-Alanine | >1000 |
The apparent inhibition by lysine is the consequence of isotopic dilution of the radiolabeled substrate.
As described in the “Results,” these samples apparently contained contaminating lysine.
The initial assumption was that the compounds in Table 3, other than lysine, were dead-end inhibitors of TilS, i.e. they formed completely nonproductive complexes with the enzyme. It was noted, however, that every compound with measurable inhibitory potency versus the enzyme contained a primary amine and, thus, was potentially a competitive alternate substrate. Because the assay relies on incorporation of radioactive lysine into tRNA, alternative substrates that replace lysine will appear to be inhibitors and require other methods to discern their true character. This question was addressed in several ways. Analogs were incubated at ≥10-fold their IC50 (see Table 4) with high concentrations of ATP, tRNAIle2, and TilS to maximize the modification of the tRNA. An aliquot of each analog reaction was then diluted into a standard assay containing tritiated lysine to assess whether any unmodified tRNAIle2 remained. As recorded in Table 4, complete conversion with most analogs could be inferred from the complete disappearance (≥99%) of the initial substrate; in reactions K and L about a quarter of the residual tRNAIle2 substrate remained. Formation of fully modified tRNA products in reactions B–J could be directly detected by gel electrophoresis. As shown in Fig. 2, reactions B–E and G–J clearly produced tRNA species with slower migration than the unmodified substrate. The migration shift was minimal for cadaverine (reaction F), possibly because this reactant uniquely added some positive charge to the polyanionic tRNA in the electrophoresis at pH 8.7, whereas the other reactants made either zwitterionic (reactions B, C, D, E, G) or neutral (reactions H, I, J) additions.
TABLE 4.
Synthesis of alternative tRNA products from TilS reactions with tRNAIle2 and lysine analogs
| Reaction | Amine substrate | Concentration | % Residual unmodified tRNA | Predicted tRNA mass | Observed tRNA mass | Mass error |
|---|---|---|---|---|---|---|
| mm | ||||||
| A | None (control) | (100) | 24,665.5 | 24,669.4 | 3.9 | |
| B | Lysine | 0.1 | 0 | 24,793.7 | 24,795.3 | 1.6 |
| C | Aminoethylcysteine | 0.1 | 0 | 24,811.7 | 24,813.2 | 1.5 |
| D | Aminoethylserine | 0.1 | 0 | 24,795.6 | 24,796.2 | 0.6 |
| E | (5R)-5-Hydroxylysine | 0.2 | 1 | 24,809.7 | 24,811.5 | 1.8 |
| F | Cadaverine | 10 | 1 | 24,749.7 | 24,752.2 | 2.5 |
| G | 4,5-trans-Dehydrolysine | 0.1 | 1 | 24,791.7 | 24,793.0 | 1.3 |
| H | Ethanolamine | 20 | 1 | 24,708.6 | 24,710.0 | 1.4 |
| I | N-Acetylethylenediamine | 0.1 | 1 | 24,748.6 | 24,750.1 | 1.5 |
| J | Histamine | 5 | 1 | 24,758.6 | 24,759.7 | 1.1 |
| K | N-α-Methyl-lysine | 20 | 27 | 24,807.7 | 24,794.9 | -12.8 |
| L | Ornithine | 10 | 22 | 24,779.6 | 24,794.3 | 14.7 |
FIGURE 2.

Analysis of TilS reactions with tRNAIle2 and lysine analogs by gel electrophoresis. Samples of the reaction products were run as described under “Experimental Procedures.” Lane headings refer to the reactions listed in Table 4; A, control; B, lysine; C, aminoethylcysteine; D, aminoethylserine, E, (5R)-5-hydroxylysine; F, cadaverine; G, 4,5-trans-dehydrolysine; H, ethanolamine; I, N-acetylethylenediamine; J, histamine.
Finally, the product tRNAs were also analyzed by electrospray ionization mass spectrometry; each reaction with lysine or an analog was predicted to generate a modified tRNA with a unique molecular weight. As seen in Table 4, tRNA products with the expected masses were indeed found in reactions A–J. In reactions K and L, however, the molecular weights of the products did not match the predictions and instead matched the product of reaction B which used lysine as substrate. This indicates that low level lysine contamination in the N-α-methyl lysine and ornithine (on the order of 0.1%) accounted for the modified tRNAs formed in reactions K and L, respectively. Trace lysine at such a level likely accounts for all of the potency of these two compounds as (apparent) inhibitors in Table 3 as well. For the eight other lysine analogs, however, the evidence is unambiguous that TilS is capable of accepting alternative substrates for modification of tRNAIle2.
Nucleotides in the anticodon loop are among the common identity elements that define specific recognition of tRNAs by aminoacyl tRNA synthetases. It has previously been reported that the lysidine modification of cytidine 34 in the anticodon of tRNAIle2 is an absolute determinant for switching recognition from MetRS to IleRS (3). We sought to verify and extend these observations using both partially purified native E. coli and purified recombinant B. subtilis tRNA synthetase enzymes. The availability of differentially TilS-modified tRNAIle2 molecules enabled us to explore in greater detail the specificity determinants of the anticodon for aminoacylation. After appropriate assay conditions were established for coupling of methionine and isoleucine to their respective tRNAs, the tRNAs from TilS reaction mixtures A–J in Table 4 were each tested as a substrate for aminoacylation by MetRS or IleRS. The results are presented in Table 5. Consistent with literature precedent, the unmodified tRNAIle2 (reaction A) showed measurable activity as a substrate for the native E. coli MetRS, although its reaction rate was slower than authentic tRNAMet. Also as expected, aminoacylation with methionine was undetectable after TilS modification of tRNAIle2 with lysine (reaction B). Table 5 newly reveals that all of the alternative modifications made to tRNAIle2 in TilS reactions C–J were equally effective at creating negative determinants for MetRS recognition.
TABLE 5.
Evaluation of alternatively TilS-modified tRNAs from Table 4 as substrates for tRNA synthetases
|
tRNA substrate (amine modifier)
|
Relative aminoacylation
ratea
|
||||
|---|---|---|---|---|---|
| E. coli MetRS | E. coli IleRS | B. subtilis IleRS | |||
| % | |||||
| tRNAMet | (100) | 7 | 32 | ||
| Reaction A (control) | 23 | 2 | 0 | ||
| Reaction B (lysine) | 0 | (100) | (100) | ||
| Reaction C (aminoethylcysteine) | 1 | 104 | 127 | ||
| Reaction D (aminoethylserine) | 0 | 44 | 95 | ||
| Reaction E ((5R)-5-hydroxylysine) | 4 | 23 | 130 | ||
| Reaction F (cadaverine) | 1 | 356 | 133 | ||
| Reaction G (4,5-trans-dehydrolysine) | 2 | 19 | 105 | ||
| Reaction H (ethanolamine) | 0 | 123 | 99 | ||
| Reaction I (N-acetylethylenediamine) | 3 | 31 | 92 | ||
| Reaction J (histamine) | 0 | 205 | 116 | ||
The 100% reference corresponds to 3240, 2210, or 4600 net cpm for E. coli MetRS, E. coli IleRS, or B. subtilis IleRS, respectively, measured after a 2-h assay as described under “Experimental Procedures”.
The patterns of substrate utilization were reversed with respect to IleRS. Whereas the rate of aminoacylation of unmodified tRNAIle2 (reaction A) with isoleucine was essentially undetectable using either native E. coli or recombinant B. subtilis IleRS, the aminoacylation rate was robust with lysine-modified tRNAIle2 (reaction B). Table 5 demonstrates that all of the alternatively modified tRNAIle2 products of TilS reactions C–J were also acceptable as IleRS substrates, although differences were apparent between the E. coli and B. subtilis enzymes. The aminoacylation rates of the alternative substrates ranged from 5-fold slower to 3.5-fold faster than the lysine-modified natural substrate with E. coli IleRS, whereas with B. subtilis IleRS all of the tRNA species were nearly equivalent as substrates. These results indicate that although both IleRS enzymes recognized a variety of positive identity determinants in the anticodon, there were differences in the extent of their interactions with the modified nucleotides.
DISCUSSION
Bacterial resistance has seriously undermined the clinical efficacy of established antibiotics. One strategy to address multidrug-resistant bacteria is to develop compounds with new modes of action that engage unexploited targets. Researchers have turned to comparative microbial genomics to identify conserved essential genes which may encode novel candidate targets. One new opportunity was identified when careful biochemical detective work connected an uncharacterized gene with the essential tRNA modification that yields lysidine (5). The broadly conserved function of TilS is unique to bacteria but is not a current target of antibacterial drugs. The action of TilS is required to provide one of two tRNA substrates for IleRS, itself an essential gene for protein synthesis. Because IleRS is the validated target of the antibiotic mupirocin (13), the proximal reaction catalyzed by TilS may also be a suitable point of intervention. Translation of a reporter gene with added AUA codons is markedly impaired by genetic depletion of TilS in E. coli (5), consistent with the essentiality of the enzyme and suggestive of its potential role in a regulatory mechanism for protein biosynthesis.
Our studies were initiated to probe the substrate specificities of TilS as part of mechanistic characterization and a search for inhibitors. We found that the enzyme is fairly strict in its recognition of ATP and tRNA but more relaxed with respect to lysine. Both inhibitors (Table 1) and alternate substrates (Table 2) were found among the small set of ATP analogs tested, suggesting that this nucleotide binding site has potential for small molecule intervention. On the other hand, there was no evidence for binding of cytidine (or CMP) alone when this pyrimidine was removed from the context of the tRNA substrate. The enzyme can tolerate the purine guanosine, however, when presented at position 34 of the tRNA substrate as seen by the potent inhibitor C34G-tRNAIle2. As explored in other published work, the binding interactions for the tRNA substrate are likely extensive throughout the C-terminal domains of the enzyme (9, 10) and, thus, may be challenging to effectively inhibit with a small molecule.
A number of potential inhibitors were initially identified among the lysine analogs tested (Table 3). A primary amine functionality was the common minimal structural feature. Upon closer examination, however, all proved to be alternate substrates rather than true dead-end inhibitors (Fig. 2 and Table 4). Incorporation of each analog into tRNA was unambiguously confirmed by mass spectrometry. TilS is evidently flexible with regard to the binding determinants recognized for the lysine substrate. Several x-ray crystallographic structures are available for TilS (8, 9), but the only one with ligands bound is for the enzyme from the thermophilic bacterium Aquifex aeolicus (8). TilS contains a globular N-terminal domain with clear sequence similarities and three-dimensional structure to “N-type” ATP pyrophosphatases. Appended to the C terminus of the N-terminal domain is a subdomain that is specific to TilS enzymes. In the A. aeolicus structure, lysine is bound in this TilS-specific domain via hydrophobic interactions at the entrance of a tunnel to the active site. The electrostatic potential of this tunnel is acidic, which may be important for selecting the amine functionality of the substrate. There is significant mobility in the TilS-specific domain as evidenced by the comparison of the apoenzyme structure with AMPPNP- and ATP/lysine-bound complexes. Additional structures with tRNA bound will be required to fully elucidate the key protein residues involved in substrate selection.
The unexpected tolerance of TilS for alternative amine substrates permitted the preparation of a series of unnaturally modified tRNAIle2 molecules. We investigated how well these tRNAs would serve as substrates for aminoacylation by MetRS and IleRS. All modifications disabled the tRNA as a substrate for MetRS while enabling to varying degrees the tRNA as a substrate for IleRS. The anticodon wobble position of the two natural tRNA substrates for IleRS contains either guanosine (in tRNAIle1 for recognition of major Ile codons AUU and AUC) or lysidine (in tRNAIle2 for recognition of minor Ile codon AUA) (2, 4). The x-ray crystal structure of Staphylococcus aureus IleRS in complex with E. coli tRNAIle1 and mupirocin shows that this synthetase uses three domains at the C terminus as well as one domain at the extreme N terminus to contact the anticodon loop of the tRNA (14). Although guanosine is structurally distinct from lysidine, the IleRS enzyme accepts both at position 34 while discriminating against the lysidine precursor cytidine (15, 16). It has been proposed that the hydrogen bonding pattern of the nucleoside base at position 34 is critical to enzyme recognition (15, 17). In one tautomer of lysidine the hydrogen bond donor and acceptor groups match guanosine while being completely opposite to cytidine (Fig. 3). Note that the reversal of the cytidine hydrogen bonding pattern in this model is dependent only upon the conversion of a ketone to an imine with a primary amine; this can be fulfilled by amines other than lysine, which is consistent with our observation that alternative TilS-mediated modifications of tRNAIle2 are able to create recognizable substrates for IleRS. There are clearly species differences among IleRS enzymes in this respect, with our data indicating greater discrimination for the modified cytidine by E. coli enzyme than by B. subtilis enzyme. Further experimentation will be required to assess the relevance of the relaxed specificity of TilS and IleRS to bacteria in their native environment.
FIGURE 3.

Comparison of potential hydrogen bonding patterns for cytidine (C), lysidine (L), and guanosine (G). The arrows denote hydrogen bond donors and acceptors.
Analysis of bacterial genomes indicates that tRNAIles with a CAU anticodon sequence are widely distributed (18, 19). The cytidines are presumptively modified in vivo to introduce the proper determinants for AUA codon recognition and aminoacylation by IleRS. The catalytic flexibility of both TilS and IleRS observed in this work indicates that lysine, at least with regard to its chemical properties, is not uniquely qualified for the modification of the wobble position of tRNAIle2. The choice of this amino acid may instead be part of a more complex mechanism of regulation of protein biosynthesis. It should be noted, however, that the presence of lysidine has been confirmed in only E. coli, B. subtilis, and Mycoplasma capricolum (2, 20, 21). The tRNA recognizing the isoleucine AUA codon in at least one archaebacterial species contains a different (non-lysidine) modification (22, 23). This raises the interesting possibility that even some eubacteria may have evolved their TilS and IleRS enzymes to recognize alternatives to lysine and lysidine, respectively.
Footnotes
The abbreviations used are: TilS, tRNAIle-lysidine synthetase; MetRS, methionyl tRNA synthetase; IleRS, isoleucyl tRNA synthetase; TCEP, tris(2-carboxyethyl)phosphine; SPA, scintillation proximity assay; TAPS, 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid.
References
- 1.Björk, G. R., Ericson, J. U., Gustafsson, C. E., Hagervall, T. G., Jonsson, Y. H., and Wikström, P. M. (1987) Annu. Rev. Biochem. 56 263–287 [DOI] [PubMed] [Google Scholar]
- 2.Muramatsu, T., Yokoyama, S., Horie, N., Matsuda, A., Ueda, T., Yamaizumi, Z., Kuchino, Y., Nishimura, S., and Miyazawa, T. (1988) J. Biol. Chem. 263 9261–9267 [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu, T., Nishikawa, K., Nemoto, F., Kuchino, Y., Nishimura, S., Miyazawa, T., and Yokoyama, S. (1988) Nature 336 179–181 [DOI] [PubMed] [Google Scholar]
- 4.Harada, F., and Nishimura, S. (1974) Biochemistry 13 300–307 [DOI] [PubMed] [Google Scholar]
- 5.Soma, A., Ikeuchi, Y., Kanemasa, S., Kobayashi, K., Ogasawara, N., Ote, T., Kato, J., Watanabe, K., Sekine, Y., and Suzuki, T. (2003) Mol. Cell 12 689–698 [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi, K., Ehrlich, S. D., Albertini, A., Amati, G., Andersen, K. K., Arnaud, M., Asai, K., Ashikaga, S., Aymerich, S., Bessieres, P., Boland, F., Brignell, S. C., Bron, S., Bunai, K., Chapuis, J., Christiansen, L. C., Danchin, A., Debarbouille, M., Dervyn, E., Deuerling, E., Devine, K., Devine, S. K., Dreesen, O., Errington, J., Fillinger, S., Foster, S. J., Fujita, Y., Galizzi, A., Gardan, R., Eschevins, C., Fukushima, T., Haga, K., Harwood, C. R., Hecker, M., Hosoya, D., Hullo, M. F., Kakeshita, H., Karamata, D., Kasahara, Y., Kawamura, F., Koga, K., Koski, P., Kuwana, R., Imamura, D., Ishimaru, M., Ishikawa, S., Ishio, I., Le Coq, D., Masson, A., Mauel, C., Meima, R., Mellado, R. P., Moir, A., Moriya, S., Nagakawa, E., Nanamiya, H., Nakai, S., Nygaard, P., Ogura, M., Ohanan, T., O'Reilly, M., O'Rourke, M., Pragai, Z., Pooley, H. M., Rapoport, G., Rawlins, J. P., Rivas, L. A., Rivolta, C., Sadaie, A., Sadaie, Y., Sarvas, M., Sato, T., Saxild, H. H., Scanlan, E., Schumann, W., Seegers, J. F., Sekiguchi, J., Sekowska, A., Seror, S. J., Simon, M., Stragier, P., Studer, R., Takamatsu, H., Tanaka, T., Takeuchi, M., Thomaides, H. B., Vagner, V., van Dijl, J. M., Watabe, K., Wipat, A., Yamamoto, H., Yamamoto, M., Yamamoto, Y., Yamane, K., Yata, K., Yoshida, K., Yoshikawa, H., Zuber, U., and Ogasawara, N. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grosjean, H., and Björk, G. R. (2004) Trends Biochem. Sci. 29 165–168 [DOI] [PubMed] [Google Scholar]
- 8.Kuratani, M., Yoshikawa, Y., Bessho, Y., Higashijima, K., Ishii, T., Shibata, R., Takahashi, S., Yutani, K., and Yokoyama, S. (2007) Structure 15 1642–1653 [DOI] [PubMed] [Google Scholar]
- 9.Nakanishi, K., Fukai, S., Ikeuchi, Y., Soma, A., Sekine, Y., Suzuki, T., and Nureki, O. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 7487–7492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikeuchi, Y., Soma, A., Ote, T., Kato, J., Sekine, Y., and Suzuki, T. (2005) Mol. Cell 19 235–246 [DOI] [PubMed] [Google Scholar]
- 11.Sampson, J. R., and Uhlenbeck, O. C. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macarrón, R., Mensah, L., Cid, C., Carranza, C., Benson, N., Pope, A. J., and Díez, E. (2000) Anal. Biochem. 284 183–190 [DOI] [PubMed] [Google Scholar]
- 13.Hughes, J., and Mellows, G. (1980) Biochem. J. 191 209–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silvian, L. F., Wang, J., and Steitz, T. A. (1999) Science 285 1074–1077 [PubMed] [Google Scholar]
- 15.Muramatsu, T., Miyazawa, T., and Yokoyama, S. (1992) Nucleosides Nucleotides 11 719–730 [Google Scholar]
- 16.Nureki, O., Niimi, T., Muramatsu, T., Kanno, H., Kohno, T., Florentz, C., Giege, R., and Yokoyama, S. (1994) J. Mol. Biol. 236 710–724 [DOI] [PubMed] [Google Scholar]
- 17.Senger, B., Auxilien, S., Englisch, U., Cramer, F., and Fasiolo, F. (1997) Biochemistry 36 8269–8275 [DOI] [PubMed] [Google Scholar]
- 18.Marck, C., and Grosjean, H. (2002) RNA 8 1189–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva, F. J., Belda, E., and Talens, S. E. (2006) Nucleic Acids Res. 34 6015–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andachi, Y., Yamao, F., Muto, A., and Osawa, S. (1989) J. Mol. Biol. 209 37–54 [DOI] [PubMed] [Google Scholar]
- 21.Matsugi, J., Murao, K., and Ishikura, H. (1996) J Biochem. 119 811–816 [DOI] [PubMed] [Google Scholar]
- 22.Gupta, R. (1984) J. Biol. Chem. 259 9461–9471 [PubMed] [Google Scholar]
- 23.Köhrer, C., Srinivasan, G., Mandal, D., Mallick, B., Ghosh, Z., Chakrabarti, J., and Rajbhandary, U. L. (2008) RNA 14 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]