

Abstract
Purpose
Cell death can be induced by exogenous reactive oxygen species (ROS). Endogenous ROS can also play a role in cell death triggered by agents that are not themselves ROS. One of the most potent ROS-generating systems is the iron-catalyzed Fenton reaction. Herein, the authors tested whether iron plays an important role in cell death induced by diverse stimuli in retinal pigment epithelial (RPE) cells.
Methods
The ability of the iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH) to chelate intracellular labile iron was tested in the human cell line ARPE-19. The ability of SIH to protect against RPE cell death induced by hydrogen peroxide, staurosporine, anti-Fas, and exposure to A2E plus blue light was determined. ROS production by staurosporine was assessed in the presence and absence of SIH. The protective activity of SIH was compared with that of other iron chelators and an antioxidant.
Results
Acute exposure to SIH was nontoxic and at least partially protective against cell death induced by all tested agents. On a molar basis, SIH was more protective against hydrogen peroxide than other iron chelators and an antioxidant. SIH decreased levels of staurosporine-induced ROS.
Conclusions
Iron chelation with SIH can decrease levels of ROS and protect RPE cells against cell death induced by diverse stimuli. These results suggest a central role for iron in cell death pathways, potentially involving the generation of oxidative stress. SIH or related iron chelators may prove useful for protection against diseases involving RPE death, such as AMD.
Iron is essential for life because of its role in one-electron redox chemistry in the electron transport chain and as a cofactor in heme and iron-sulfur cluster–containing proteins. However, it also represents a potentially dangerous electron-transporting catalytic system that is able to induce oxidative damage. In the Fenton reaction, iron reacts with hydrogen peroxide (H2O2) to produce hydroxyl radical, the most reactive and toxic of the reactive oxygen species (ROS). Iron is prevented from reacting with H2O2 by storage within proteins such as ferritin. At the same time a small amount of redox-active iron exists in the intracellular labile iron pool, making this accessible ferrous iron dangerous under conditions of cellular oxidative stress. Moreover, superoxide and H2O2 are able to release iron from its storage proteins, increasing the labile iron pool and creating a vicious circle of ROS production.1-3
Iron homeostasis is controlled at the level of intestinal iron absorption because there is no known iron excretion mechanism.4 Hereditary diseases causing impaired iron homeostasis, such as the common recessive disease hereditary hemochromatosis, result in iron-induced oxidative damage to organs. Patients with the rare hereditary disease aceruloplasminemia have iron overload of the brain, retina, and pancreas, leading to degeneration in these organs.5 The retinal degeneration in these patients resembles an early-onset form of the blinding disease age-related macular degeneration.6 Moreover, elevated iron levels have been detected in Alzheimer and Parkinson disease–affected brains, suggesting its contribution to these neurodegenerations.3,7
Patients with iron overload resulting from multiple blood transfusions require treatment with iron chelators to prevent damage to the heart and liver. For decades patients have been successfully treated by infusion of deferoxamine (DFO), which is given by slow subcutaneous infusion. Recently, a new crop of chelators, some of which can be taken orally and are more cell and blood-brain barrier permeable, have been developed.8 Salicylaldehyde isonicotinoyl hydrazone (SIH) is among these lipophilic chelators.9 It has been shown that SIH can be nontoxic in animals10 and very effective in protecting cultured cells from oxidant-induced death.11 SIH given intravenously to mice prolonged survival after injection of hepatotoxic and lethal doses of an anti–Fas antibody.12 The mechanism of this protection is hypothesized to be based on SIH blockage of ROS induced by anti–Fas antibody. It was also found that SIH protects against H2O2-induced lysosomal rupture and loss of mitochondrial membrane potential in murine macrophage-like J774 cells, thus providing protection against apoptosis and necrosis.13
In recent years a large body of evidence has accumulated to suggest that ROS may play a role as common mediators of apoptosis.14-18 Many chemotherapeutic agents inducing apoptosis simultaneously induce intracellular production of ROS. For this reason, we tested in the present study whether SIH can protect cells not just against H2O2 but also against cell death inducers that are not themselves ROS.
We tested this hypothesis in retinal pigment epithelial (RPE) cells, a monolayer of cells that support the photoreceptors of the retina. RPE cells are subject to oxidant insult from superoxide and H2O2 produced by mitochondrial respiration and by photo-oxidation. This photo-oxidation is exacerbated by the accumulation of age pigment, or lipofuscin, in these postmitotic cells. A major fluorophore of RPE lipofuscin is A2E, which is generated in photoreceptor cells through the reaction of all-trans-retinal and phosphatidylethanolamine, producing the precursor A2PE. RPE cells then ingest the A2PE through phagocytosis of photoreceptor outer segments. Within lysosomes A2PE undergoes phosphate cleavage to generate A2E.19,20 A2E acts as a photosensitizer, causing the production of ROS when exposed to blue light and inducing the apoptosis of cultured RPE cells.21
Two other agents that induce cell death in RPE cells are staurosporine22 and Fas.23 Staurosporine is a protein kinase C inhibitor that increases mitochondrial production of ROS soon after administration.16 Several lines of evidence suggest that mitochondrial production of H2O2 is an important component of staurosporine-induced cell death. RNAi-mediated knockdown of the mitochondrial peroxidase PrxIII increases susceptibility to staurosporine-induced death. Conversely, ectopic expression of the H2O2-degrading enzyme catalase in the mitochondria protects against staurosporine-induced cell death.15 Antioxidant compounds can also protect against staurosporine-induced death.24,25 Thus, reaction of this staurosporine-induced H2O2 with iron to produce damaging hydroxyl radical becomes a plausible mechanism for staurosporine-induced cell death.
Activation of the cell surface receptor Fas also induces apoptosis in RPE cells,23 and it may play a role in age-related macular degeneration.26,27 H2O2 levels are increased within 15 minutes in cells treated with a Fas-activating antibody, suggesting that the H2O2 may play a role early in the apoptotic pathway induced by Fas. Consistent with this idea, the antioxidant glutathione can protect cells from Fas-triggered death, and a B-cell line that survives despite Fas activation does so because of a failure to produce H2O2 when exposed to anti–Fas antibodies.14 Furthermore, treatment of cells with the antioxidant N-acetyl cysteine blocks Fas-induced death by blocking ROS-mediated assembly of the apoptosome.28
Because iron-mediated Fenton chemistry is an important mechanism of H2O2 toxicity, we tested whether SIH can protect not only against death induced by exogenous oxidants or photo-oxidation but also against death triggers that induce apoptosis accompanied by increased endogenous production of ROS.
Materials and Methods
Cell Culture
Human adult RPE cells (ARPE-19 cell line; ATCC, Manassas, VA)29 were cultured until confluent (unless otherwise indicated) in 24-well Falcon plates in 1:1 DMEM/F12 with high glucose and l-glutamine (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (Hyclone, Logan, UT).
Cell Death Induction and SIH Protection
To evalute the cytoprotective properties of SIH, SIH at different concentrations was applied to confluent ARPE-19 cells 4 hours before the addition of H2O2, staurosporine, or anti–Fas antibody. Hydrogen peroxide (30% H2O2; Sigma, St. Louis, MO) at a final concentration ranging from 1 to 5 mM, staurosporine (1 mM solution; Sigma) at a final concentration of 100 to 500 nM, or anti–Fas antibody (Upstate-Millipore, Billerica, MA) at a final concentration of 1 μg/mL was mixed with the indicated concentrations of SIH immediately before the application of ARPE-19 cells. All reagents were dissolved in minimal essential medium (MEM; Invitrogen), as indicated, and were applied rapidly and uniformly after two washing steps in Hanks balanced salt solution (Invitrogen).
Viability Assays
Cell death was assayed after 24 hours (LDH Release Assay Kit; Roche, Basel, Switzerland). Cytotoxicity was calculated using the ratio between maximum LDH release in 5 mM hydrogen peroxide–treated cells and minimum LDH release in cells incubated in MEM. The percentage of cytotoxicity was calculated as (experimental value for LDH release − minimum LDH release)/(maximum LDH release − minimum LDH release) × 100. Visualization of cell death was performed (Live/Dead Viability/Cytotoxicity kit; Invitrogen). Epifluorescence microscopy was performed with a microscope (TE-300; Nikon, Tokyo, Japan), camera (SpotRT Slider; Diagnostic Instruments, Sterling Heights, MI), and software (ImagePro Plus, version 4.1; Media Cybernetics, Silver Spring, MD).
A2E/Blue Light
For blue light irradiation experiments, ARPE-19 cells devoid of endogenous lipofuscin30 were grown in complete medium (DMEM with 10% fetal calf serum and supplements), as previously described.30 At confluence, the cells were allowed to accumulate A2E from a 10-μM concentration delivered in the culture medium over a 2-week period.21,30,31 Cells that had accumulated A2E were treated with SIH in MEM for 4 hours unless otherwise indicated. SIH/MEM was subsequently exchanged for DPBS, and cells were irradiated at 430 nm for 20 minutes, as previously described,32 returned to complete medium, and incubated for 24 hours Variations on this paradigm included experiments in which exposure to SIH/MEM was continued during illumination at 430 nm and for 24 hours thereafter and experiments in which cells were not preincubated with SIH but instead were treated with SIH/MEM beginning immediately after irradiation and continuing for 24 hours or beginning 1 hour after irradiation and continuing for 23 hours. In all experiments, controls included A2E-laden irradiated cells not treated with SIH, cells that received no treatment (A2E-free, non-irradiated, incubation with MEM in the absence of SIH), and cells treated only with SIH.
MTT Assay
In blue light irradiation experiments, cell death was assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, a measure of the ability of healthy cells to cleave the yellow tetrazolium salt MTT to purple formazan crystals. Briefly, after the postillumination incubation described, 20 μL MTT labeling reagent (Roche Diagnostics, Indianapolis, IN) was added to 0.2 mL culture medium in each well. After 4-hour incubation, the labeling reagent was replaced with 200 μL solubilization solution for overnight incubation. After centrifugation at 13,000 rpm for 2 minutes, supernatants were measured spectrophotometrically. A decrease in the absorbance (570 nm) of reduced MTT was indicative of diminished cellular viability.
Quantitative Real-Time PCR
ARPE-19 cells were cultured until confluent in 24-well Falcon plates in 1:1 DMEM/F12 with high glucose and l-glutamine (Invitrogen) supplemented with 10% FBS (Hyclone). Cells were analyzed with quantitative RT-PCR for gene expression after exposure to 5 μM SIH, BIH, and 100 μM H2O2 in MEM solution. Treatments were applied 5 hours before RNA extraction and reapplied 1 hour before RNA extraction, the same timing used for SIH application in the cell viability experiments. RNA isolation was performed (RNeasy Mini Kit; Qiagen, Valencia, CA) according to the manufacturer's protocol. cDNA was synthesized with reverse transcription reagents (TaqMan; Applied Biosystems, Darmstadt, Germany) according to the manufacturer's protocol. Gene expression assays were obtained from Applied Biosystems (TaqMan) and were used for relative-quantitative PCR analysis with the transferrin receptor (Hs00174609m1) probe. Eukaryotic 18S rRNA (Hs99999901s1) served as an internal control because of its constant expression level across the studied sample set. Real-time PCR (TaqMan; Applied Biosystems) was performed (ABI Prism 7500 Sequence Detection System; Applied Biosystems) according to the ΔΔCT method, which provides normalized expression values. All reactions were performed in triplicate, and water was used as a negative control.
Statistical Analysis for LDH and MTT Assays
Within each experiment, the mean absorbance value of duplicated wells was obtained. We then calculated the mean ± SEM for each condition across replicate experiments conducted on different days. Across replicates on different days, experimental samples were compared with controls using the two group t-test. The n was either 3 or 4 for each experimental condition. P <0.05 was considered statistically significant. All statistical analysis was performed on statistical software (SAS, version 9.1; SAS Institute, Cary, NC).
Statistical Analysis for Quantitative PCR
Mean ± SEM of target mRNA relative quantity in ARPE-19 cells were calculated for all treatment groups (n = 3). Means for all treatment groups were compared with those of untreated controls using the two group t-test. P < 0.05 was considered statistically significant. All statistical analysis was performed on statistical software (GraphPad; GraphPad Software, San Diego CA).
Rhodamine 123
Intracellular ROS generation was measured in cultured cells with the fluorescent probe dihydrorhodamine 123 (DHR123). Mitochondria were localized with a red fluorescent mitochondrial marker (Mitotracker Red 580; Molecular Probes, Eugene, OR). After 30 minutes of staurosporine exposure with or without SIH, cells were incubated with staurosporine with or without SIH in the presence of 10 μM DHR123 and 2 μM dye (Mitotracker; Molecular Probes) at 37°C for 15 minutes. DHR fluoresces and localizes to mitochondria when oxidized by ROS to the positively charged rhodamine 123.33 Red fluorescent mitochondrial marker (Mitotracker; Molecular Probes) passively diffuses across the plasma membrane and specifically accumulates in active mitochondria.
Results
SIH Protection of ARPE-19 from Cell Death Induced by Lethal Doses of H2O2
Confluent ARPE-19 cells treated with 1 and 5 mM H2O2 for 24 hours exhibited increasing amounts of cell death (Fig. 1). H2O2 (5 mM) caused 100% cell death assessed by both the LDH release assay and the fluorescent assay (Live/Dead; Invitrogen). When cells were treated with 5 μM SIH before the application of H2O2, followed by treatment by H2O2 mixed with SIH, 5 mM H2O2 cytotoxicity was completely abolished. Cell viability detected by both assays correlated well in all experiments. Diminished cytoprotection was observed when SIH was applied to cells together with H2O2 but without SIH pretreatment, and no protection was found when SIH was applied to the cells 1 hour after H2O2 or after the SIH had been preincubated with ferric ammonium citrate at a 2:1 molar ratio (data not shown). To rule out the possibility of a chemical interaction between SIH and H2O2, we examined UV/VIS spectra of both chemicals and its mixture but did not detect any changes in the spectroscopic properties of the SIH-H2O2 mixture compared with SIH and H2O2 alone (data not shown).
Figure 1.
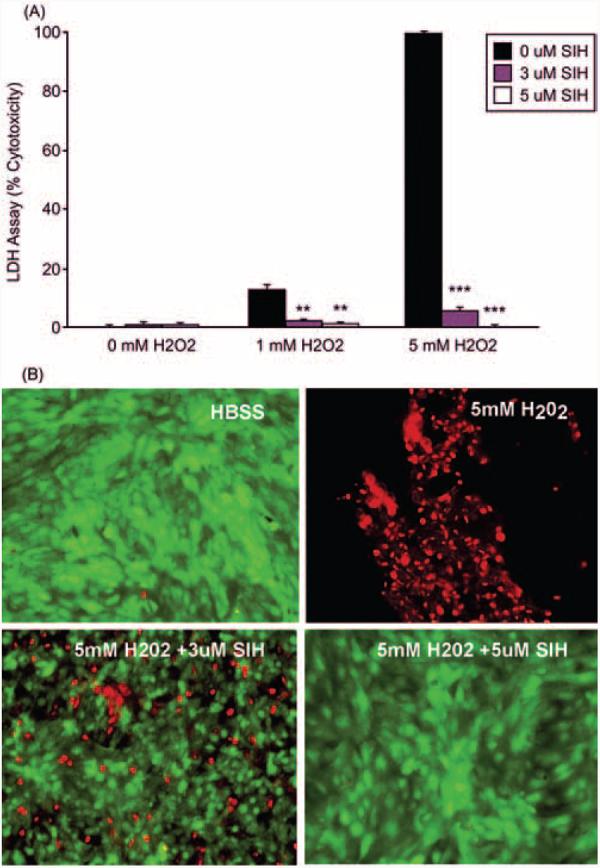
Graph of cytotoxicity measured by LDH release into the medium showing SIH-protection of ARPE-19 cells against H2O2-induced cell death. (A) Cytotoxicity of 1 and 5 mM H2O2 exposure for 24 hours when coincubated 4 hours before and during H2O2 exposure with the indicated concentrations of SIH. **P < 0.01 relative to no SIH at the corresponding H2O2 level. ***P < 0.001. (B) Fluorescence photomicrographs of ARPE-19 cells labeled with a fluorescent assay. ARPE-19 cells were treated for 24 hours with HBSS (control), 5 mM H2O2 alone, or 5 mM H2O2 with SIH, as indicated. Red: dead cells as detected by ethidium bromide homodimer fluorescence. Green: live cells as detected by calcein fluorescence.
SIH More Effectively Protects ARPE-19 from Cell Death Induced by Lethal Doses of H2O2 Than Other Iron Chelators or an Antioxidant
The ability of SIH to protect against H2O2-induced death was compared with that of other compounds (Fig. 2). In contrast to the previous experiment, which was conducted in confluent cells, this experiment was conducted in subconfluent cells, which are more susceptible to H2O2-induced death than confluent cells (data not shown). Under these conditions, 10 μM SIH treatment reduced H2O2-induced cytotoxicity to 45.9% compared with H2O2 without SIH. At increased SIH concentrations of 100 and 1000 μM, H2O2-induced cytotoxicity was further reduced to 8.7% compared with H2O2 without SIH. Compared with the iron chelator DFO, SIH provided significantly more protection against H2O2-induced death at concentrations of 10, 100, and 1000 μM chelator. Equimolar concentrations of SIH also provided better protection against H2O2-induced death than did the extracellular iron chelator diethylenetriaminepentaacetic acid (DTPA) or the antioxidant N-acetyl cysteine (NAC).
Figure 2.
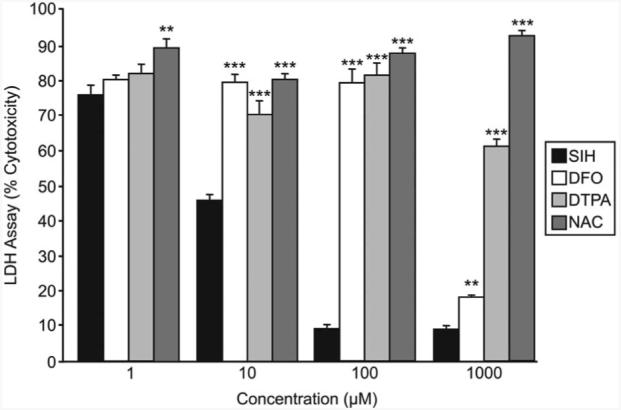
Graph of cytotoxicity measured by LDH release into the medium showing SIH protection of subconfluent ARPE-19 cells against H2O2-induced cell death compared with other iron chelators and an antioxidant. Cytotoxicity of 5 mM H2O2 exposure for 24 hours when coincubated 4 hours before and during H2O2 exposure with the indicated concentration of SIH, DFO, DTPA, or NAC. **P < 0.01 relative to SIH at the same concentration of iron chelator or antioxidant. ***P < 0.001.
SIH-Mediated Protection of ARPE-19 against Fas-Induced Cell Death
Fas is a cell surface receptor known to initiate cell death when activated in a number of cell types, including ARPE-19 cells.23 The receptor can be activated by exposure to an anti-Fas antibody that acts like Fas-ligand. Treatment of ARPE-19 cells with SIH beginning 4 hours before treatment with anti-Fas antibody reduced cell death caused by Fas activation, as indicated by the LDH release assay (Fig. 3).
Figure 3.
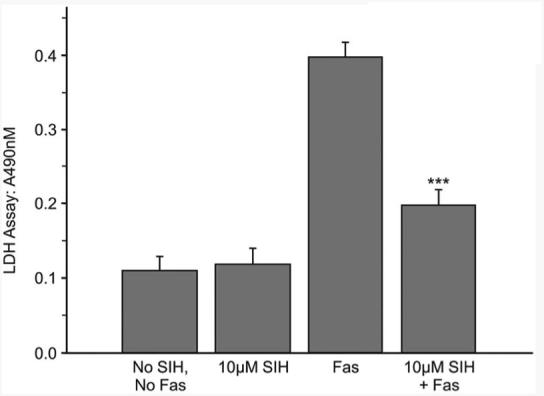
Graph of LDH release into the medium after anti–Fas antibody treatment with or without SIH, as indicated. P < 0.001 relative to Fas without SIH.
SIH-Mediated Protection of ARPE-19 Cells from Cell Death Induced by Staurosporine
Staurosporine at concentrations of 100 to 500 nM causes apoptosis of ARPE-19 cells.22 When tested herein, 200 nM and 500 nM staurosporine caused significant cell death (Fig. 4A). When SIH was applied to cells 4 hours before treatment with 200 nM staurosporine (Fig. 4B) or 500 nm staurosporine (not shown) and was present through 24 hours of staurosporine treatment, cell viability was increased in an SIH concentration-dependent manner (Fig. 4B). The greatest SIH cytoprotective effect was achieved at 10 μM SIH. The visualization of dead cells using live/dead fluorescent imaging correlated with data from LDH release assay (not shown).
Figure 4.
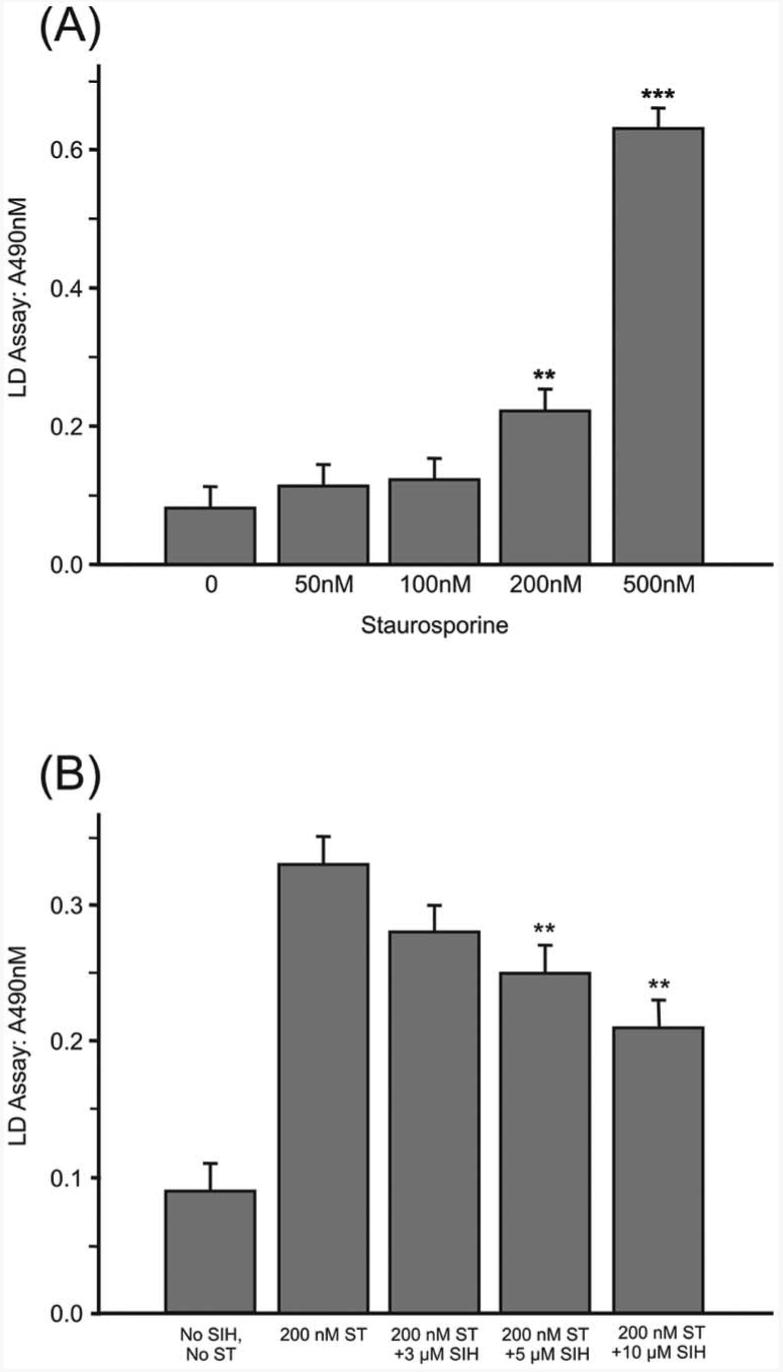
Analysis of the protective effects of SIH against staurosporine-induced cell death. (A) Graph of LDH release after exposure of ARPE-19 cells to varying concentrations of staurosporine. **P < 0.01 relative to no staurosporine. ***P < 0.001. (B) Graph of LDH release after exposure of ARPE-19 cells to 200 nM staurosporine plus different concentrations of SIH. **P < 0.01 relative to staurosporine with no SIH.
SIH-Mediated Decrease in ROS Production Induced by Staurosporine Treatment
Mitochondrial ROS production is an important component of the cell death mechanism of staurosporine.15 To test whether iron contributes to ROS production and whether a decrease in mitochondrial ROS might be a mechanism of SIH protection, ROS were detected with rhodamine 123 after 30 minutes of staurosporine treatment. In the presence of ROS, nonfluorescent dihydrorhodamine 123 is converted to green fluorescent rhodamine 123. Increased fluorescence of rhodamine 123 co-localized with the red fluorescent mitochondrial marker (Mitotracker) in ARPE-19 cells treated with 200 nM staurosporine for 30 minutes, followed by incubation with dihydrorhodamine 123 for 15 minutes (Fig. 5). A greater increase in fluorescence, in addition to more nonmitochondrial fluorescence, was observed in cells treated with 500 nM staurosporine. In the presence of 3 μM SIH, staurosporine-induced rhodamine 123 fluorescence was diminished.
Figure 5.

Fluorescence photomicrographs of ARPE-19 cells. Level of ROS generation is detected by rhodamine 123 (green) in cells treated with staurosporine and 3 μM SIH, as indicated. Red: location of mitochondria. Yellow: colocalization of activated rhodamine 123 and mitochondria.
SIH Cytoprotection against Cell Death by Irradiation with Blue Light in the Presence of A2E
Human RPE cells accumulate lipofuscin with age. A major component of this lipofuscin is A2E, a blue light–absorbing compound that we have shown to potentiate blue light–induced photooxidative stress and apoptosis of ARPE-19 cells. When A2E-loaded ARPE-19 cells were treated with SIH before, during, or even as late as 1 hour after blue light exposure, cell viability was increased (Fig. 6). No SIH-mediated toxicity was observed in cells incubated with SIH but not exposed to A2E or blue light.
Figure 6.

Protective effects of SIH against cell death induced by blue light in the presence of A2E were assessed. Graphs show ARPE-19 cell viability measured by the MTT assay. Increased absorbance indicates increased viability. (A) Cells were incubated with the indicated concentrations of SIH-MEM for 4 hours, irradiated with blue light for 20 minutes in DPBS-SIH, and incubated in MEM-SIH for 24 hours before MTT assay. (B) Cells were pretreated with MEM-SIH for 4 hours, irradiated with blue light for 20 minutes in DPBS, and incubated in MEM for 24 hours. (C) Cells were irradiated by blue light for 20 minutes in DPBS, and SIH-MEM was applied immediately after irradiation and maintained for 24 hours. (D) Cells were irradiated with blue light for 20 minutes in DPBS, and SIH-MEM was applied 1hour after irradiation and maintained for 23 hours before MTT assay. For all graphs, relative to no SIH: *P < 0.05, **P < 0.01, ***P < 0.001
SIH-Mediated Increase in Transferrin Receptor mRNA Levels
Transferrin receptor (TfR) mRNA levels increase in response to chelation-induced reduction of the intracellular labile iron pool.34-36 To better elucidate the mechanism of SIH cytoprotection, ARPE-19 cells were exposed to 5 μM SIH and BIH (isonicotinic acid benzylidene-hydrazide) and 100 μM H2O2. BIH (a gift from Louise Charkoudian and Katherine Franz, Duke University) is similar to SIH but has a substituted hydrazone functionality that lacks the key metal-binding phenol group of SIH and, subsequently, has no iron chelation activity. Low-concentration H2O2 was also used to cause a stress response in the RPE cells without eliciting cell death. TfR mRNA levels were comparable to those of controls (Fig. 7) in BIH- and H2O2-treated ARPE-19 cells. SIH caused a greater than twofold increase in TfR mRNA levels, indicating that it decreased intracellular labile iron levels.
Figure 7.
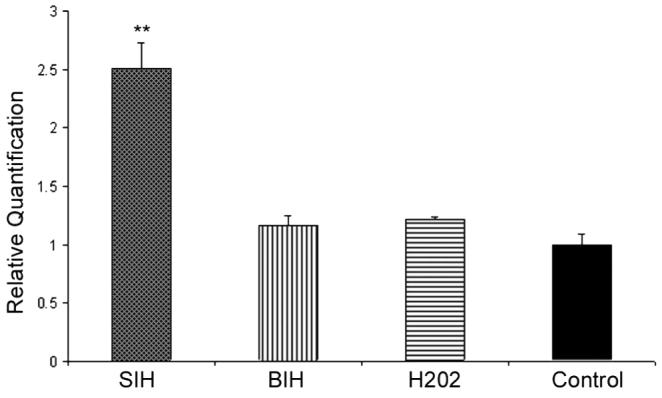
Bar graph showing quantitative PCR results indicating TfR mRNA levels from ARPE-19 cells treated with 5 μM SIH and BIH and 100 μM H2O2. There was a greater than twofold increase in TfR mRNA levels after chelation of intracellular iron pools with SIH. Negative controls (the non–iron-chelating SIH analog BIH and H2O2) show TfR mRNA levels similar to those of untreated controls. **P < 0.05.
Discussion
Generation of ROS has been associated with cell death, and some reports suggest that ROS may function as effectors in apoptotic pathways.37-39 Iron is a potent generator of ROS, and iron-generated ROS within mitochondria and lysosomes may promote cell death.40 Iron chelators can protect cells from death induced by exogenous oxidants. The lipophilic, cell membrane-permeable chelator SIH is particularly effective at protecting cultured cardiomyoblasts from death induced by the addition of H2O2 to the medium.11 Yet it was unknown whether SIH might protect RPE cells from death induced by H2O2 or by agents that are not ROS themselves but may cause a cell to produce iron-catalyzed ROS as part of the mechanism of cell death induction.
In confluent cells, SIH in the 3- to 10-μM range completely protected cells from otherwise lethal concentrations of H2O2. The mechanism of protection is likely to be iron chelation because SIH induced increases in TfR mRNA levels, indicating that it decreases the intracellular labile iron pool. Further, the observed cytoprotective effect of SIH was markedly diminished by SIH preincubation with Fe (III) in the molar ratio 2:1, which would prevent SIH from chelating intracellular iron. This result suggests the necessity for iron as a catalyst in the transformation of H2O2 into highly toxic hydroxyl radicals. Thus, chelation of labile iron blocks the production of hydroxyl radicals and effectively prevents cell death induced by a lethal dose of H2O2. Although cytoprotection was most complete when cells were incubated with SIH before and during H2O2 treatment, incubation with SIH only before but not during H2O2 was partially protective (not shown).
Compared with SIH, the iron chelators DFO and DTPA poorly protected ARPE-19 against H2O2-induced cytotoxicity. Under subconfluent conditions, 100 μM SIH reduced cytotoxicity to 8.7% in response to 5 mM H2O2 treatment. In comparison, cytotoxicity of ARPE-19 in equimolar concentrations of DFO or DTPA was approximately 80%. Further, DFO at a concentration of 1 mM reduced cytotoxicity to 18%, which was significantly less protective than SIH at the same concentration. Thus, on a molar basis, SIH provides more protection against H2O2-induced cytotoxicity than other iron chelators. The thiol-containing antioxidant NAC, which, in addition to scavenging free radicals, helps regenerate intracellular glutathione, was minimally protective against H2O2-induced cell death.
The pyridinium bisretinoid A2E, an autofluorescent pigment that accumulates in RPE with age and in some retinal disorders, can mediate light-induced damage to the cell. We have shown that cell death triggered by blue light irradiation in A2E-laden ARPE-19 cells was decreased in the presence of SIH. Interestingly, SIH displayed a cytoprotective effect not only when cells were preincubated with SIH before blue light irradiation but also when SIH was applied 1 hour after blue light irradiation. Although the mechanism of cell death induced by irradiation by blue light in the presence of A2E is based on ROS generation, singlet oxygen may be more important as an initial insult than superoxide21,30,41-43. Thus, labile iron may not play a role in the initial insult but, rather, may amplify the cell death signal.
Several recent reports suggest a role for ROS in drug-induced apoptosis involving the engagement of downstream proteins involved in the execution of apoptosis,44 including the assembly of the apoptosome.28 Intracellular generation of H2O2, relatively inert and the most stable of the ROS, has been considered an important mediator of apoptosis, and, based on our results, Fenton chemistry is likely to be important in this pathway. We used two different reagents to induce cell death in ARPE-19 cells: anti-Fas antibody to activate Fas receptor and staurosporine, a chemotherapeutic agent. Iron chelation by SIH significantly decreases cell death in cultured ARPE-19 cells on treatment by these apoptotic stimuli. Detection of an SIH-mediated decrease in rhodamine 123 fluorescence induced by staurosporine supports the hypothesis that Fenton chemistry-induced ROS are important in the cell death pathway induced by this agent. Although rhodamine 123 fluorescence has been used to monitor mitochondrial ROS production,45-47 in the current context, rhodamine 123 fluorescence could result from oxidation of the dye either in the mitochondria or in the cytosol, with subsequent accumulation in the mitochondria.15 In the context of A2E/blue light–induced ROS, because A2E localizes to lysosomes, it is possible that the primary site of action of SIH is lysosomal. Alternatively, it may be mitochondrial, because lysosomal A2E can inhibit oxidative phosphorylation, an effect that is mitigated by antioxidants.48 Further, because RPE photodamage associated with A2E accumulation involves the formation of A2E photooxidation products,42 cleavage of oxidized-A2E (for instance, at the  bonds of endoperoxide moieties) followed by diffusion of fragments could cause cellular damage at sites other than the lysosomal compartment.32 Similarly, the formation of reactive cleavage products of photooxidized-A2E would explain the observation that complement can be activated in serum overlying irradiated A2E-laden RPE.49
bonds of endoperoxide moieties) followed by diffusion of fragments could cause cellular damage at sites other than the lysosomal compartment.32 Similarly, the formation of reactive cleavage products of photooxidized-A2E would explain the observation that complement can be activated in serum overlying irradiated A2E-laden RPE.49
In conclusion, SIH protects against exogenous ROS and endogenous ROS produced by cell death-inducing agents. These results suggest that iron-catalyzed production of ROS is important in these pathways. Our finding that SIH can protect cells from a wide range of insults emphasizes its therapeutic potential. While monitoring for potential toxicity from inhibition of iron-requiring proteins, we will test it in preclinical models of retinal degeneration. In these short-term experiments, SIH was nontoxic. This may be because SIH preferentially chelates the accessible, potentially dangerous labile iron without depriving a cell of iron more tightly bound to iron-dependent proteins. Given that long-term administration of SIH may be more toxic, intermittent SIH dosing and iron prochelators, active only when they are needed in the presence of H2O2,50,51 will be tested.
Acknowledgment
The authors thank Prem Ponka (McGill University, Montreal, QC, Canada) for the generous gift of SIH.
Supported by National Institutes of Health Grants EY018922 and T32-EY007035, the International Retina Research Foundation, the University of Pennsylvania Vision Science Training Grant, the F.M. Kirby Foundation, and the Paul and Evanina Bell Mackall Foundation Trust. JLD is a Research to Prevent Blindness William and Mary Greve Scholar.
Footnotes
Disclosure: N. Lukinova, None; J. Iacovelli, None; T. Dentchev, None; N. Wolkow, None; A. Hunter, None; D. Amado, None; G.-S. Ying, None; J.R. Sparrow, None; J.L. Dunaief, P
References
- 1.Reif DW. Ferritin as a source of iron for oxidative damage. Free Radic Biol Med. 1992;12:417–427. doi: 10.1016/0891-5849(92)90091-t. [DOI] [PubMed] [Google Scholar]
- 2.Chevion M, Jiang Y, Har-El R, Berenshtein E, Uretzky G, Kitrossky N. Copper and iron are mobilized following myocardial ischemia: possible predictive criteria for tissue injury. Proc Natl Acad Sci U S A. 1993;90:1102–1106. doi: 10.1073/pnas.90.3.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rouault TA, Cooperman S. Brain iron metabolism. Semin Pediatr Neurol. 2006;13:142–148. doi: 10.1016/j.spen.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Buss JL, Neuzil J, Gellert N, Weber C, Ponka P. Pyridoxal isonicotinoyl hydrazone analogs induce apoptosis in hematopoietic cells due to their iron-chelating properties. Biochem Pharmacol. 2003;65:161–172. doi: 10.1016/s0006-2952(02)01512-5. [DOI] [PubMed] [Google Scholar]
- 5.Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A. 1995;92:2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunaief JL, Richa C, Franks EP, et al. Macular degeneration in a patient with aceruloplasminemia, a disease associated with retinal iron overload. Ophthalmology. 2005;112:1062–1065. doi: 10.1016/j.ophtha.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 7.Lee DW, Andersen JK, Kaur D. Iron dysregulation and neurodegeneration: the molecular connection. Mol Interv. 2006;6:89–97. doi: 10.1124/mi.6.2.6. [DOI] [PubMed] [Google Scholar]
- 8.Brittenham GM. Iron chelators and iron toxicity. Alcohol. 2003;30:151–158. doi: 10.1016/s0741-8329(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 9.Ponka P, Schulman HM. Acquisition of iron from transferrin regulates reticulocyte heme synthesis. J Biol Chem. 1985;260:14717–14721. [PubMed] [Google Scholar]
- 10.Klimtova I, Simunek T, Mazurova Y, et al. A study of potential toxic effects after repeated 10-week administration of a new iron chelator—salicylaldehyde isonicotinoyl hydrazone (SIH) to rabbits. Acta Medica (Hradec Kralove) 2003;46:163–170. [PubMed] [Google Scholar]
- 11.Simunek T, Boer C, Bouwman RA, et al. SIH—a novel lipophilic iron chelator—protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. J Mol Cell Cardiol. 2005;39:345–354. doi: 10.1016/j.yjmcc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Lesnikov VA, Abbasi N, Lesnikova MP, et al. Protection of human and murine hepatocytes against Fas-induced death by transferrin and iron. Apoptosis. 2006;11:79–87. doi: 10.1007/s10495-005-3086-2. [DOI] [PubMed] [Google Scholar]
- 13.Kurz T, Gustafsson B, Brunk UT. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J. 2006;273:3106–3117. doi: 10.1111/j.1742-4658.2006.05321.x. [DOI] [PubMed] [Google Scholar]
- 14.Devadas S, Hinshaw JA, Zaritskaya L, Williams MS. Fas-stimulated generation of reactive oxygen species or exogenous oxidative stress sensitize cells to Fas-mediated apoptosis. Free Radic Biol Med. 2003;35:648–661. doi: 10.1016/s0891-5849(03)00391-5. [DOI] [PubMed] [Google Scholar]
- 15.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 16.Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res. 1998;51:293–308. doi: 10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 17.Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions: evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- 18.Goossens V, Grooten J, De Vos K, Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Itagaki Y, Ben-Shabat S, Nakanishi K, Sparrow JR. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J Biol Chem. 2000;275:29354–29360. doi: 10.1074/jbc.M910191199. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Shabat S, Parish CA, Vollmer HR, et al. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J Biol Chem. 2002;277:7183–7190. doi: 10.1074/jbc.M108981200. [DOI] [PubMed] [Google Scholar]
- 21.Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Invest Ophthalmol Vis Sci. 2002;43:1222–1227. [PubMed] [Google Scholar]
- 22.Mao YW, Liu JP, Xiang H, Li DW. Human αA- and αB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- 23.Lincoln JE, Boling M, Parikh AN, Yeh Y, Gilchrist DG, Morse LS. Fas signaling induces raft coalescence that is blocked by cholesterol depletion in human RPE cells undergoing apoptosis. Invest Ophthalmol Vis Sci. 2006;47:2172–2178. doi: 10.1167/iovs.05-1167. [DOI] [PubMed] [Google Scholar]
- 24.Verhaegen S, McGowan AJ, Brophy AR, Fernandes RS, Cotter TG. Inhibition of apoptosis by antioxidants in the human HL-60 leukemia cell line. Biochem Pharmacol. 1995;50:1021–1029. doi: 10.1016/0006-2952(95)00233-p. [DOI] [PubMed] [Google Scholar]
- 25.Slater AF, Stefan C, Nobel I, van den Dobbelsteen DJ, Orrenius S. Signalling mechanisms and oxidative stress in apoptosis. Toxicol Lett. 1995;82–83:149–153. doi: 10.1016/0378-4274(95)03474-9. [DOI] [PubMed] [Google Scholar]
- 26.Hinton DR, He S, Lopez PF. Apoptosis in surgically excised choroidal neovascular membranes in age- related macular degeneration. Arch Ophthalmol. 1998;116:203–209. doi: 10.1001/archopht.116.2.203. [DOI] [PubMed] [Google Scholar]
- 27.Dunaief JL, Dentchev T, Ying G-S, Milam AH. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 28.Sato T, Machida T, Takahashi S, et al. Fas-mediated apoptosome formation is dependent on reactive oxygen species derived from mitochondrial permeability transition in Jurkat cells. J Immunol. 2004;173:285–296. doi: 10.4049/jimmunol.173.1.285. [DOI] [PubMed] [Google Scholar]
- 29.Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res. 1996;62:155–169. doi: 10.1006/exer.1996.0020. [DOI] [PubMed] [Google Scholar]
- 30.Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999;40:2988–2995. [PubMed] [Google Scholar]
- 31.Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:1981–1989. [PubMed] [Google Scholar]
- 32.Sparrow JR, Zhou J, Cai B. DNA is a target of the photodynamic effects elicited in A2E-laden RPE by blue-light illumination. Invest Ophthalmol Vis Sci. 2003;44:2245–2251. doi: 10.1167/iovs.02-0746. [DOI] [PubMed] [Google Scholar]
- 33.Rothe G, Oser A, Valet G. Dihydrorhodamine 123: a new flow cytometric indicator for respiratory burst activity in neutrophil granulocytes. Naturwissenschaften. 1988;75:354–355. doi: 10.1007/BF00368326. [DOI] [PubMed] [Google Scholar]
- 34.Rouault T, Klausner R. Regulation of iron metabolism in eukaryotes. Curr Top Cell Regul. 1997;35:1–19. doi: 10.1016/s0070-2137(97)80001-5. [DOI] [PubMed] [Google Scholar]
- 35.Reznichenko L, Amit T, Zheng H, et al. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (−)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- 36.Hadziahmetovic M, Dentchev T, Song Y, et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–2736. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carmody RJ, Cotter TG. Signalling apoptosis: a radical approach. Redox Rep. 2001;6:77–90. doi: 10.1179/135100001101536085. [DOI] [PubMed] [Google Scholar]
- 38.Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–33. doi: 10.1016/s0891-5849(00)00302-6. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson MD. Reactive oxygen species and programmed cell death. Trends Biochem Sci. 1996;21:83–86. [PubMed] [Google Scholar]
- 40.Terman A, Gustafsson B, Brunk UT. The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact. 2006;163:29–37. doi: 10.1016/j.cbi.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 41.Ben-Shabat S, Itagaki Y, Jockusch S, Sparrow JR, Turro NJ, Nakanishi K. Formation of a nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew Chem Int Ed Engl. 2002;41:814–817. doi: 10.1002/1521-3773(20020301)41:5<814::aid-anie814>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 42.Sparrow JR, Vollmer-Snarr HR, Zhou J, et al. A2E-epoxides damage DNA in retinal pigment epithelial cells: vitamin E and other antioxidants inhibit A2E-epoxide formation. J Biol Chem. 2003;278:18207–18213. doi: 10.1074/jbc.M300457200. [DOI] [PubMed] [Google Scholar]
- 43.Jang YP, Matsuda H, Itagaki Y, Nakanishi K, Sparrow JR. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J Biol Chem. 2005;280:39732–39739. doi: 10.1074/jbc.M504933200. [DOI] [PubMed] [Google Scholar]
- 44.Oh SH, Lim SC. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol Appl Pharmacol. 2006;212:212–223. doi: 10.1016/j.taap.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 45.Rothe G, Emmendorffer A, Oser A, Roesler J, Valet G. Flow cytometric measurement of the respiratory burst activity of phagocytes using dihydrorhodamine 123. J Immunol Methods. 1991;138:133–135. doi: 10.1016/0022-1759(91)90074-p. [DOI] [PubMed] [Google Scholar]
- 46.Cai J, Jones DP. Superoxide in apoptosis: mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 47.Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T. Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol. 2000;20:7311–7318. doi: 10.1128/mcb.20.19.7311-7318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vives-Bauza C, Anand M, Shirazi AK, et al. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–24780. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charkoudian LK, Pham DM, Franz KJ. A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. J Am Chem Soc. 2006;128:12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 51.Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, Franz KJ. Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. J Inorg Biochem. 2008;102:2130–2135. doi: 10.1016/j.jinorgbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]