

Abstract
Grid computing offers the powerful alternative of sharing resources on a worldwide scale, across different institutions to run computationally intensive, scientific applications without the need for a centralized supercomputer. Much effort has been put into development of software that deploys legacy applications on a grid-based infrastructure and efficiently uses available resources. One field that can benefit greatly from the use of grid resources is that of drug discovery since molecular docking simulations are an integral part of the discovery process. In this paper, we present a scalable, reusable platform to choreograph large virtual screening experiments over a computational grid using the molecular docking simulation software DOCK. Software components are applied on multiple levels to create automated workflows consisting of input data delivery, job scheduling, status query, and collection of output to be displayed in a manageable fashion for further analysis. This was achieved using Opal OP to wrap the DOCK application as a grid service and PERL for data manipulation purposes, alleviating the requirement for extensive knowledge of grid infrastructure. With the platform in place, a screening of the ZINC 2,066,906 compound “druglike” subset database against an enzyme's catalytic site was successfully performed using the MPI version of DOCK 5.4 on the PRAGMA grid testbed. The screening required 11.56 days laboratory time and utilized 200 processors over 7 clusters.
Keywords: Grid computing, virtual screening, molecular simulation, DOCK, Opal OP
1 Introduction
The continual progression of computational capabilities has removed countless barriers in the scientific community in terms of allowing new computer simulation-based experimental techniques, providing more and more detailed descriptions of complex systems. Yet supercomputers with the purpose of providing the substantial computational and storage resources needed by scientists are not commonplace among all universities and research institutions due to many restricting factors such as cost and maintenance.
Grid computing can supply any party with access to this level of computing by sharing hardware resources across institutions and using sophisticated middleware to solve problems such as resource heterogeneity, job scheduling, data management, and security [1]. Specific software toolkits and protocols ensure interoperability between resources for the development and deployment of applications in a grid environment. These grid technologies have been integrated with Web services [2] that define interfaces and follow specific conventions of the distributed computational model to create what are called grid services. This is the basis of the Open Grid Services Architecture (OGSA) [3], a standards-based distributed service system that has been widely adopted by the grid computing community. Due to the nature of Service-Oriented Architectures (SOA), they are very suitable for creating general workflows to automate multi-step processes [4].
Access to these resources is a major issue for biomedical scientists who would like to take advantage of computational models as tools to gain insight into biological mechanisms, but their work setting and expertise is focused on the traditional wet-bench science. To fill this need, there are a number of grid projects with the goal of helping biomedical scientists bring novel, as well as legacy, applications into the grid environment [5]. The Pacific Rim Grid Middleware and Application Assembly (PRAGMA) [6] is an organization with the aim of building scientific collaboration on an international scale and has developed and implemented a multinational grid testbed in order to run scientific applications spanning fields such as high energy physics, computational biology, and molecular dynamics simulations. The virtual screening platform described in this paper was created and used on the PRAGMA grid testbed.
By simulating the interactions between proteins and small molecules on a computer, virtual screening experiments have become an integral part of the drug discovery process [7]. This alternative to a blind search provides a smaller subset of compounds possessing properties suitable for binding to the active site of a target protein molecule, thereby streamlining the high throughput screening (HTS) experiments. Testing compounds identified by virtual screening has been shown to enrich hit rates over HTS by 1,700-fold [8]. This is especially important when a database size reaches millions of compounds making the HTS experiments too costly to perform [9].
A notable limitation of virtual screening is the computational cost due to the complex calculations being performed with such a large number of compounds. Ideally, biomedical scientists would not be forced to limit the size of the compound database or how extensive the simulations are. Previous virtual screening experiments with the interest of identifying inhibitory compounds typically have used only a single docking algorithm and screened databases containing around 50,000-200,000 compounds [8, 10, 11]. But by taking advantage of grid computing to significantly reduce the constraints of required time and resources, the use of multiple, computationally expensive docking and scoring algorithms, such as calculating water solvation energies, when screening databases containing millions of compounds becomes a viable strategy.
Our method to deploy a large, grid-enabled screening experiment couples the DOCK application [12] with a combination of grid middleware and simple PERL scripts to create a powerful, automated, and scalable virtual screening platform. This paper provides in-depth descriptions of some of the technologies used to create the platform, details on how it performs the virtual screening, an account of its implementation to screen the ZINC [13] 2,066,906 compound “druglike” subset database against an enzyme's catalytic site, and conclusions on its utility.
2 Dock and Related Technologies
The platform described in this paper is not a stand-alone application, but rather an integration of technologies that enable the desired features of a virtual screening experiment while taking advantage of grid resources. Variances in administrative policies, hardware/software configurations, and resource availability are typical of a grid environment and making these complexities transparent to the user, whose main concern is the scientific results of an experiment, is an important priority. A number of parties are attempting to provide this type of user experience by developing and standardizing the grid middleware layer of infrastructure. Aligning this middleware with the parallelization methods of DOCK enables performing distributed virtual screening experiments with expansive compound libraries.
Dock
Using a molecular docking method for virtual screening requires a solved crystal structure of the target molecule's binding site. Compounds from a database are then oriented within the site to calculate the most energetically favorable fit along with a binding score. These scores are used to rank the compounds according to predicted binding affinities relative to each other and provide the subset of compounds to test experimentally [14]. This is the process performed by the open source molecular docking software DOCK, which was used in the platform described in this paper.
DOCK has the ability to treat each compound as a flexible structure in order to explore a series of possible conformations as they are oriented in the target molecule's binding site. First introduced in DOCK 4 [15], the flexible docking feature is performed by the “anchor-and-grow” algorithm that chooses a rigid portion of a compound to be oriented into the binding site, then the remaining portions of the compound are added to find the most stable conformation [16]. Screening a database with flexible docking has been shown to improve accuracy in predicting biologically active molecules compared to a rigid, single conformation docking method [17], yet requires a significant increase in computational time. A typical flexible method with minimization steps can take five seconds to over a minute long docking time per compound and target molecule pair. These time scales make the screening of a large database on the order of a million compounds unreasonable without the use of a significantly large pool of computational resources.
For the purpose of reducing the computational time, a MPI (Message Passing Interface) version of DOCK has been developed and available as a part of DOCK since version 5.0 [18, 19]. MPI is a library specification for message-passing programming and is widely used for parallel computing in a single computing cluster system [20]. The MPI version of DOCK distributes the workload of screening a large library of compounds against a receptor protein over a specified number of processors available within a single cluster. Each compound from the database is coupled to the receptor data by the master node of the cluster and then the pair is passed to a slave node within the cluster to be oriented and scored as its own separate process. Once this finishes, the results are sent back to the master node that performs the bookkeeping task of ranking the compounds according to energy scores while simultaneously sending the next compound in the list to an available slave node [21].
To carry out the screening process over a computational grid consisting of numerous clusters, the DOCK distribution on its own is not sufficient. Using the MPI implementation of DOCK to screen a database of compounds is ideal in a single computing cluster environment since all the data, input and results, is localized within that cluster. Increasing the performance of DOCK would then require adding computational resources to the single cluster, a demand that may not be financially or administratively feasible for most biomedical labs. This is where grid computing becomes an attractive solution for computationally intensive experiments by distributing the workload over shared resources, providing large amounts of computational resources without the need for a centralized supercomputer.
Zinc
Screening a large and diverse compound database increases the chances of finding compounds that bind with high affinity and specificity to the protein of interest. The set of compounds screened in this paper came from the freely available ZINC database that contains over 4.6 million compounds with charge information and hydrogens already added, making them ready for use with DOCK [22]. Every compound that is available commercially has the purchasing information linked to it in the database. Other features include pre-processed subsets of compounds and sub-structure searching. The continual growth of the ZINC database and its open access make it an important resource of the virtual screening toolset [23]. One example of the ZINC database fulfilling this role was aiding in the identification of novel inhibitors for zinc β-lactamase [11].
Grid Middleware
Grid middleware can be described as the layer of infrastructure between the network and applications with the purpose of sharing resources and enabling collaboration among users by managing security, access, and data exchange [24]. For a scientific application to take advantage of grid resources, it typically must be integrated with the middleware or be written specifically for a grid environment. Most build on the Globus Toolkit [25], a “base” set of services and libraries created to provide many of the desired capabilities of an application running in a grid environment and can be categorized as lower middleware [26]. These capabilities include data management, communication, security, and fault detection for a process, along with resource monitoring, management, and discovery [3]. The latest incarnation of Globus, version 4.x (GT4) [27], uses Web services heavily in structuring its components and defining its interfaces.
Upper middleware consists of methods for an application to take advantage of the basic services provided by a toolkit such as Globus. Scheduling and distributing the workload while providing the user an indication of a computational experiment's progress are typical services provided by this layer of infrastructure. One example of this is Nimrod/G [28], an application that enables user level scheduling to provide transparent access to grid resources and utilizes Globus components such as the Monitoring and Discovery System (MDS) for resource discovery. Nimrod/G is the grid-enabled version of the parametric modeling application Nimrod [29] whose purpose is to explore the parametric space of a complex system described by a computational experiment. An earlier version of DOCK was implemented on grid resources by Buyya et al. using Nimrod/G to create a “Virtual Laboratory” for molecular docking [30]. This system makes very efficient use of grid resources, but is not as flexible as a Web service-based system when building a virtual screening solution that incorporates a number of different docking and scoring methods, each requiring their own procedures for input preparation and output collection.
Opal and Opal OP
Opal is a method of wrapping a legacy scientific application as a Web service, exposing it via a SOAP API and hosted inside a Jakarta Tomcat container [4]. This gives the user access to an installation of a scientific application on a remote cluster through HTTP. With the application living on the grid environment, access through the use of Web service architecture is referred to as a grid service. Each submitted job is assigned a job ID and its own working directory where input and output files are stored. This same ID is used for status query on the running job. Due to the required preparation of the input files for each DOCK job in the screening process, a grid service that performs this prep work, as well as executes DOCK is preferable over a service that only performs a direct execution of DOCK.
Opal Operation Provider (Opal OP) [31] utilizes Opal's technology, but wraps the legacy application as an operation provider, or a programming module, which can be accessed through a grid service. This gives application developers more flexibility by allowing extension of their grid service. The Opal OP toolkit creates a grid service quickly by automating the complex task of writing the web service description language (WSDL) file and building and deploying the service into a GT4 container.
3 Virtual Screening Platform
Overview
Starting with a target protein and large, diverse compound database, a virtual screening experiment could be comprised of the following steps:
Preliminary screening: Screen the original, full-size database with a fast docking method for filtering purposes
Data management: Collect docking results and rank them according to energy score
Rebuilding database: Build a new database consisting of a user defined top percentage of ranked compounds
Refined screening: Rescreen the new, smaller database using multiple scoring methods that perform more computationally expensive docking methods such as calculation of solvation energies, a more extensive conformational search, or molecular dynamics simulations.
Data management: Collect results from each of these additional screenings
From this point, a consensus of the results from all scoring methods can be consulted to determine which compounds have the highest binding affinity for the protein's binding site and ultimately the compounds chosen to be tested in the lab. Using a number of scoring methods, versus a single method, has been shown to result in a more accurate identification of strongly binding compounds by removing false positives [32]. The overall process is illustrated in Figure 1.
Figure 1. The Platform Workflow.
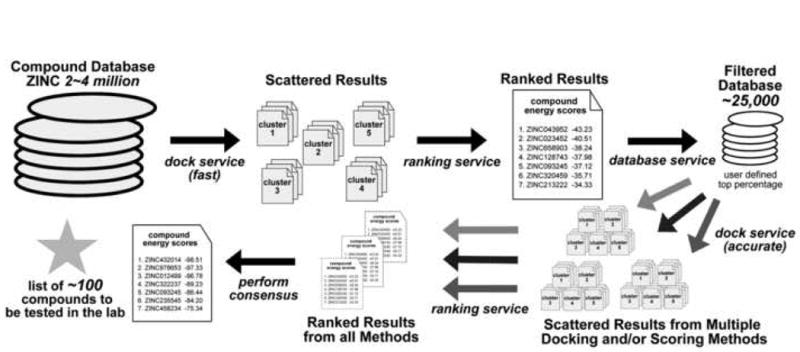
Starting with a large database of compounds, such as the ZINC library, a rapid docking method is used for the initial screening and is distributed via the dock service. Results data, consisting of compound lists ranked from best to worst energy score, are scattered across the grid resources. The ranking service searches for and gathers this data to construct a list of results encompassing the entire database of compounds. From this list, a top percentage of compounds can be selected to make up a new, smaller database of around 25,000 compounds. Conformational data output by the screening for each compound is retrieved by the database service and is used to build this new database. A number of different, more stringent parameters are then used to rescreen these compounds, distributed again by the dock service and results gathered by the ranking service. After performing a consensus amongst all scoring methods, a short list of the best potential binding compounds is generated to test in the lab in vitro.
To achieve desired features such as automation, reusability for future and different types of screenings, and the presentation of results in the experiment, simple PERL scripts were written and make up the grid services. Grid services were created for three major functions in the virtual screening experiment and these services are installed and hosted on the master node of each remote cluster. The dock service submits DOCK MPI jobs to the scheduling software of its respective cluster, the ranking service searches for and organizes results generated by DOCK MPI, and the database service reconstructs conformational data of scored compounds to be re-screened in further experiments.
Each of these grid services is accessed through the Opal OP toolkit by its own PERL script running on the client machine to manage job distribution, input data delivery, and the collection of results. Opal OP was used to wrap the DOCK application as program module that is accessed by a grid service. This provides access to the submission and status query of DOCK jobs, with simple command-line tools, on any remote cluster that hosts the dock service container. Input files and arguments required by the application can be sent to the remote cluster as well, and are defined in the job submission command-line arguments. Results data can be retrieved directly with HTTP (wget) or, in the case of this paper, the ranking and database grid services were created to sort and manipulate results on the remote clusters and then retrieve them for simplified interpretation. These relationships are outlined in Figure 2.
Figure 2. Platform Scripts.
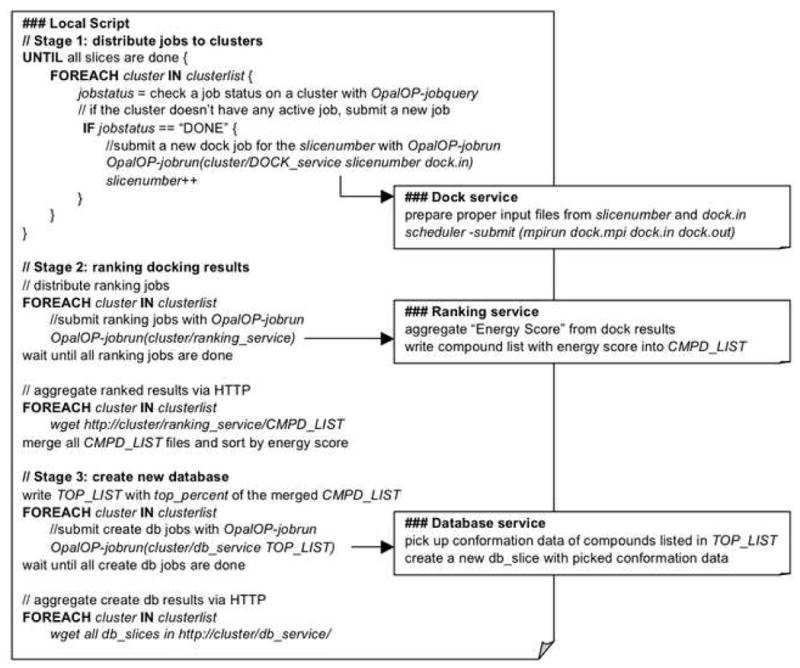
Pseudocode outline of the local and remote scripts that make up the virtual screening platform. The automated platform consists of three stages that perform the distribution of DOCK jobs, the ranking screening results, and the building new databases from the results. Each stage in the local script makes requests to the grid services installed and hosted on the master node of each remote cluster for job submission and status querying.
Since the DOCK software suite includes a number of screening methods, different sets of input files may be required for different types of screenings. The grid services were written specific to the DOCK application and support all of the included docking and scoring methods. Differences in input files, database size, and docking method used between screenings require only small alterations in environment variables and a few lines of code. Versions 5 and 6 of DOCK are supported by the platform and can be used interchangeably.
Distributed Screening (dock service)
The ZINC database “druglike” subset is split into 88 separate files, referred to as “slices”. A method that distributes the 88 separate DOCK MPI jobs was developed and resembles how the DOCK MPI implementation organizes its process management within a single cluster. The distribution method involves a script on the client machine, referred to as the local script, proceeding through a queue of database “slices” and calling the dock service on each remote cluster through Opal OP's job-run command, as displayed in Figure 3. The dock service consists of a script that processes the received input files and arguments needed for the specific DOCK MPI job, checks architecture, and then submits the job to the scheduler of that cluster. After job submission, the local script periodically checks job status for each cluster, this time through the use of Opal OP's job-query command, until a ‘DONE’ flag is received, which indicates that the next database “slice” in the queue should be launched on that cluster. With this model, the division of the database into a greater number of “slices” increases the resource allocation efficiency. This is due to the fact that a higher ratio of “slices” to number of clusters reduces the chance of a significant portion of the database being held up on a slow cluster and extending the total experiment time.
Figure 3. Distributed Screening through the Dock Service.
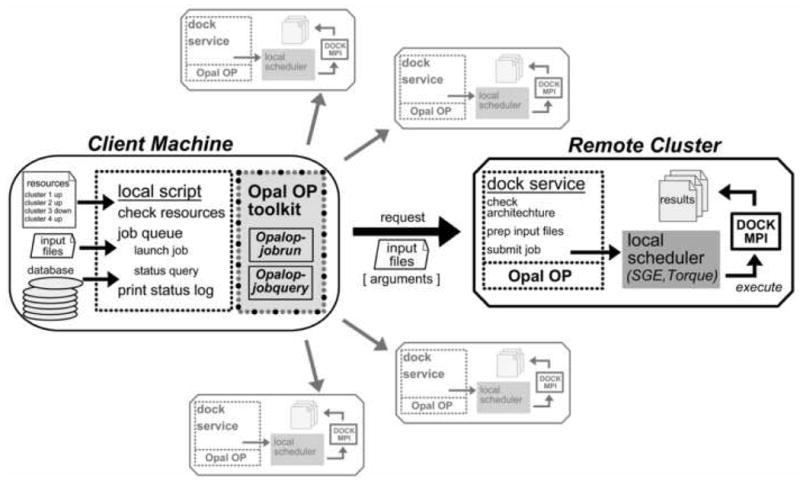
Job distribution is handled by the local script on the client machine that delivers required input through Opal OP toolkit and makes calls to the dock service hosted on each remote cluster. Resources can be added or removed by editing the resource list on the client machine. The database can be stored and accessed on the client machine or any other location that can be reached via HTTP for file transfer. Requests to launch DOCK MPI on each remote cluster are sent to the dock service with the Opalop-jobrun command. After preparing received input files according to the given arguments, the dock service submits the DOCK MPI job to the scheduling software to be executed. The status of each job is checked using the Opalop-jobquery command. Each remote cluster then has its own screening results that must be collected and sorted once the screening reaches completion.
In order to manage resources used for a screening experiment, the local script first reads a user-created list of clusters with hostname and availability information. Clusters are represented as hash keys with an array of values to keep track of the hostname, database “slice” number, job status, job duration, and the unique jobID assigned by Opal OP. The user-created cluster list is accessed after each cycle of job-status checks in order to allow the addition or removal of resources while the screening is being performed. The ease of grid service creation makes it very attractive to add resources that are hosting the dock service, in the event they become available during a screening, by simply editing the cluster list file.
The distributed nature of a Grid environment makes it naturally fault-prone due to power failures, resource maintenance, or other factors outside the control of the user. This platform strove for a simple architecture, thus minimizing the number of possible sources of error. As the screening proceeds, the local script keeps a log of its progress with the jobID and cluster information for each “slice” that is submitted. This log allows the user to check the progress of screening experiments and provides useful information in the case of investigating errors and job failure on remote clusters since the jobID corresponds to the directory of the submitted job.
Clusters that go down mid-job will not receive a ‘DONE’ flag for their current job so they will not be given any more jobs from the queue, thus providing a natural fault handling ability. Each finished job has its time recorded in the log and the time for the entire screening is recorded when the local script finishes.
One other notable error caused by the heterogeneity of the Grid is the discrepancy between results generated by DOCK executables compiled on different resources. Comparing results from a test screening included in the DOCK software package across all resources ensured that there were no variations in the results before performing screening experiments.
Rebuilding Results (ranking service and database service)
Each DOCK MPI job produces a ranked list of results according to energy scores. But since each database “slice” is run as its own job, compounds are compared only to those within their own individual “slice”. A method to rank the results of the screening as if they were one single DOCK MPI job was created by pairing another local script to the ranking service in order to bring together the data located on a number of remote clusters in an organized fashion.
The local script reads a cluster list file just as the distributed screening method does in order to specify the resources to gather results from. Opal OP job-run command invokes the ranking service on each remote cluster. The ranking service searches for completed DOCK results among the unique directories created during the screening process for each submitted DOCK MPI job. Energy scores of all compounds are saved to a file along with cluster and directory information in order to provide access to the compound structural conformation data that is output by the screening. Job status is checked repeatedly and when all remote clusters return a “DONE” state, the created lists of results are gathered over HTTP. These files are read by the local script and used to create a list of all the compounds from the database, ranked according to energy score, from the most favorable to least favorable binding compounds. The user would use this list as the presentable results of the screening. Figure 4 shows the ranking process.
Figure 4. Retrieving Ranked Results.
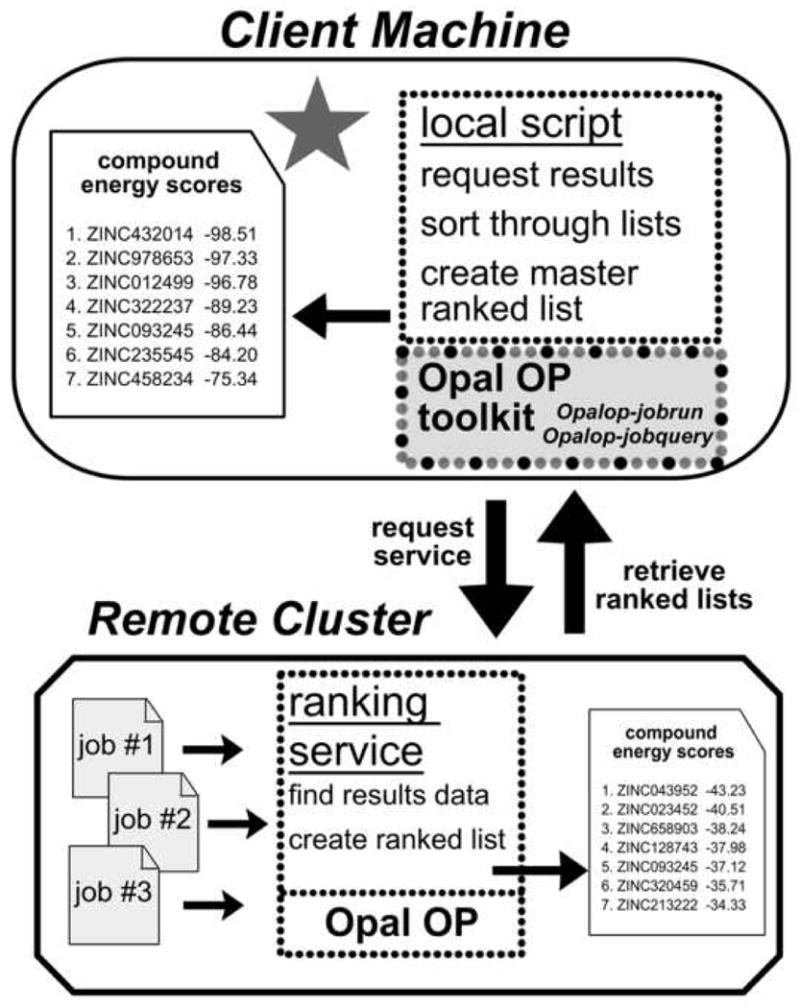
The local script on the client machine calls the ranking service running on each remote cluster. The ranking service searches for screening results and puts together a list of compounds and their calculated energy scores. When every remote cluster finishes its search, the compound lists are retrieved via HTTP by the local script. Compounds from all the lists are sorted according to energy score to produce a ranked list of the best binding compounds. Additional remote clusters are not included in the diagram for clarity.
In addition to bringing together energy score rankings, a grid service was developed to automate building a new database consisting of the conformational data output by the initial screening. This becomes useful when initially performing a fast docking method to remove those compounds that have undesirable chemical and geometrical properties, and then re-screening the remaining compounds in more stringent screening experiments.
Once results are collected by the ranking service, the user is queried by the local script what top percentage, or best binding compounds, of the initial screening results are to be used to construct a new filtered, database. After determining which compounds make-up this top percentage, the local script invokes the database service on each remote cluster while providing the list of compounds. Conformational data of these compounds are gathered together by the database service and used to construct “slices” of the new database. After all the Opal OP job status queries return a “DONE” state, the remotely scattered “slices” are retrieved by the local script via HTTP. The end product is a filtered database consisting of the best binding compounds from the initial screening. This process is shown in Figure 5. Since this filtered database's structure is similar to that of the original database, further screenings can be carried out using the dock service distribution method described above.
Figure 5. Building a Filtered Database.
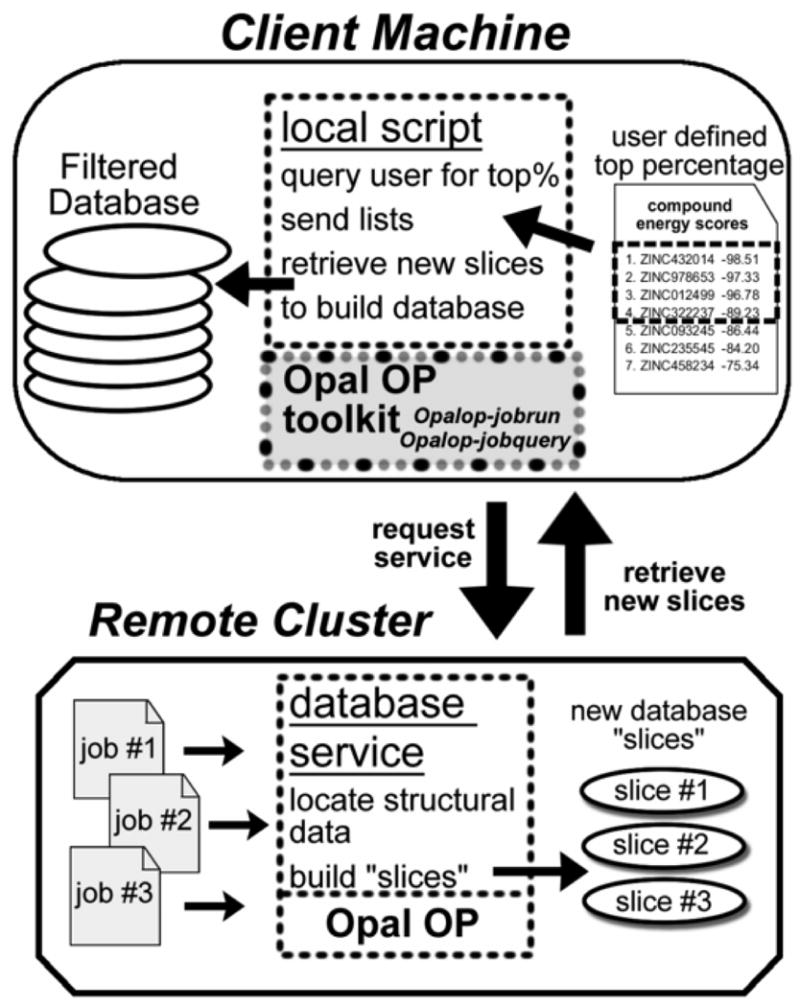
The local script on the client machine first queries the user for a desired percentage of the best binding compounds from the screening results. This generates lists of compounds whose conformational data needs to be retrieved to build a new database. These lists are sent to the database service on each remote cluster and the conformational data is pulled from the previous screenings results and used to build the “slices” of the new database. All the “slices” are retrieved by the local script via HTTP to create a filtered database consisting of a user-defined top percentage of compounds from the original database. This smaller database can then be used in more stringent docking and/or scoring methods that require more time per compound.
4 Application of the Platform to Identify Inhibitors
With the purpose of both testing the platform and searching for inhibition leads for a protein of interest, a screening experiment was performed on the PRAGMA grid testbed. The grid services and DOCK5.4 application were installed on the seven clusters outlined in Table 1. The average time per slice of database, ∼25,000 compounds, is listed in Table 1 but does not reflect the maximum performance of the cluster because resource availability fluctuates throughout the screening depending on Grid workload. Scheduling software can also put priority on job execution requests, allowing for dedicated jobs that queue other lower priority jobs until the dedicated jobs complete. This is exemplified by the small number of database slices completed by the TRECC cluster listed in Table 1, despite having a comparable average completion time per slice.
Table 1. PRAGMA Grid testbed resources used in the virtual screening experiment.
| Host | Location | Resources | Average time per slice (hr) | Number of slices finished |
|---|---|---|---|---|
| CAFÉ | Osaka U. Japan | 10-20 nodes i686 2800MHz SGE | 30.5 | 9 |
| TEA | Osaka U. Japan | 10-40 nodes i686 1400MHz SGE | 16.5 | 16 |
| ROCKS-52 | SDSC, CA | 15-20 nodes i686 2388MHz SGE | 32.7 | 8 |
| F32 | AIST Japan | 20-60 nodes i686 3060MHz SGE | 10.7 | 24 |
| X | AIST Japan | 10-20 nodes i686 2800MHz SGE | 17.0 | 15 |
| TRECC | Univ. of Illinois | 10-20 nodes i686 2400MHz Torque | 19.8 | 3 |
| LZU | LanZhou U. China | 10-20 nodes i686 2993MHz Torque | 21.4 | 12 |
From a crystal structure of the catalytic domain of a protein, hydrogen atoms and charge were added to the protein molecule in Chimera [33] using the included “Dock Prep” feature. The relatively large active site of the protein to be explored was represented by 40 “spheres” generated using SPHGEN [34], which is part of the DOCK suite. A chemical and energy grid was generated on this active site with a grid spacing of 0.5 angstroms to be used by DOCK in its energy calculations, including van der Waals and electrostatic forces [19]. The bump filter [21] feature was also utilized with an overlap ratio of 0.75 as a means of identifying the orientations of a compound that result in deep penetration into the receptor molecule by the compound's atoms and preventing them from being scored and minimized during docking.
DOCK requires the compounds screened to have hydrogens added, contain charge information, and be stored in the SYBYL mol2 format [35]. The ZINC compound library was chosen as the database to be screened because of its exhastive collection of compounds, free access, and availability in a preprocessed mol2 format. Specifically, the 2,066,906 compound “druglike” subset of the ZINC library was screened and is made up of compounds of molecular weights between 150 and 500 Da and have no more than 10 hydrogen donors [36].
DOCK 5.4's grid energy scoring method was used for the screening and compounds were treated as flexible through DOCK's anchor-and-grow algorithm when docked into the active site. The maximum number of orientations to be scored was set to 500 and minimum anchor size was defined as six heavy atoms. The number of anchor orientations and conformations per growth cycle were set to a maximum of 100 and 20, respectively. Ten cycles of simplex minimization were performed on each rigid anchor orientation and twenty cycles during the flexible growth stage.
Many of these variable parameters go into determining how long DOCK takes to find the most energetically favorable conformation for each compound in the active site of the receptor molecule. The combination of the large active site represented by the 40 spheres and the extensive compound conformation search with flexible docking required an average docking time of 82 seconds per compound per processor, which is fairly long when performing such a large-scale screening. These docking parameters were chosen in order to have a screening that both exemplifies a moderately extensive search and tests the platform's ability to adjust over time to changes in the grid testbed's workload. With a number of simultaneous users, not all processors are available within each cluster at one time, so the scheduling software allocates proper resources to each job.
Over the course of the screening, 150-200 processors were being used at one time depending on grid workload and averaged 180 processors. The screening took a total of 11.56 days of laboratory time, which is the equivalent of 2081 CPU days (over 5 1/2 years). Segmentation faults occurred during the first attempted screening of the “druglike” subset of the ZINC library. Examination of the fault causing compounds revealed 136 compounds with similar structures. Once these compounds were removed from the database, segmentation faults did not reoccur. Due to the possibility that a different type of structural moiety in a compound database would cause segmentation faults in future screenings, a method of automatically resubmitting a failed database slice was not pursued to avoid wasting the CPU time leading up to the consistent fault. With these 136 compounds removed, using a test screen before starting large screening experiments to ensure proper setup on each cluster resulted in 100% success-rate experiments in this study.
The retrieval and sorting of results data, which was scattered across the seven clusters, into a single ranked list and the rebuilding of a new database consisting of the top 2.5% of the ∼2 million compounds required only an additional ten minutes. Although the percentage of compounds used in forming the new database was arbitrary, this method has been used by others [37, 38]. In this study, one factor that was used to determine the percentage was the screening time required by the more computationally expensive secondary phase scoring methods. The number of compounds was selected to limit the time of the rescreen to 5-7 days.
This filtered database was then rescreened using a more stringent energy scoring method in DOCK 5.4 through the dock service. Parameters that were altered to perform a more rigorous screening experiment included a denser chemical and energy grid sampled at 0.3 angstroms and increased simplex minimization. The required time per compound was 10 minutes during this stage for a total laboratory time of 1.5 days. Once this finished, the same ∼40,000 compound database was screened again using the AMBER score method of DOCK 6. The AMBER score's abilities to calculate solvation energies and perform molecular dynamics simulations, treating both the compound and receptor as flexible for an “induced fit,” make it a very robust and computationally expensive screening method. One caveat is that AMBER score requires preprocessing of the compounds with a provided Perl script. This step was easily incorporated into the automated workflow with the addition of two lines in the dock service script to run and verify the results of the preparatory AMBER Perl script. Molecular dynamics simulation was performed on each compound-receptor complex in 4500 steps with 250 cycles of minimization before and after. Screened compounds required up to 30 minutes each to complete the scoring process, yielding a total laboratory time of about 5 days. It is important to note that the times reported here are highly dependent on the DOCK input parameters chosen for each scoring method.
These second phase screenings were distributed through the DOCK service with their own respective input parameters, just as the first phase screening was performed. Results from both the stringent energy score and AMBER score second phase screenings were collected from the distributed Grid resources and sorted using the ranking service. A consensus was taken between the results of the two scoring methods by simply summing the compound rankings from each scoring. A number of compounds identified by the virtual screening experiment will be obtained and tested to determine their inhibitory effects on the activity of the protein of interest. This will not only confirm the computational results obtained, but provide important biochemical tools to facilitate ongoing biomedical research and potential pharmaceutical reagents.
5 Conclusions
With the grid services in place on the remote clusters, the user on the local cluster can execute an entire virtual screening experiment with only a few commands since the processes of job distribution, monitoring progress, data manipulation, and retrieval of results has been automated by the platform. The large pool of resources offered by grid computing enables biomedical scientists to take advantage of thorough, yet computational expensive, flexible docking methods when screening a very large database of compounds. In practice, use of the platform orchestrated the screening of over two million compounds on seven different computer clusters in under two weeks, and then rebuilt a centralized database of results for analysis using a total of only three commands.
As described throughout the paper, the platform does not serve as a stand-alone application. Tying together a number of grid middleware and information technologies, such as the Globus Toolkit and Opal OP, provided a base to build an automated, custom, virtual screening platform that takes advantage of grid resources and maintains the ability to adjust to different types of molecular docking experiments. The standardization of communication provided by the Web service model of grid middleware allowed for portability between different architectures. Thus, the addition of a newly available cluster to a growing pool of resources became a simple process, requiring minimal time and effort.
Further development plans include building a portal to access the grid-enabled implementation of DOCK through the web, similar to Australian BioGrid Portal (ABGP) [39] that was built on top of the virtual screening system developed by Buyya et al. The step-by-step, GUI access to grid resources provided by the ABGP produces a very positive quality of service for the user, but again offers limited flexibility when using several different docking methods. Using Opal OP for the simple creation and deployment of extensible grid services is very fitting since a number of grid services can be created to handle the differing pre- and post-processing requirements of the numerous docking and scoring methods available. Displaying results is also handled by grid services and is a rapid method for sorting through data stored on scattered resources and presenting a single ranked list of energy scores.
To our knowledge, the platform described in this paper is the first effort to combine complex molecular docking methods with the well-suited, service-based grid environment to provide a complete solution capable of screening extensive compound databases. We feel that this work provides a powerful tool to perform virtual screening experiments and serves as a model for bringing legacy scientific applications to a grid environment with a custom workflow. Two separate projects that utilize the system described in this paper are underway with the interest of finding inhibitory compounds to investigate the enzymatic activity of intracellular signaling proteins, one of them being the follow-up to the results of the screening performed in this paper.
Acknowledgments
We thank Dr. Peter Arzberger and Dr. David Abramson for critical reading of the manuscript. We are grateful to the PRAGMA sites for providing the computational resources. This project was supported by PRIME (NSF 0407508), PRIUS, PRAGMA, and Calit2.
Footnotes
Conflict of interest statement: The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Foster I, Kesselman C, Tuecke S. The anatomy of the grid: Enabling scalable virtual organization. J-IJHPCA. 2001;15(3):200–222. [Google Scholar]
- 2.W3C Web Services Architechture. 2004 http://www.w3.org/TR/ws-arch/.
- 3.Foster I, Kesselman C, Nick JM, et al. In: The physiology of the grid, in Grid Computing. Berman Fran, Fox Geoffrey, Hey Tony., editors. John Wiley & Sons, Ltd; West Sussex, England: 2003. pp. 217–249. [Google Scholar]
- 4.Krishnan S, Stearn B, Bhatia K, et al. Opal: Simple web services wrappers for scientific applications. ICWS. 2006:823–832. [Google Scholar]
- 5.Stevens R. Trends in cyberinfrastructure for bioinformatics and computational biology. CTWatch Quarterly. 2006;2(3):1–5. Available: http://www.ctwatch.org/quarterly.
- 6.Zheng C, Abramson D, Arzberger P, et al. The PRAGMA testbed - building a multi-application international grid. Ccgrid. 2006;2:57. [Google Scholar]
- 7.Walters WP, Stahl MT, Murcko MA. Virtual screening - an overview. Drug Discovery Today. 1998;3(4):160–178. [Google Scholar]
- 8.Doman TN, McGovern SL, Witherbee BJ, et al. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45(11):2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 9.Sheridan RP, Kearsley SK. Why do we need so many chemical similarity search methods? Drug Discov Today. 2002;7(17):903–911. doi: 10.1016/s1359-6446(02)02411-x. [DOI] [PubMed] [Google Scholar]
- 10.Enyedy IJ, Ling Y, Nacro K, et al. Discovery of small-molecule inhibitors of bcl-2 through structure-based computer screening. J Med Chem. 2001;44(25):4313–4324. doi: 10.1021/jm010016f. [DOI] [PubMed] [Google Scholar]
- 11.Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry. 2005;44(37):12316–12328. doi: 10.1021/bi050801k. [DOI] [PubMed] [Google Scholar]
- 12.UCSF DOCK. http://dock.compbio.ucsf.edu/.
- 13.Irwin JJ, Shoichet BK. ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lyne PD. Structure-based virtual screening: An overview. Drug Discov Today. 2002;7(20):1047–1055. doi: 10.1016/s1359-6446(02)02483-2. [DOI] [PubMed] [Google Scholar]
- 15.Ewing TJA, Makino S, Skillman AG, et al. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des. 2001;15(5):411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 16.Makino S, Kuntz ID. Automated flexible ligand docking method and its application for database search. Journal of Computational Chemistry. 1997;18(14):1812–1825. [Google Scholar]
- 17.Knegtel RM, Wagener M. Efficacy and selectivity in flexible database docking. Proteins. 1999;37(3):334–345. doi: 10.1002/(sici)1097-0134(19991115)37:3<334::aid-prot3>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Moustakas D, Lang P, Pegg S, et al. Development and validation of a modular, extensible docking program: DOCK 5. J Comput Aided Mol Des. 2006;20(10):601–619. doi: 10.1007/s10822-006-9060-4. [DOI] [PubMed] [Google Scholar]
- 19.Lang PT, Moustakas D, et al. DOCK 6.1 Users Manual. 2007 Available: http://dock.compbio.ucsf.edu/
- 20.Gropp W, Lusk E, Skjellum A. In: Using MPI: Portable parallel programming with the message-passing interface. , editor. MIT Press; Cambridge, Massachusetts: 1999. p. 371. [Google Scholar]
- 21.Kuntz ID, Moustakas DT, Lang PT. DOCK 5.4 User Manual. 2006 doi: 10.1007/s10822-006-9060-4. Available: http://dock.compbio.ucsf.edu/ [DOI] [PubMed]
- 22.Irwin JJ, Shoichet BK. ZINC is not commericial - A free database for virtual screening. 2006 http://zinc.docking.org/
- 23.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432(7019):862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laszewski Gv, Amin K. In: Grid middleware, in Middleware for Communications. Mahmoud Quasay H, Wiley John., editors. John Wiley & Sons, Ltd; West Sussex, England: 2004. pp. 109–130. [Google Scholar]
- 25.Foster I, Kesselman C. Globus: A metacomputing infrastructure toolkit. International J Supercomputer Applications. 1997;11(2):115–128. [Google Scholar]
- 26.Li WW, Krishnan S, Mueller K, et al. Building cyberinfrastructure for bioinformatics using service oriented architecture. Ccgrid. 2006;2:39. [Google Scholar]
- 27.Foster I. Globus toolkit version 4: Software for service-oriented systems. Journal of Computer Science and Technology. 2006;21(4):513–520. [Google Scholar]
- 28.Abramson D, Giddy J, Kotler L. High performance parametric modeling with Nimrod/G: Killer application for the global grid? Ipdps. 2000;00:520. [Google Scholar]
- 29.Abramson D, Sosic R, Giddy J, et al. Nimrod: A tool for performing parametrised simulations using distributed workstations. Hpdc. 1995:112–121. [Google Scholar]
- 30.Buyya R, Branson K, Giddy J, et al. The virtual laboratory: A toolset to enable distributed molecular modeling for drug design on the world-wide grid. Concurrency and Computation: Practice and Experience. 2003;15(1):1–25. [Google Scholar]
- 31.Ichikawa K, Date S, Krishnan S, et al. Opal OP: An extensible grid-enabling wrapping approach for legacy applications. GCA. 2007:117–127. [Google Scholar]
- 32.Charifson PS, Corkery JJ, Murcko MA, et al. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 33.Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 34.Kuntz ID, Blaney JM, Oatley SJ, et al. A geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161(2):269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 35.Tripos Inc. SYBYL Mol2 File Format. 2005 http://www.tripos.com/data/support/mol2.pdf.
- 36.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 37.Graves AP, Shivakumar DM, Boyce SE, et al. Rescoring docking hit lists for model cavity sites: Predictions and experimental testing. J Mol Biol. 2008;377(3):914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang N, Kalyanaraman C, Irwin JJ, et al. Physics-based scoring of protein-ligand complexes: Enrichment of known inhibitors in large-scale virtual screening. Journal of Chemical Information and Modeling. 2006;46(1):243–253. doi: 10.1021/ci0502855. [DOI] [PubMed] [Google Scholar]
- 39.Gibbins H, Nadiminti K, Beeson B, et al. The Australian BioGrid portal: Empowering the molecular docking research community. APAC'05. 2005 [Google Scholar]