

Abstract
AIM: To study the expression and phosphorylation of extracellular signal-regulated kinase (ERK) 1 and ERK2 in multidrug resistant (MDR) hepatocellular carcinoma (HCC) cells.
METHODS: MDR HCC cell lines, HepG2/adriamycin (ADM) and SMMC7721/ADM, were developed by exposing parental cells to stepwise increasing concentrations of ADM. MTT assay was used to determine drug sensitivity. Flow cytometry was employed to analyze cell cycle distribution and measure cell P-glycoprotein (P-gp) and multidrug resistant protein 1 (MRP1) expression levels. ERK1 and ERK2 mRNA expression levels were measured by quantitative real-time PCR (QRT-PCR). Expression and phosphorylation of ERK1 and ERK2 were analyzed by Western blot.
RESULTS: MTT assay showed that HepG2/ADM and SMMC7721/ADM were resistant not only to ADM, but also to multiple anticancer drugs. The P-gp expression was over 10-fold higher in HepG2/ADM cells than in HepG2 cells (8.92% ± 0.22% vs 0.88% ± 0.05%, P < 0.001) and over 4-fold higher in SMMC7721/ADM cells than in SMMC7721 cells (7.37% ± 0.26% vs 1.74% ± 0.25%, P < 0.001). However, the MRP1 expression was not significantly higher in HepG2/ADM and SMMC7721/ADM cells than in parental cells. In addition, the percentage of MDR HepG2/ADM and SMMC7721/ADM cells was significantly decreased in the G0/G1 phase and increased in the the S phase or G2/M phase. QRT-PCR analysis demonstrated that the ERK1 and ERK2 mRNA expression increased apparently in HepG2/ADM cells and decreased significantly in SMMC7721/ADM cells. Compared with the expression of parental cells, ERK1 and ERK2 protein expressions were markedly decreased in SMMC7721/ADM cells. However, ERK2 protein expression was markedly increased while ERK1 protein expression had no significant change in HepG2/ADM cells. Phosphorylation of ERK1 and ERK2 was markedly decreased in both HepG2/ADM and SMMC7721/ADM MDR cells.
CONCLUSION: ERK1 and ERK2 activities are down-regulated in P-gp-mediated MDR HCC cells. ERK1 or ERK2 might be a potential drug target for circumventing MDR HCC cells.
Keywords: Multidrug resistance, Extracellular signal-regulated MAP kinases, Hepatocellular carcinoma, P-glycoprotein, Multidrug resistance-associated protein
INTRODUCTION
Hepatocellular carcinoma (HCC) is the third cause of cancer-related death[1,2]. Drugs used in the treatment of HCC are cytotoxic with a high risk of side effects and none of them is specific for HCC[3]. Moreover, HCC is a hypervascular solid cancer characterized by a high degree of drug resistance[4]. Multidrug resistance (MDR) to chemotherapeutic agents plays a major role in the failure of cancer therapy[5]. MDR phenotype, an intrinsic or acquired cross-resistance to a variety of structurally and functionally unrelated drugs, is almost constantly expressed in HCC and represents one of the major problems in cancer eradication by limiting the efficacy of chemotherapy[6]. Resistance to therapy can result from decreased drug uptake, increased DNA repair or drug inactivation[7].
Mitogen-activated protein kinase (MAPK) pathway is an attractive target or therapeutic intervention in cancer due to its integral role in the regulation of cancer cell proliferation, invasiveness, and survival. Pharmaceutical agents can inhibit various kinases and GTPases comprising the pathway[8,9]. Extracellular signal-regulated kinase (ERK) 1/2 is a member of the MAPK family. ERK1 and ERK2 are isoforms of the “classical” MAPK[10]. The activity of ERK1/2 has been implicated in the regulation of embryonic morphogenesis, cell proliferation, tumor transformation, and apoptosis[11]. It has been recently found that P-gp expression in the MDR1-transduced human breast cancer cell lines MCF-7/MDR and MDA-MB-231/MDR is positively regulated by the ERK pathway and blockade of the MEK-ERK-RSK pathway can suppress cell surface P-gp expression by promoting its degradation[12]. In addition, there are several lines of evidence that modulation of ERK activation may reverse MDR in prostatic, gastric and hematopoietic cancers[13–16]. However, there is little evidence that ERK activity is related to MDR of HCC.
The aim of this research was to study the crucial kinases of ERK pathway, including expression and phosphorylation (activity) of ERK1 and ERK2 in MDR HCC cell lines, and to explore whether the relationship between MDR and ERK1/2 kinases involves specific molecular aspects of these cell lines. Once they are well characterized, the ERK pathway might be exploited for overcoming MDR of HCC.
MATERIALS AND METHODS
Cell culture
Human HCC cell lines, HepG2 and SMMC7721, were purchased from Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Science, Chinese Academy of Sciences. HepG2 was cultured with DMEM (HyClone, Logan, UT, USA) and SMMC7721 was cultured with RPMI-1640 (HyClone, Logan, UT, USA). Both media were supplemented with 10% calf serum (HyClone, Logan, UT, USA) and maintained at 37°C in a humidified atmosphere containing 50 mL/L CO2 and 950 mL/L air. Multidrug resistant human HCC cell lines, HepG2/adriamycin (ADM) and SMMC7721/ADM, were developed by our group. To develop the HepG2/ADM and SMMC7721/ADM cells, ADM (Pharm-sh Pharmaceutical Co., Ltd., Shanghai, China) was added respectively to HepG2 and SMMC7721 cells at a stepwise increasing concentration from 0.01 to 0.2 mg/L. Resistant cells were selected by removing the non-resistant dead cells. Multidrug resistance was maintained by culturing the cells with 0.2 mg/L ADM and MDR cells were named HepG2/ADM and SMMC7721/ADM.
Measurement of cellular sensitivity to anticancer drugs
MTT (Sigma-Aldrich, St. Louis, MO, USA) assay was used to determine drug sensitivity. Sensitivity of cultured HepG2/ADM and SMMC7721/ADM cells to anticancer drugs, including ADM, fluorouracil, cisplatin, cyclophosphamide, mitomycin and vincristine (Pharm-sh Pharmaceutical Co., Ltd., Shanghai, China), was detected, respectively[17]. IC50 value was assessed by probit regression analysis using SPSS11.5 statistical software. Resistance index (RI) was calculated according to the formula: RI = IC50 for MDR cells IC50/IC50 for parental cells.
Flow cytometric analysis of cell cycle distribution
Cultured HepG2/ADM and SMMC7721/ADM cells and their parental cells were collected respectively through trypsinization, washed with ice-cold PBS, centrifuged at 500 × g for 5 min at 4°C, washed twice with ice-cold PBS and fixed in 70% ethanol for 2 h at 4°C. Samples were rehydrated with PBS and the cells were incubated for 30 min at room temperature with a propidium iodide stating solution in PBS containing 0.2 mg/mL propidium iodide, 0.2 mg/mL DNAse-free RNAse A (Roche, Basel, Switzerland), and 0.1% Triton X-100. Using red propidium-DNA fluorescence, 4000 events were acquired with a Epics@ XL Beckman Coulter FACS machine (Beckman Coulter Inc., Fullerton, CA, USA)for each sample and the percentage of cells in G0/G1, S and G2/M phases of the cell cycle was calculated using the Systerm IITM software (Beckman Coulter Inc., Fullerton, CA, USA).
Flow cytometric analysis of cell p-gp and mrp1 expression level
Cultured MDR and parental cells were collected as above. Then, samples were immunostained with P-glycoprotein antibody (FITC) (Cat.ab66250, Abcam plc, Cambridge, UK) and MRP1 antibody (FITC) (Cat.No.557593, BD Biosciences Pharmingen, San Diego, CA, USA), respectively, according to the proper protocol. The cells were fixed and permeabilized with a BD Cytofix/CytopermTM solution (Cat.No.554722, BD Biosciences, San Jose, CA, USA) before they were immunostained with MRP1 antibody (FITC). Flow cytometry was carried out with a fluorescent-activated cell scan (FACS) using the Systerm IITM software. Fluorescence of the cells treated with fluorescent isotype control IgG (Cat.ab18455, Abcam plc) was evaluated in each experiment to measure the level of background fluorescence of negative cells. Mean fluorescence intensity (MFI) of positively stained cells was determined.
RNA extraction and quantitative real-time polymerase chain reactions (QRT-PCR)
ERK1 and ERK2 mRNA expression levels were measured by QRT-PCR. Total RNA was extracted using the TRIzol reagent (GIBCO BRL, Life Technologies Inc., Rockville, MD, USA) following the constructions of its manufacturer, and reverse transcripted to cDNA using the Gene Amp RNA PCR kit in a DNA thermal cycler (Bio-Rad). QRT-PCR was performed with SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA) in an ABI Prism 7700 real time PCR machine (Applied Biosystems).
The synthesized cDNA served as a template in a (25 μL) reaction. A non-template control was included in all experiments. Primer sequences are as follows: ERK1 GenBank: NM_002746 forward, 5'-TCAACACCACCTGCGACCTT-3', and reverse, 5'-GCGTAGCCACATACTCCGTCA-3'; ERK2 GenBank: NM_002745 forward, 5'-GTTCCCAAATGCTGACTCCAA-3', and reverse, 5'-CTCGGGTCGTAATACTGCTCC-3'; β-actin GenBank: NM_001101 forward, 5'-TGACGTGGACATCCGCAAAG-3', and reverse, 5'-CTG- GAAGGTGGACAGCGAGG-3'.
QRT-PCR was performed at 94°C for 4 min, followed by 40 cycles at 94°C for 15 s, at 60°C for 25 s, and at 72°C for 25 s. Oligonucleotides and reagents for PCR assay were purchased from Qiagen GmbH, Hilden, Germany. Data were analyzed with the Sequence detector software (v1.9, Applied Biosystems). The mean Ct value for duplicate measurements was used to detect the expression of target gene with normalization to a housekeeping gene used as an internal control (β-actin) according to the 2−ΔCt formula.
Western blot analysis
Protein was collected from cultured HepG2, SMMC7721, HepG2/ADM and SMMC7721/ADM cells and its concentration was measured (protein assay dye, Bio-Rad). Then, the protein was denatured in a LDS sample buffer for 5 min at 95°C, run on SDS-PAGE (NUPAGE, 4%-12% Bis-Tris, Invitrogen, Carlsbad, CA, USA) and blotted onto PVDF membranes (0.2 μm, Invitrogen). Membranes were blocked with 5% dry milk in TBS-T (TBS containing 0.05% Tween 20) for 1 h at room temperature and incubated overnight at 4°C with antibodies against ERK1, ERK2, or phospho-ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, Inc., Danvers, MA, USA). After incubation with the respective primary antibodies, the membranes were exposed to species-specific horseradish peroxidase-labeled secondary antibodies at room temperature, and developed using the ECL plus Western blotting reagent (GE Healthcare, Little Chalfont, UK) and Fuji Film LAS-1000 equipment (Fuji Film, Tokyo, Japan). Parallel membranes were incubated with 1:5000 rabbit monoclonal antibodies to GAPDH (Cell Signaling Technology, Inc.) and HRP-coupled rabbit anti-mouse secondary antibody. Primary and secondary antibody solutions were prepared in a PBS solution containing 2% bovine serum albumin and 0.1% Tween-20. After incubation with antibodies, the membranes were washed 3 times for 5 min in PBS containing 0.1% Tween-20. Calculation and statistics were performed using the ImageJ 1.37 software.
Statistical analysis
Statistical analysis was performed using Student’s t-test to compare the two groups and ANOVA was used with Dunnett’s post-test for multiple comparisons when the three groups or more were compared. P < 0.05 was considered statistically significant. The results were expressed as mean ± SE. Values were analyzed using the statistical package SPSS for Windows Ver.11.5 (SPSS Inc., Chicago, IL, USA).
RESULTS
Determination of MDR
Each step of developing MDR HepG2/ADM and SMMC7721/ADM cells took 7-8 wk. MDR was maintained by culturing the cells with 0.2mg/L ADM. Cytotoxicity assay found that HepG2/ADM and SMMC7721/ADM were resistant not only to ADM but also to multiple anticancer drugs. Among them, fluorouracil (5-FU), cyclophosphamide (CTX), cisplatin (CDDP), mitomycin (MMC), and vincristine (VCR) were tested in our study. Their lethal dose (IC50) was significantly higher for HepG2/ADM and SMMC7721/ADM cells than for non-resistant parental cells (Figure 1 and Table 1).
Figure 1.

Measurement of cellular sensitivity to anticancer drugs and parental cells. Cytotoxicity assay for adriamycin, fluorouracil, cyclophosphamide, tested cisplatin, mitomycin, and vincristine was performed to evaluate the IC50 for HepG2 and HepG2/ADM cells (A), SMMC7721 and SMMC7721/ADM cells (B). Dose response curves were derived from five independent experiments using MTT assay. The data were shown as mean ± SE.
Table 1.
Determination of IC50 and resistance index of different anticancer drugs (mean ± SD)
|
IC50 |
Resistance index |
IC50 |
Resistance index | |||
| HepG2 | HepG2/ADM | SMMC7721 | SMMC7721/ADM | |||
| Adriamycin (mg/L) | 0.0063 ± 0.0022 | 0.135 ± 0.053 | 21.43 | 0.0139 ± 0.008 | 0.266 ± 0.036 | 19.14 |
| Fluorouracil (μmol/L) | 1.114 ± 0.271 | 25.34 ± 2.38 | 22.75 | 0.689 ± 0.082 | 48.5 ± 2.57 | 70.39 |
| Cyclophosphamide (mg/L) | 2.902 ± 0.369 | 32.68 ± 4.962 | 11.26 | 2.315 ± 0.279 | 60.08 ± 4.93 | 25.95 |
| Cisplatin (mg/L) | 0.0527 ± 0.013 | 1.084 ± 0.0749 | 20.57 | 0.0483 ± 0.011 | 1.637 ± 0.172 | 33.89 |
| Mitomycin (mg/L) | 0.061 ± 0.017 | 1.085 ± 0.246 | 17.79 | 0.032 ± 0.013 | 0.644 ± 0.168 | 20.13 |
| Vincristine (mg/L) | 0.0093 ± 0.0035 | 0.086 ± 0.0098 | 9.25 | 0.006 ± 0.004 | 0.247 ± 0.023 | 41.16 |
Cell cycle distribution
Cell cycle phase distribution was detected by flow cytometry to determine whether there is any difference in cell cycle kinetics between MDR HepG2/ADM and SMMC7721/ADM cells and their parental cells. The percentage of HepG2/ADM and SMMC7721/ADM cells was significantly decreased at the G0/G1 phase and increased at the S phase or G2/M phase (Figure 2 and Table 2).
Figure 2.
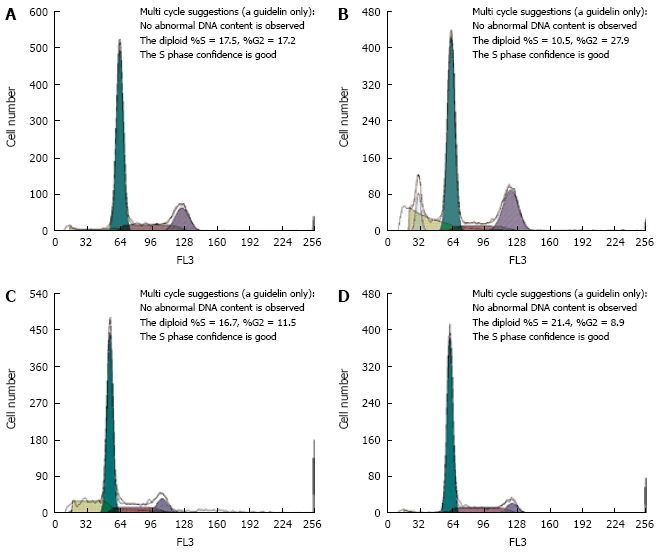
Cell cycle distributions in MDR and parental cells. Cell cycle distributions of HepG2 (A), HepG2/ADM (B), SMMC7721 (C) and SMMC7721/ADM (D) cells were analyzed by flow cytometry as described in Materials and Methods.
Table 2.
Cell cycle distribution of parental and MDR HCC cells (mean ± SD)
| Cells (n = 5) | G0/G1 | S | G2/M |
| hepG2 | 65.08 ± 1.61 | 18.32 ± 1.37 | 16.58 ± 0.65 |
| hepG2/ADM | 62.44 ± 1.77a | 12.24 ± 1.21b | 25.36 ± 2.12c |
| SMMC7721 | 71.12 ± 1.38 | 17.86 ± 1.91 | 11.02 ± 1.95 |
| SMMC7721/ADM | 67.8 ± 2.15a | 23.6 ± 0.93b | 8.62 ± 2.74a |
P < 0.05,
P < 0.01,
P < 0.001 vs control cells.
P-gp and MRP1 expression
Protein expression of P-gp and MRP1 in HepG2/ADM, SMMC7721/ADM and their parental cells was evaluated. P-gp expression was over 10-fold higher in HepG2/ADM cells than in HepG2 cells, and over 4-fold higher in SMMC7721/ADM cells than in SMMC7721 cells. However, the MRP1 expression was not significantly higher in HepG2/ADM and SMMC7721/ADM cells than in parental cells (Figure 3 and Table 3).
Figure 3.

Expression of P-gp and MRP1 in MDR and parental cells. Histograms showing that P-gp expression was over 10-fold higher in HepG2/ADM cells than in HepG2 cells, and over 4-fold higher in SMMC7721/ADM cells than in SMMC7721 cells, but MRP1 expression had no significant difference (A, D). Corresponding flow histograms for P-gp (B, E) and MRP1 (C, F) are presented. The results are shown as mean ± SE (n = 5). Statistical analyses comparing MDR cells with parental cells were performed using Student's t-test. bP < 0.001 vs SMMC7721 cells.
Table 3.
P-gp and MRP1 expression in MDR and parental cells (mean ± SD)
| Cells (n = 5) | P-gp (%) | MRP1 (%) |
| HepG2 | 0.88 ± 0.05 | 0.93 ± 0.15 |
| HepG2/ADM | 8.92 ± 0.22a | 0.9 ± 0.18 |
| SMMC7721 | 1.74 ± 0.25 | 1.21 ± 0.35 |
| SMMC7721/ADM | 7.37 ± 0.26a | 0.79 ± 0.02 |
P < 0.001 vs parental cells.
ERK1 and ERK2 mRNA expression
To examine the role of ERK signaling pathway in the development of MDR, ERK1 and ERK2 mRNA expression in parental and MDR cells was assessed, respectively (Figure 4). QRT-PCR analysis demonstrated that ERK1 and ERK2 mRNA expression was apparently higher in HepG2/ADM cells and significantly lower in SMMC7721/ADM cells.
Figure 4.
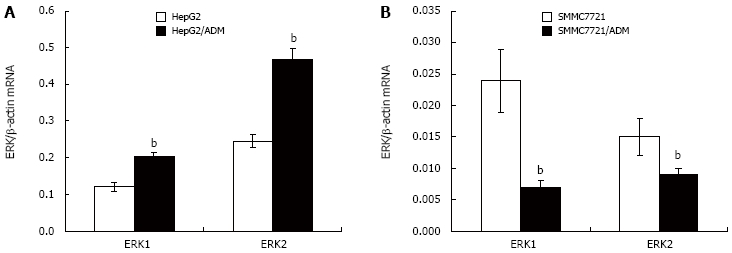
ERK1 and ERK2 mRNA expression in MDR and parental cells. ERK1 and ERK2 mRNA levels were measured by QRT-PCR. A: HepG2/ADM and HepG2 cells; B: SMMC7721/ADM and SMMC7721 cells. Results were normalized by β-actin mRNA expression and compared with the levels in parental cells (n = 3). The results are shown as mean ± SE. Statistical analyses comparing MDR cells with parental cells were performed using Student's t-test. bP < 0.01 vs parental cells (data not shown).
Expression and phosphorylation of ERK1 and ERK2
The expression of ERK1, ERK2, p-ERK1 and p- ERK2 in HepG2/ADM, SMMC7721/ADM and their parental cells, was detected by Western blot analysis, respectively. The expression of ERK1 and ERK2 protein was markedly lower in SMMC7721/ADM cells than in their parental cells. However, the expression of ERK2 protein was significantly higher in SMMC7721/ADM cells than in their parental cells while the expression of ERK1 protein had no significant change in HepG2/ADM cells. The phosphorylation of ERK1 and ERK2 was markedly lower in HepG2/ADM and SMMC7721/ADM MDR cells than in their parental cells (Figure 5).
Figure 5.

Expression and phosphorylation of ERK1 and ERK2 in MDR and parental cells. Western blot analysis of the ERK1 (A), ERK2 (B), p-ERK1 and p-ERK2 (C) expression in HepG2/ADM, SMMC7721/ADM as well as in HepG2 and SMMC7721 cells (n = 3) was performed. The expression of ERK1 and ERK2 was markedly lower in SMMC7721/ADM cells than in parental cells. However, the ERK2 expression was markedly increased and the ERK1 expression had no significant change in HepG2/ADM cells. The phosphorylation of ERK1 and ERK2 was lower in MDR cells than in parental cells (D). The results are shown as mean ± SE. Statistical analyses comparing MDR cells with parental cells were performed using Student's t-test. aP < 0.05, cP < 0.01 vs parental cells (data not shown).
DISCUSSION
Multidrug resistant cancer cells may develop in patients upon prolonged treatment with anti-cancer drugs. MDR poses a great obstacle to chemotherapy for cancer because a higher dosage of drugs is needed to be administered to patients, which will cause severe adverse effects[18]. Most strategies developed to reverse MDR phenotype involve use of resistance modulators, which have the common ability to reverse the phenotype by inhibiting MDR transporter function[19,20]. A more efficient strategy to circumvent MDR is to down-regulate the expression of genes coding transporters. However, regulation of MDR-related gene expression is highly complex. For instance, such complexity is embodied in multiple transcription-regulatory elements in the 5’ and 3’ flanking sequences of the mdr-1 gene, and numerous protein factors involved in transcription-regulatory processes in a cell type- and stimulus-dependent manner[21]. The mechanism underlying the expression of MDR-related genes has not yet been fully understood. Thus, the molecular mechanism and signal-transduction pathways involved in regulation of MDR-related genes should be further studied in order to overcome MDR and improve chemotherapeutic efficacy.
The ERK signaling pathway mediates a number of cellular processes, including cell differentiation, growth, survival, and apoptosis. Several growth factors stimulate a protein kinase cascade that sequentially activates Raf, MEK, and ERK1/2. The role of ERK pathway in the generation of MDR has received more consideration than before, and some specific blocking agents of the ERK pathway have been found. Recent studies demonstrated that modulation of ERK activation may be a new method to reverse MDR[15,22]. However, whether ERK activation is positively or negatively correlated with MDR still remains controversial. Furthermore, the relationship between ERK activity and MDR of HCC is unknown. In this study, we found that ERK activity was down-regulated in MDR HCC Cells.
ADM is a chemotherapeutic agent principally used in treatment of solid tumors, including HCC[23]. Although ADM can work through various mechanisms, it is still not immune to the MDR phenotype, and consequently, development of resistance to ADM has been well-documented in a broad range of cell lines[24–26]. We established two MDR HCC cell lines, HepG2/ADM and SMMC7721/ADM. In this study, death of MDR cells with a lower drug resistance occurred at about 24-48 h after a higher concentration of ADM was added in the medium, and reached the peak at about 72 h. To recover the proliferation ability, the culture of remained adherent cells took at least 7-8 wk. The whole development of MDR HCC cell lines lasted ten months. The IC50 of anticancer drugs was much higher for HepG2/ADM and SMMC7721/ADM cells than for their parental cells, suggesting that the acquired MDR of HepG2/ADM and SMMC7721/ADM is reliable.
To elucidate the mechanism involved in the development of acquired MDR of HepG2/ADM and SMMC7721/ADM cells, cell cycle distribution and expression of MDR-related proteins (P-gp and MRP1) were analyzed by FCM. The percentage of MDR HepG2/ADM and SMMC7721/ADM cells was significantly decreased in the G0/G1 phase and increased in the S phase or G2/M phase than that of their parental cells, which probably contributes to the lower ability of cells to proliferate. Moreover, this kind of delayed cell cycle can result in cellular escape of cytotoxicity of cell cycle specific agents (e.g. vincristine, fluorouracil, etc.) and generate MDR[27,28]. P-gp and MRP1 are members of the ATP-binding cassette transporter proteins. Over-expression of ATP-binding cassette transporter proteins represents one of the major mechanisms that contribute to the MDR phenotype. Both P-gp and MRP1 function as a drug efflux pump that actively transports drugs from the inside to the outside of cells and causes a defect in the intracellular accumulation of drugs necessary for cancer cell killing. The results of our study show that P-gp was much higher in MDR HepG2/ADM and SMMC7721/ADM cells than in their parental cells, but the expression of MRP1 was low in both MDR and parental cells, indicating that MDR of HepG2/ADM and SMMC7721/ADM cells mainly attribute to the over-expression of P-gp but not MRP1. This phenomenon can partially be explained by the high expression of P-gp and low expression of MRP1 in liver tissue or HCC cell lines[29].
ERK1 and ERK2, isozymes of ERK, are extensively expressed in cultured cell lines and mammalian tissues[30]. To answer the question of whether ERK1 and ERK2 are involved in P-gp-mediated MDR in HCC cells, we detected the expression of ERK1 and ERK2 mRNA in parental and MDR cells, and the expression and phosphorylation (activity) of ERK1 and ERK2 protein. The results showed that the expression of ERK1 and ERK2 mRNA was increased in HepG2/ADM cells and decreased in SMMC7721/ADM cells. However, ERK1 protein expression had no significant change in HepG2/ADM cells. The phosphorylation of ERK1 and ERK2 was markedly decreased in HepG2/ADM and SMMC7721/ADM MDR cells, suggesting that ERK1 and ERK2 activity is down-regulated in P-gp-mediated MDR HCC cells. However, the decreased activity was not in accordance with mRNA and protein expression in HepG2/ADM cells. The expression of ERK1 and ERK2 protein was diverse, which may contribute to the augments on whether ERK activation is positively or negatively correlated with MDR.
In summary, MDR of HepG2/ADM and SMMC7721/ADM cells mainly attribute to the over-expression of P-gp but not MRP1. ERK1 and ERK2 activity is down-regulated in P-gp-mediated MDR HCC cells, providing new insights into the complicated regulatory mechanism of MDR phenotype. ERK1 and ERK2 might be potential drug targets for circumventing MDR HCC cells. In vivo studies are warranted to examine whether ERK1 and ERK2 have a clinical potential in modulating the MDR phenotype during HCC chemotherapy.
COMMENTS
Background
The development of multidrug resistance (MDR) to chemotherapeutic agents plays a major role in the failure of cancer therapy, including hepatocellular carcinoma (HCC). Recent studies have shown that modulation of extracellular signal-regulated kinase (ERK) activation may reverse MDR in prostatic, gastric and hematopoietic cancers. However, there is little evidence that ERK activity is related with MDR of HCC cells.
Research frontiers
Multidrug resistant cancer cells may develop in patients upon prolonged treatment with anti-cancer drugs. Most strategies developed to reverse the MDR phenotype involve use of resistance modulators. A more efficient strategy to circumvent MDR is to down-regulate the expression of genes coding for transporters. Thus, to overcome MDR and improve chemotherapeutic efficacy, the molecular mechanism and signal-transduction pathway involved in the regulation of MDR-related genes should be further studied.
Innovations and breakthroughs
In this study, the MDR of HepG2/ADM and SMMC7721/ADM cells could attribute to the over-expression of P-gp but not MRP1, ERK1 and ERK2 activity was down-regulated in P-gp-mediated MDR HCC cells, thus providing new insights into the complicated regulatory mechanism of MDR phenotype.
Applications
ERK1 and ERK2 might be used as potential drugs targets for circumventing HCC MDR.
Terminology
Multidrug resistance: an intrinsic or acquired cross-resistance to a variety of structurally and functionally unrelated drugs, which is almost constantly expressed in cancer and represents one of the major problems in cancer eradication by limiting the efficacy of chemotherapy, and resistance to therapy can result from decreased drug uptake, increased DNA repair or drug inactivation.
Peer review
The authors examined the expression and phosphorylation of ERK1/2 in MDR HCC cell lines, and demonstrated that ERK1 and ERK2 activity was down-regulated in P-gp-mediated MDR HCC cells, indicating that ERK1 or ERK2 might be used as a potential drug target for circumventing MDR HCC cells.
Supported by Innovation Fund of Fujian Province, No. 2007-CXB-7, Key Science and Technology Project of Xiamen, No. 3502Z20077045
Peer reviewer: Seyed-Moayed Alavian, MD, Professor, Gastroenterology and Hepatology, Department of Internal Medicine, Baqiyatallah University of Medical Sciences and Tehran Hepatitis Center, PO Box 14155-3651-Tehran, Iran
S- Editor Tian L L- Editor Wang XL E- Editor Ma WH
References
- 1.Okuda K. Hepatocellular carcinoma. J Hepatol. 2000;32:225–237. doi: 10.1016/s0168-8278(00)80428-6. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Estimating the world cancer burden: Globocan 2000. Int J Cancer. 2001;94:153–156. doi: 10.1002/ijc.1440. [DOI] [PubMed] [Google Scholar]
- 3.Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866–3884. doi: 10.1038/sj.onc.1209550. [DOI] [PubMed] [Google Scholar]
- 4.Wakamatsu T, Nakahashi Y, Hachimine D, Seki T, Okazaki K. The combination of glycyrrhizin and lamivudine can reverse the cisplatin resistance in hepatocellular carcinoma cells through inhibition of multidrug resistance-associated proteins. Int J Oncol. 2007;31:1465–1472. [PubMed] [Google Scholar]
- 5.Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–1876. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 6.Folmer Y, Schneider M, Blum HE, Hafkemeyer P. Reversal of drug resistance of hepatocellular carcinoma cells by adenoviral delivery of anti-ABCC2 antisense constructs. Cancer Gene Ther. 2007;14:875–884. doi: 10.1038/sj.cgt.7701082. [DOI] [PubMed] [Google Scholar]
- 7.Modok S, Mellor HR, Callaghan R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr Opin Pharmacol. 2006;6:350–354. doi: 10.1016/j.coph.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Panka DJ, Atkins MB, Mier JW. Targeting the mitogen-activated protein kinase pathway in the treatment of malignant melanoma. Clin Cancer Res. 2006;12:2371s–2375s. doi: 10.1158/1078-0432.CCR-05-2539. [DOI] [PubMed] [Google Scholar]
- 9.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 10.Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 11.Avruch J. MAP kinase pathways: the first twenty years. Biochim Biophys Acta. 2007;1773:1150–1160. doi: 10.1016/j.bbamcr.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katayama K, Yoshioka S, Tsukahara S, Mitsuhashi J, Sugimoto Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol Cancer Ther. 2007;6:2092–2102. doi: 10.1158/1535-7163.MCT-07-0148. [DOI] [PubMed] [Google Scholar]
- 13.Kisucka J, Barancik M, Bohacova V, Breier A. Reversal effect of specific inhibitors of extracellular-signal regulated protein kinase pathway on P-glycoprotein mediated vincristine resistance of L1210 cells. Gen Physiol Biophys. 2001;20:439–444. [PubMed] [Google Scholar]
- 14.Lin JC, Chang SY, Hsieh DS, Lee CF, Yu DS. Modulation of mitogen-activated protein kinase cascades by differentiation-1 protein: acquired drug resistance of hormone independent prostate cancer cells. J Urol. 2005;174:2022–2026. doi: 10.1097/01.ju.0000176476.14572.39. [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li S, Han Y, Liu J, Zhang J, Li F, Wang Y, Liu X, Yao L. Calebin-A induces apoptosis and modulates MAPK family activity in drug resistant human gastric cancer cells. Eur J Pharmacol. 2008;591:252–258. doi: 10.1016/j.ejphar.2008.06.065. [DOI] [PubMed] [Google Scholar]
- 16.McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006;46:249–279. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Chen B, Sun Q, Wang X, Gao F, Dai Y, Yin Y, Ding J, Gao C, Cheng J, Li J, et al. Reversal in multidrug resistance by magnetic nanoparticle of Fe3O4 loaded with adriamycin and tetrandrine in K562/A02 leukemic cells. Int J Nanomedicine. 2008;3:277–286. doi: 10.2147/ijn.s2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stavrovskaya AA. Cellular mechanisms of multidrug resistance of tumor cells. Biochemistry (Mosc) 2000;65:95–106. [PubMed] [Google Scholar]
- 19.Angelini A, Iezzi M, Di Febbo C, Di Ilio C, Cuccurullo F, Porreca E. Reversal of P-glycoprotein-mediated multidrug resistance in human sarcoma MES-SA/Dx-5 cells by nonsteroidal anti-inflammatory drugs. Oncol Rep. 2008;20:731–735. [PubMed] [Google Scholar]
- 20.Yan F, Jiang Y, Li YM, Zhen X, Cen J, Fang WR. Reversal of P-glycoprotein and multidrug resistance-associated protein 1 mediated multidrug resistance in cancer cells by HZ08 Isomers, tetrataisohydroquinolin derivatives. Biol Pharm Bull. 2008;31:1258–1264. doi: 10.1248/bpb.31.1258. [DOI] [PubMed] [Google Scholar]
- 21.Stierle V, Duca M, Halby L, Senamaud-Beaufort C, Capobianco ML, Laigle A, Jolles B, Arimondo PB. Targeting MDR1 gene: synthesis and cellular study of modified daunomycin-triplex-forming oligonucleotide conjugates able to inhibit gene expression in resistant cell lines. Mol Pharmacol. 2008;73:1568–1577. doi: 10.1124/mol.107.042010. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y, Bally M, Dragowska WH, Mayer L. Inhibition of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase enhances chemotherapeutic effects on H460 human non-small cell lung cancer cells through activation of apoptosis. Mol Cancer Ther. 2003;2:641–649. [PubMed] [Google Scholar]
- 23.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 24.Barrand MA, Heppell-Parton AC, Wright KA, Rabbitts PH, Twentyman PR. A 190-kilodalton protein overexpressed in non-P-glycoprotein-containing multidrug-resistant cells and its relationship to the MRP gene. J Natl Cancer Inst. 1994;86:110–117. doi: 10.1093/jnci/86.2.110. [DOI] [PubMed] [Google Scholar]
- 25.Mehta K. High levels of transglutaminase expression in doxorubicin-resistant human breast carcinoma cells. Int J Cancer. 1994;58:400–406. doi: 10.1002/ijc.2910580316. [DOI] [PubMed] [Google Scholar]
- 26.Shen DW, Cardarelli C, Hwang J, Cornwell M, Richert N, Ishii S, Pastan I, Gottesman MM. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem. 1986;261:7762–7770. [PubMed] [Google Scholar]
- 27.Rebbaa A. Targeting senescence pathways to reverse drug resistance in cancer. Cancer Lett. 2005;219:1–13. doi: 10.1016/j.canlet.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt CA. Cellular senescence and cancer treatment. Biochim Biophys Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–127. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 30.Sugden PH, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein-coupled receptors. Cell Signal. 1997;9:337–351. doi: 10.1016/s0898-6568(96)00191-x. [DOI] [PubMed] [Google Scholar]