

Abstract
Constitutive activation of the mitogen-activated protein kinase (MAPK) pathway is implicated in the development and progression of many human cancers, including melanoma. Mutually exclusive activating mutations in NRAS or BRAF have been identified in ∼85% of melanomas and components of this pathway have been developed as molecular targets for therapeutic intervention. We and others have demonstrated that inhibition of this pathway with specific small molecule MEK inhibitors induces a wide range of apoptotic responsiveness in human melanoma cells both in vitro and in vivo. To define the molecular mechanism underlying variable apoptotic sensitivity of melanoma cells to MEK inhibition, we examined the expression and subcellular localization of Bcl-2 family members in a comprehensive set of human melanoma cell lines. While the proapoptotic protein Bim was activated and localized to the mitochondrial membrane in all cell lines regardless of apoptotic sensitivity, Bmf activation and cytosolic translocation was exclusive to sensitive cells. In resistant cells, Bmf remained sequestered to the cytoskeleton through dynein light chain 2 (DLC2) binding. Overexpression of Bmf in resistant cells did not enhance apoptosis, whereas expression of mutant BmfA69P, which has decreased binding to DLC2, promoted cell death. Expression of BmfA69P mutants possessing the BH3 domain mutation L138A, which impairs BH3 interactions, did not enhance apoptosis in resistant cells. RNA interference targeting Bim and Bmf provided protection from apoptosis induced by MEK inhibition. These results demonstrate a novel role for Bmf in promoting apoptosis and provide insight into the mechanism of apoptotic resistance to MEK inhibition in melanoma.
Introduction
Skin cancer is the most common malignancy in the United States, representing nearly one third of all newly diagnosed cancers. Melanoma accounts for only 4% of all skin cancers, but is responsible for 79% of skin cancer deaths (1). During the last twenty years, the incidence of melanoma has more than tripled in Caucasian Americans with an estimated 62,480 new cases diagnosed in 2008 (1). Melanoma is the most common cancer in men and women ages 20−29 and is the leading cause of cancer death in women between the ages of 25−29 (2). In 1940, the lifetime risk of developing melanoma was 1 in 1,500 which contrasts sharply with current estimates of 1 in 61 (1, 2). Currently, the alkylating agent dacarbazine (DTIC) is the only FDA approved chemotherapeutic agent for treating melanoma. Clearly, current therapies used to treat metastatic melanoma are inadequate as the 5-year survival rate has remained at less than 15% for decades with over 8,000 deaths annually (1). With mutually exclusive mutations in NRAS and BRAF, this pathway is constitutively activated in most cases of sporadic malignant melanomas (3) and several clinical trials of MEK-targeted therapies in melanoma have focused on exploiting the dependence of these tumors on MAPK signaling. Additionally, about 50% of those with BRAF mutations also have AKT3 amplification or PTEN loss, resulting in deregulation of AKT signaling (4).
Recent in vitro data suggests that MAPK and AKT signaling promotes melanoma cell survival through the regulation of Bcl-2 protein family members (5-7), which are essential regulators of the apoptotic pathway. The anti-apoptotic members Bcl-2, Bcl-XL, Mcl-1, Bcl-w and Bfl-1 each possess 4 domains termed Bcl-2 homology domains (BH) and are often overexpressed in many cancer types including melanoma. They regulate the release of cytochrome c from the mitochondria by sequestering proapoptotic Bcl-2 family members. The proapoptotic members fall into two subgroups; those containing BH domains 1−3 (Bak, Bax and Bok) and those possessing BH3 domains only (Bad, Bim, Bmf, Bik, Hrk, Bid, Puma, and Noxa). Upon release from antiapoptotic members (e.g., Bcl-2), Bax and/or Bak oligomerize to induce release of apoptosis-promoting proteins (including cytochrome c) from the mitochondrial intermembrane space. In response to various death-promoting stimuli, BH3-only members are activated by multiple means, including post-translational modification, transcriptional upregulation as well as subcellular localization (8). BH3-only proteins exhibit selective binding affinities for antiapoptotic Bcl-2 proteins, therefore activation of two or more can enhance apoptosis depending on the repertoire of antiapoptotic proteins that are present (9).
Increasing evidence suggests that BH3 only members act as sentinels of cellular stress throughout a cell and are regulated by components of the RAS/MAPK and/or PI3K/AKT pathways. BAD is a pro-apoptotic BH3-only member of the Bcl-2 family of proteins that heterodimerizes with and antagonizes pro-survival proteins such as Bcl-2 and Bcl-xL. This interaction triggers the release of cytochrome c from the mitochondria, which activates a caspase cascade leading to apoptosis (8). Several kinases including AKT, p70S6K, PKA, JNK, and RSK (a direct downstream target of ERK) have been shown to phosphorylate and inactivate BAD, thereby promoting survival (5, 10-14). In pancreatic cancer cells, RSK activates the transcription factor CREB, which promotes survival by increasing the expression of the anti-apoptotic proteins Bcl-2, Bcl-xL, and Mcl-1 (15). The pro-apoptotic protein Bmf is transcriptionally repressed by both the RAS/MAPK and PI3K/AKT pathways in breast cancer cells (16). In addition, ERK directly phosphorylates the pro-apoptotic protein Bim, which leads to its rapid degradation via the proteasome pathway (17, 18).
In this study, we classified a comprehensive panel of human melanoma cell lines as either sensitive (≥ 30 % cell death) or resistant (≤ 30% cell death) to MEK inhibition to determine the mechanism of resistance to cell death in this context. Following MEK inhibition, we compared the expression, activation, and sub-cellular localization of Bcl-2 family members between resistant and sensitive cell lines to identify potential differences.
Materials and Methods
Cell culture
Human melanoma cell lines C8161 (19) and CHL-1 (20) (wild-type BRAF and NRAS), A375, C32, CACL, LOX-IMVI, M14-Mel, MALME-3M, SK-MEL-5, SK-MEL-28, UACC-62 and UACC-257 (BRAF mutants) (21), SK-MEL-2, SK-MEL-103 and SK-MEL-147 (NRAS mutants) were cultured in RPMI 1640 (Gibco, Rockville, MD) supplemented with 5% fetal bovine serum (Hyclone, Logan, UT). Human neonatal epidermal melanocytes (Cascade Biologics, Portland, OR) were maintained in Medium 254 supplemented with melanocyte growth factors (Cascade Biologics, Portland, OR).
Reagents
The MEK inhibitor CI-1040 (PD184352) was obtained from Pfizer Global Research and Development. PD98059, an additional MEK inhibitor, was purchased from Cell Signaling Technology (CST, Danvers, MA). The BH3 mimetic ABT-737 along with an inactive control molecule was acquired from Abbott (Abbott Park, IL). The caspase inhibitors Z-VAD-FMK, Z-DEVD-FMK, and Z-IETD-FMK were purchased from Becton Dickinson (BD, Franklin Lakes, NJ). Drugs were dissolved in DMSO, aliquoted, and stored at −20 °C.
Drug treatments and cell viability
All cell viability assays were initiated 24 hours post melanocyte or melanoma cell line seeding and were carried out for the times indicated. Single treatment dosing with CI-1040 was 2 μmol/L based on titration studies examining phospho-ERK levels and minimal melanocyte toxicity. PD98059 required a daily dosing schedule of 20 μmol/L to inhibit ERK phosphorylation. DMSO treated controls were vehicle control samples that were treated with an equal volume of DMSO. Cell viability and apoptosis was verified by multiple methods including ViaCount (Guava Technologies, Hayward, CA), Annexin V binding (Guava Technologies, Hayward, CA), cell cycle analysis with propidium iodide staining (Roche, Basel, Switzerland), and DAPI staining as previously described (6). Samples were prepared per the manufacturers specifications at select time points and assayed promptly. Experiments were done in triplicate and data are presented as the mean with standard error of the mean (SEM).
Subcellular fractionation
Initial mitochondrial and cytosolic fractionation was carried out with M14-MEL and SK-MEL-28 cells over 0, 48 and 72 hours post CI-1040 treatment using a commercially available mitochondrial isolation kit (Pierce, Rockford, IL). Subsequent cytosolic fractional isolations for additional cell lines were carried out in a similar manner using the nuclear and cytosolic fraction kit (BioVision, Mountain View, CA) over multiple time points. Each protocol was followed as per the manufacturer's instructions with the exception of transferring the cell pellets to 2 ml microcentrifuge tubes after the first centrifugation followed by washing the cell pellets 3 times therein with ice cold PBS to adequately remove residual trypsin.
Western Blotting
For direct immunoblot analysis, adherent and nonadherent cells from each plate or well were lysed in SDS sample buffer and boiled prior to SDS-PAGE. Protein concentration was determined using the Bio-Rad Dc Protein Assay (Bio-Rad, Hercules, CA) per the manufacturer's specifications. Typically, 30 μg of protein was loaded per well in 4−20% gradient polyacrylamide gels (Invitrogren, Carlsbad, CA) and transferred onto nitrocellulose (Bio Rad, Hercules, CA). Equal volumes of 2× SDS sample buffer was added to native lysates from subcellular fractions followed by boiling and SDS-PAGE followed by immunoblotting. For a complete list of all antibodies used see Supplementary data.
Stable Bmf and Bim constructs
V5 N-terminal tagged and wild-type Bmf and Bim were cloned from M14-Mel cells by RT-PCR into pCR8/GW/TOPO (Invitrogen, Carlsbad, CA) and sequence verified with Genbank NM_033503 for Bmf and Genbank NM_138621 for Bim. PCR-based site directed mutagenesis was used to generate Bmf mutants. Through LR Clonase reactions (Invitrogen, Carlsbad, CA) these genes were then recombined into a Gateway® compatible modified lentiviral vector, pDEST-FG12-cmv, for subsequent expression analysis. The pDEST-FG12-cmv vector has been described (22). This vector also harbors an independent GFP reporter gene allowing easy detection of infection efficiency. Lentiviral production and infections were carried out as previously described (23). Viral gene delivery was confirmed by Western blot analysis. Details regarding primer sequences, cloning strategies, and lentiviral infection are available upon request.
Stable pro-survival Bcl-2 family member constructs
HA N-terminal tagged and wild-type Bcl-xL and Bfl-1 were cloned from NHEM cells by RT-PCR into pCR8/GW/TOPO (Invitrogen, Carlsbad, CA) and sequence verified with Genbank NM_009743 for Bcl-xL and Genbank NM_004049 for Bfl-1. Bcl-2 and Mcl-1 cDNAs (Origene, Rockville, MD) were PCR amplified and cloned into pCR8/GW/TOPO (Invitrogen, Carlsbad, CA) and sequence verified with Genbank NM_000633 for Bcl-2 and Genbank NM_021960 for Mcl-1. These genes were then recombined into the pDEST-FG12-cmv lentiviral vector using LR clonase (Invitrogen, Carlsbad, CA) for subsequent viral production, infection, and stable expression (23). Infection efficiency was verified by GFP expression and gene expression was confirmed by Western blot analysis. Primer sequences and cloning strategies are available upon request.
Stable Bmf and Bim short hairpin RNA (shRNA) constructs and Bad short interfering RNA (siRNA)
Short 19 to 22 bp Bmf (Genbank NM_033503 nucleotides 326−344, 116−134, 213−235 and 393−414) and Bim (Genbank NM_138621 nucleotides 436−454 and 820−841) specific oligos (IDT, Coralville, IA) were annealed and cloned into the lentiviral vector KH1-LV (24) which contains an H1 promoter and independent GFP expression driven by a ubiquitin-C promoter. An additional scrambled shRNA was designed and used as a control. Subsequent viral production and infection efficiency was verified by GFP expression while stable shRNA potency was determined by Western blot analysis. Short hairpin oligo sequences and cloning strategies are available upon request. 5μM SMARTpools (Dharmacon, Lafayette, Colorado) targeting Bad were electroporated (200v, 750μF in Opti-MEM medium) into 5×106 melanoma cells using a Gene Pulser II (Bio Rad, Hercules, CA) and knockdown was verified by Western blot at the times indicated.
RT-PCR Analysis
RNA was isolated (Invitrogen, Carlsbad, CA) from CI-1040 treated and untreated melanoma cell lines over times indicated. Reverse transcription (Invitrogen, Carlsbad, CA) of 1μg of total RNA was followed by 25 cycles of PCR using GAPDH and Bmf specific primers (IDT, Coralville, IA). The following primers were used to generate 425 (GAPDH) and 254 (Bmf) base pair fragments; GAPDH Fwd primer 5′-CTACCCACGGCAAGTTCAAT-3′, GAPDH Rev primer 5′-TACTCAGCACCAGCATCACC-3′, Bmf Fwd primer 5′-GGCTATCGGCTTCCTCTCCC-3′ and Bmf Rev primer 5′-TCACCTAGGGCCTGCCCC-3′.
Results
Melanoma cell lines exhibit variable sensitivity to the MEK inhibitor PD184352 (CI-1040) (Figure 1A). It has been reported that the response of melanoma cells to MEK inhibition is largely due to BRAF (sensitive) and NRAS (resistant) mutation status (25, 26). To define the mechanism underlying the variable apoptotic response of melanoma cells to MEK inhibition, 15 human melanoma cell lines along with normal human epidermal melanocytes (NHEM) were treated with the highly specific small molecule MEK inhibitor CI-1040 (27, 28). NHEM and melanoma cells that possess wild type NRAS and BRAF are resistant to the apoptotic effects of MAPK inhibition while variable apoptotic sensitivity was not significantly different between BRAF, BRAF/PTEN and NRAS mutant cell lines (Figure 1A). CI-1040 potently inhibited ERK phosphorylation for the duration of the study (Figure 1B). Although baseline phosphorylated ERK levels were variable, there was no correlation between cell death and ERK activation. CI-1040 promotes G1 cell cycle arrest in NHEM and melanoma cells followed by significant apoptosis in sensitive melanoma cells by 72 hours (Figure 1C).
Figure 1. Cytotoxic effect of the MEK inhibitor CI-1040 in a panel of melanoma cell lines.
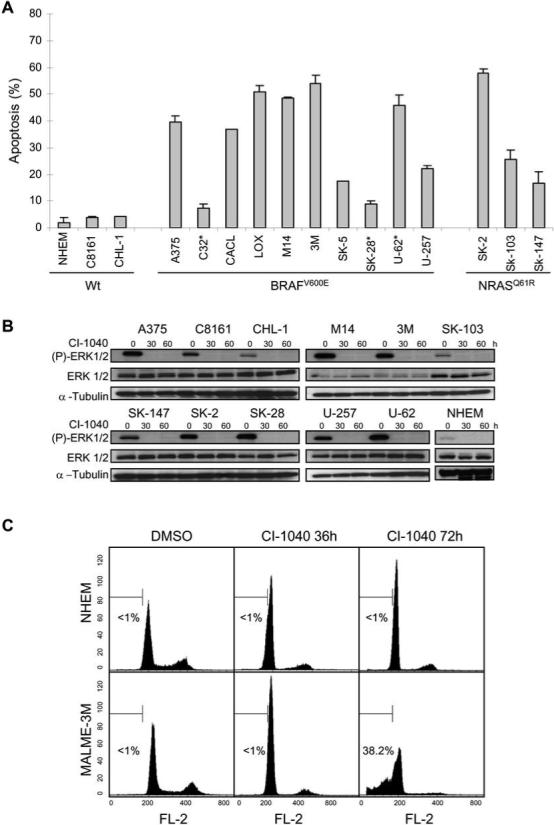
(A) Apoptotic sensitivity of normal human epidermal melanocytes (NHEM) and melanoma cell lines to the MEK inhibitor CI-1040. Cells were treated in triplicate with either DMSO as a control or 2 μM CI-1040 for 72 h and apoptosis (% sub G1 fraction) was determined by cell cycle analysis using flow cytometry. Data was normalized to the control and expressed as the mean ± SEM. Cell lines are grouped according to BRAF or NRAS mutation status. Astericks (*) indicate PTEN mutation. (B) Western blot analysis of phosphorylated ERK1/2 (P-ERK 1/2), and total ERK 1/2 expression in NHEM and melanoma cell lines treated as in (A). Whole cell lysates (30 μg each) were subjected to Western blotting using the indicated phospho-specific antibodies to detect activation of the kinases. The blots were re-probed with their respective total protein antibodies. The blots were re-probed with α-tubulin as a loading control. (C) Induction of G1 arrest in NHEM (top panels) and apoptosis in MALME-3M melanoma cells (lower panels) by CI-1040. Response of cells to DMSO (left panels) or CI-1040 for 36 hours (middle panels) or 72 hours (right panels) was analyzed by flow cytometry. A percentage of apoptosis quantified independently is indicated in each histogram.
Melanoma cells exhibit variable sensitivity to the BH3 mimetic ABT-737
It was previously reported that inactivation of the BH3-only protein Bad by MAPK signaling promotes survival in melanoma cells (5). The small molecule BH3-mimetic ABT-737 mimics Bad by binding to the same subset of Bcl-2 pro-survival proteins Bcl-2, Bcl-xL, and Bcl-w, but not Mcl-1 or Bfl-1 (29). Unlike Bad, however, ABT-737 cannot be inactivated by MAPK signaling. Therefore, ABT-737 functions in a similar manner as constitutively active Bad and was used to further evaluate the role of Bad in promoting apoptosis in melanoma cells. ABT-737 dosing was determined by single agent titration ranging from 100 nM to 5 μM on 10 melanoma cell lines out to 72 hours. The enantiomer (the stereoisomer having the mirror image of the dimethylaminoethyl group) was used as a loss-of-function control (29). The range where the enantiomer had negligible cytotoxic effects and ABT-737 induced varying degrees of cytotoxicity was between 1 and 2.5 μM for most of the cell lines (Figure 2A and data not shown). Single and combination treatments with ABT-737 and the MEK inhibitor PD98059 were carried out with a final concentration of 2.5 μM and 20 μM, respectively. As a control, 2.5 μM enantiomer was also tested and the resulting cytotoxicity was subtracted from the corresponding ABT-737 induced cytotoxicity. DMSO was used as a control for PD98059. ABT-737 exhibited significant single agent activity in many of the cell lines with greater than 50% cell death in CHL-1 and SK-MEL-103 (Figure 2A). However, the sensitivity profile for ABT-737 did not mirror that of the MEK inhibitors. The CHL-1 cell line was highly resistant to MEK inhibition but was sensitive to ABT-737 and the M14-MEL cell line was highly sensitive to MEK inhibition but relatively resistant to ABT-737. While this demonstrates that the apoptotic machinery is intact and resistance to MEK inhibition is not due to defects in the apoptotic pathway (e.g., loss of bak or bax functionality), it suggests that cell death induced by MEK inhibition is not mediated by the BH3-only protein Bad.
Figure 2. Biological effects of the ‘Bad-like’ BH3 mimetic ABT-737 and Bad RNAi in melanoma cells.
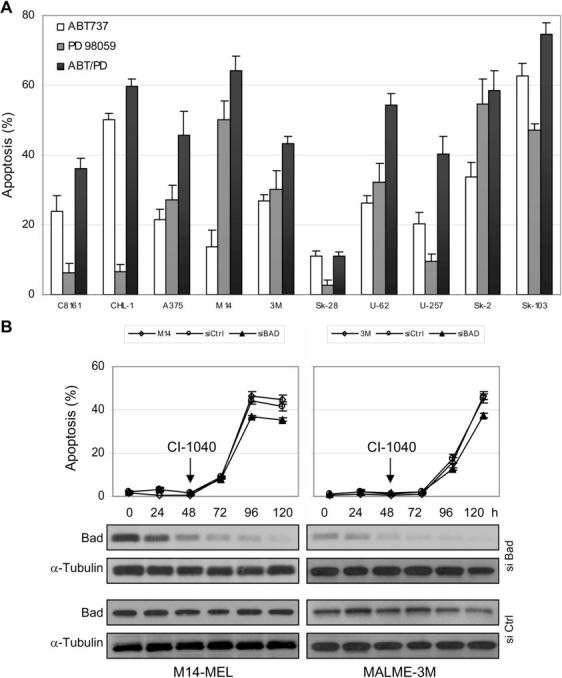
(A) Apoptotic sensitivity of melanoma cell lines to ABT-737 alone and in combination with the MEK inhibitor PD98059. Cells were treated in triplicate with either the inactive enantiomer, 2.5 μM ABT-737, DMSO, or 20 μM of the MEK inhibitor PD98059 (added every 24 h) for 72 h. Apoptosis (% sub G1 fraction) was determined by cell cycle analysis using flow cytometry. Data was normalized to the control and expressed as the mean ± standard error. (B) Analysis of the requirement of the BH3-only protein Bad in CI-1040 induced apoptosis in M14-MEL and MALME-3M cell lines. Cells were treated with DMSO or 2 μM CI-1040 48 h after transfection for a duration of 72 h (120 h post-transfection). The arrow indicates the start of CI-1040 treatment. Apoptosis was determined as described in (A). Data was normalized to the control and expressed as the mean − SEM. Untransfected (◇), siCtrl (○), siBad (▲). Immunoblots demonstrate reduced expression of Bad following transfection of siRNA SMARTpool oligonucleotides targeting Bad (Dharmacon).
Apoptosis of melanoma cells induced by MEK inhibition does not require the BH3-only protein Bad
To further examine the requirement of Bad in apoptosis induced by MEK inhibition, M14-MEL and MALME-3M cells were transfected with synthetic short interfering (si) RNA SMARTpools to specifically reduce Bad expression by RNA interference (RNAi). Expression of Bad was assessed by immunoblot analysis over time post-transfection. Levels of Bad protein were effectively reduced greater than 90% in both cell lines by 120 hours post-transfection compared with the levels expressed in cells transfected with the control siRNAs (Figure 2B). Cells were treated with CI-1040 48 hours post-transfection and assessed for cell death after 72 hours of treatment (120 hours post-transfection). Loss of Bad expression reduced apoptosis by only ∼1.2-fold in both cell lines suggesting that Bad is not required for apoptosis induced by MEK inhibition in melanoma cells (Figure 2B).
MEK inhibition leads to a dramatic increase in Bim levels in all cell lines regardless of apoptotic sensitivity
To further explore the mechanism of resistance to MEK inhibition, comparative expression analysis of Bcl-2 and IAP family members, which tightly regulate cell survival, in a subset of both sensitive and resistant melanoma cell lines was performed. Immunoblot analysis of apoptotic regulatory factors in 8 melanoma cell lines treated with CI-1040 for 72 hours was carried out to determine if alterations in any of these proteins correlated with apoptotic sensitivity. Although expression of pro-survival factors was highly variable among these cell lines, there was no correlation between expression and cell death (Figure 3). Furthermore, MEK inhibition did not impact the expression of Bcl-xL or Mcl-1. Interestingly, Bcl-2 expression was increased in response to MEK inhibition in several cell lines, but this expression did not correlate with resistance. Additionally, IAP expression did not influence apoptotic sensitivity (Figure 3). High expression of the pro-apoptotic multi-domain proteins Bax and Bak was observed, suggesting that there is no disconnect with proximal death effectors in these melanoma cell lines. Upon MEK inhibition, p53 responsive pro-apoptotic proteins Noxa and Puma β expression is reduced in several cell lines (Figure 3). ERK directly phosphorylates and targets Bim for proteolytic degradation (18) and as expected inhibition of MEK and subsequent inhibition of ERK leads to a dramatic increase in Bim levels (Figure 3). Interestingly, active Bim is present in all cell lines in response to MEK inhibition regardless of the amount of cell death induced.
Figure 3. Effect of CI-1040 on Bcl-2 and IAP family member proteins.
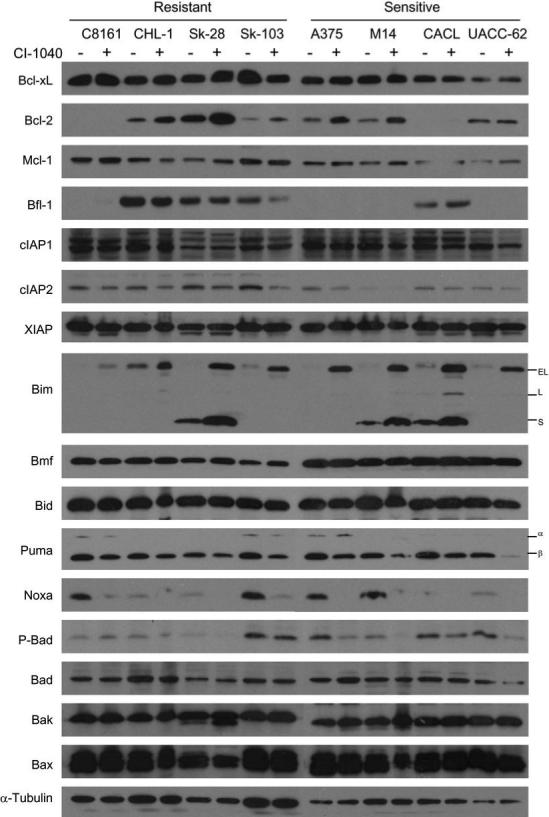
The cell lines indicated above the blots were treated with either DMSO (−) or 2 μM CI-1040 (+) for 72 h and whole cell lysates were prepared. Western blotting was performed using the antibodies indicated to the left of the blots. Puma shows two isoforms, α and β, as does Bim, EL and L, which are indicated to the right of their corresponding blots. A representative α-tubulin re-probed blot is shown in the lower panel as a loading control.
Comparative compartmental analysis between a sensitive and resistant cell line reveals differences in Bmf localization
Cellular localization of Bcl-2 family members may be more relevant to cell survival than overall expression as many of these proteins are inactivated through sequestration. To interrogate possible differences between a CI-1040 sensitive (M14-MEL) and resistant (SK-MEL-28) cell line, subcellular fractions were collected from untreated and CI-1040 treated cells over time and compared by immunoblot (Figure 4A). Bim rapidly accumulates to the outer mitochondrial membrane in both cell lines, but cytosolic accrual of Bmf was exclusive to the sensitive M14-MEL cell line (Figure 4A). Analysis of whole cell lysates revealed that expression of Bmf remains relatively constant over time in each cell line (Figure 3). While nearly all Bcl-2 family members were analyzed in this context, only Bmf localization was dramatically divergent. Further compartmental analysis of Bmf localization in additional cell lines with variable apoptotic sensitivities revealed that Bmf cytosolic localization correlated exquisitely with cell death (Figure 4B). Resistant cell lines such as C8161 and SK-MEL-28 retain Bmf in the cytoskeletal compartment. Conversely, Bmf is released into the cytosolic fraction, which is proportional to levels of apoptosis, in CI-1040 sensitive cell lines (Figure 4B). Interestingly, MEK inhibition results in enhanced Bmf transcription in melanoma cells regardless of mutation status or apoptotic sensitivity (Figure 4C).
Figure 4. Localization of Bmf correlates with resistance to MEK inhibition in a panel of melanoma cell lines.
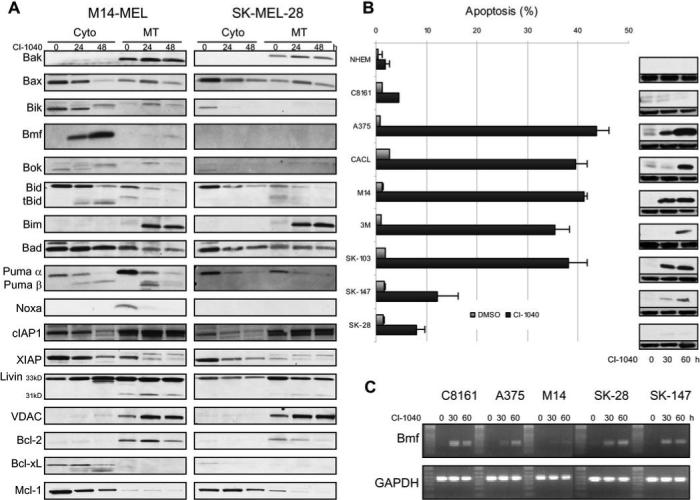
(A) A comparison of the cytosolic (Cyto) and mitochondrial (MT) fractions between a CI-1040 sensitive (M14-MEL) and resistant (SK-MEL-28) cell line reveals differences in Bmf localization. Cells were treated with DMSO (0 h) or 2 μM CI-1040 for the times indicated. Representative Western blots of fractionated lysates probed with antibodies directed against the proteins indicated are shown. The blots were re-probed with VDAC as both a loading control and confirmation of purity of the mitochondrial fraction. (B) Correlation between apoptotic sensitivity of normal human epidermal melanocytes (NHEM) and melanoma cell lines to CI-1040 and cytosolic Bmf protein levels. Cells were treated in triplicate with either DMSO or 2 μM CI-1040 for 60 h and apoptosis (% sub G1 fraction) was determined by cell cycle analysis using flow cytometry. Data is expressed as the mean ± SEM. Duplicate samples were treated with 2 μM CI-1040 for the times indicated and the cytosolic fraction was isolated. Western blots of fractionated lysates probed with an antibody directed against Bmf are shown (upper blot). The blots were re-probed with α-tubulin as both a loading control and confirmation of purity of the cytosolic fraction (lower blot). (C) Up-regulation of Bmf RNA levels after treatment with CI-1040. RT-PCR products for Bmf and GAPDH were derived from DMSO treated cells (0 h) or cells treated with 2 μM CI-1040 for the times indicated. The cell lines analyzed are indicated above each corresponding gel.
Apoptosis of melanoma cells induced by MEK inhibition requires the BH3-only proteins Bim and Bmf
To further define the roles of Bim and Bmf in promoting apoptosis, short hairpin (sh) RNAs were stably expressed in melanoma cells using a lentiviral mediated approach. The KH1-LV lentivirus (24), which coexpresses GFP driven by the UbC promoter, was VSV-G pseudotyped and used to infect the sensitive cell line M14-MEL. Since Bim is not detectable when MEK is active, levels of Bim were assessed in the presence of MEK inhibition. Infected M14-MEL cells were treated with DMSO or CI-1040 for 72 hours and expression of Bim and Bmf was assessed by immunoblot analysis. Protein levels of Bim in the CI-1040 treated samples were reduced by 20% and 80% using shRNA 1 and 2, respectively as compared with the levels expressed in cells infected with the scrambled control shRNA lentivirus (Figure 5A). Bmf protein levels were unchanged when only one shRNA target was used but a pool of 3 shRNAs reduced Bmf protein levels by 64% compared with the levels expressed in cells infected with the control shRNA lentivirus (Figure 5A). Reduced expression of either Bim or Bmf dramatically reduced CI-1040 directed apoptosis in these cells (Figure 5A), whereas reduction of other BH3-only proteins such as Bad or Bid did not (Figure 2B and data not shown). This data provides compelling evidence that both Bim and Bmf are required for promoting apoptosis in response to MEK inhibition in melanoma cells.
Figure 5. Bmf or Bim RNAi protects melanoma cells from CI-1040 induced apoptosis.
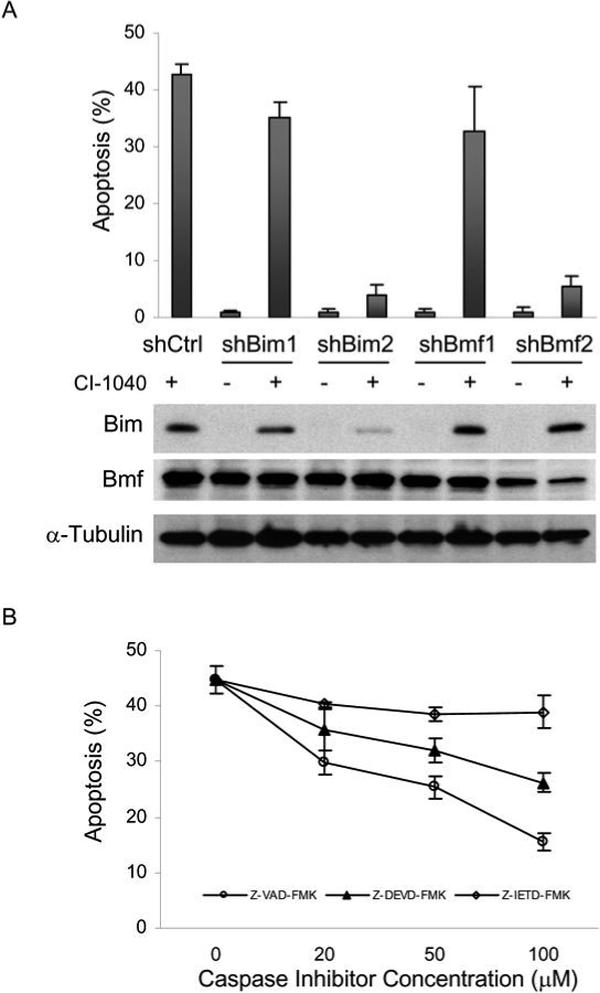
(A) Following stable lentiviral shRNA expression targeting Bim, Bmf, or scrambled control (Ctrl) in the sensitive M14-MEL cell line, cells were treated with 2μM CI-1040 or equal volume DMSO for 72 h and apoptosis (% sub G1 fraction) was determined by flow cytometry. Data is expressed as the mean ± SEM. Two different shRNAs with different efficiencies were used. Reduced expression of Bim and Bmf was confirmed by Western blot analysis with the antibodies indicated to the left of each panel. Blots were stripped and reprobed with α-Tubulin as a loading control. (B) Apoptotic sensitivity of M14-MEL cells treated with 2μM CI-1040 and increasing levels of the pan caspase inhibitor ZVAD-FMK (○), the caspase 3 inhibitor Z-DEVD-FMK (▲), or the caspase 8 inhibitor Z-IETD-FMK(◇). Apoptosis was determined as described in (A). Data is expressed as the mean ± SEM.
CI-1040 directed apoptosis demonstrates partial caspase dependence
Truncated Bid (tBid), an effector of the extrinsic apoptotic pathway, was present in M14-MEL (Figure 4A) and other sensitive cell lines (data not shown) during active apoptosis. To determine the relative contribution of the extrinsic pathway in promoting apoptosis induced by MEK inhibition, caspase inhibitors with differing specificities were employed. The pan caspase inhibitor Z-VAD-FMK and the caspase 3 directed Z-DEVD-FMK provided dose responsive protection from CI-1040 induced apoptosis (Figure 5B) while Z-IETD-FMK, the caspase 8 inhibitor, did not. This suggests that apoptosis is primarily driven by the intrinsic pathway and is dependent on caspase activation. In support of this, Bid RNAi (data not shown) had no impact on CI-1040 induced apoptosis in 3 cell lines examined.
BmfA69P confers CI-1040 sensitivity to resistant SK-MEL-28 cells
To assess the impact of Bim, Bmf and BmfL138A over-expression in M14-MEL and SK-MEL-28 cells, viral mediated delivery was employed using the FG12-CMV lentivirus (22). Viral delivered V5 N-terminal tagged Bim and Bmf or HA N-terminal tagged Bcl-xL and Mcl-1 in either cell line was expressed at high levels (Figure 6A). Despite high expression levels neither Bim nor Bmf (wt or mutant) over-expression induced cell death in either cell line in the absence of CI-1040. Overxpression of Bim or Bmf had little overall impact on cell death in the sensitive M14-MEL cell line (Figure 6). Altering critical amino acids in the BH3 domain (L138A) in Bmf diminishes its ability to promote apoptosis (30). Over-expression of Bmf or BmfL138A had no impact on cell death in SK-MEL-28 or M14-MEL cells (Figure 6). Release of Bmf from cytoskeletal components such as DLC2 is crucial for Bmf to promote apoptosis (30, 31). To promote Bmf disassociation from DLC2, we generated BmfA69P(30) and BmfA69P/L138A mutants and evaluated their effect on cell death in response to MEK inhibition. Over-expression of either Bmf mutant in the absence of CI-1040 had no impact on cell death (Figure 6), but upon MEK inhibition, only BmfA69P dramatically enhanced apoptosis in resistant SK-MEL-28 cells to a level comparable to the sensitive cell line M14-MEL (Figure 6A and B). Increased amounts of cleaved PARP, an indicator of caspase 3 activation and apoptosis, was observed in CI-1040 treated SK-MEL-28 cells expressing BmfA69P compared with the same cells expressing wt Bmf, and was comparable to CI-1040 treated M14-MEL cells (Figure 6A). BmfA69P and BmfA69P/L138A mutants each localize to the cytosolic fraction in SK-MEL-28 cells (data not shown), but the BH3 domain impaired BmfA69P/L138A mutant does not promote apoptosis. Therefore the apoptotic effects of Bmf require cytoskeletal disassociation and an intact BH3 domain.
Figure 6. BmfA69P expression in combination with CI-1040 promotes apoptosis in resistant SK-MEL-28 cells.
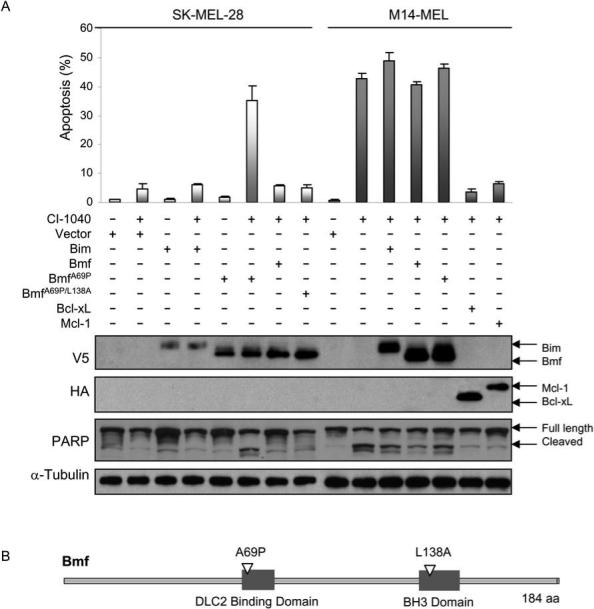
(A) Lentiviral delivery of wt and modified Bcl-2 family members in SK-MEL-28 and M14-MEL cells as indicated by +. Expression was confirmed by Western blot analysis with the labels on the left indicating specific antibodies used while labels on the right are used to further distinguish proteins or modifications (PARP). PARP is a nuclear polymerase and is a primary cleavage target of active caspase 3 – this ab recognizes both full length as well as cleaved PARP. Each cell line expressing various members of the Bcl-2 family were treated with CI-1040 (+) or DMSO vehicle control (−) for 72 h followed by cell death analysis via flow cytometry. Data is expressed as the mean ± SEM. (B) Schematic diagram of Bmf including the location of the mutations used in this study.
Pro-survival Bcl-2 family member overexpression and antagonism impact melanoma survival
To evaluate the contribution of Bcl-2 pro-survival members in resistance to MEK inhibition, lentiviral vectors (23, 24) were used to deliver and express Bcl-2, Bcl-xL, Bfl-1 and Mcl-1. Over-expression of any of these proteins in the sensitive cell line M14-MEL resulted in significant protection from apoptosis induced by CI-1040 at 72 hours. (Figure 6 and data not shown). PARP cleavage was not observed in Mcl-1 or Bcl-xL over-expressing cells (Figure 6). Expression of Bcl-xL in additional sensitive cell lines A375 and MALME-3M efficiently abrogated apoptosis (data not shown). Apoptosis induced by MEK inhibition is significantly enhanced by RNAi-mediated reduction in Bcl-2, Bcl-xL, or Mcl-1 levels in SK-MEL-103 and SK-MEL-147 cells (23). This demonstrates that these cell lines can effectively disseminate intrinsic apoptotic signals when the protective effects of Bcl-2 pro-survival members are removed. However, since no correlation between endogenous expression (Figure 3) and apoptotic sensitivity was observed, it is unlikely that these proteins are responsible for the variable sensitivity to MEK inhibition observed in melanoma cells (Figure 1A).
Discussion
By analyzing a large panel of melanoma cells with varying sensitivity to MEK inhibition, we have identified a role for the pro-apoptotic BH3-only protein Bmf in promoting cell death in response to MEK inhibition. Striking differences in Bmf activation and localization distinguished sensitive and resistant cell lines exquisitely. Unlike Bmf, Bim translocation to the outer mitochondrial membrane occurred in all melanoma cell lines regardless of sensitivity or resistance to MEK inhibition. Interestingly, both Bmf and Bim are necessary but not sufficient to promote significant apoptosis. These data suggest that strategies that induce the activation of additional pro-apoptotic proteins or those designed to abrogate the protective effects of most of the pro-survival proteins are more likely to achieve success in combination with MEK inhibition in melanoma.
There has been considerable debate regarding the mechanism underlying variable apoptotic sensitivity of melanoma cells to MEK inhibition, most notably NRAS or BRAF mutation status. Recently it was reported that the IC50 values of current generation MEK inhibitors were substantially lower in BRAF mutant melanoma cell lines compared with those harboring activating NRAS mutations suggesting that mutation status is predictive of response (26). We analyzed the apoptotic response of an inclusive melanoma cell line panel in the presence of complete MEK inhibition for the duration of our studies (Figure 4B). The results demonstrate that the apoptotic sensitivity of melanoma cells correlates with Bmf subcellular translocation in response to MEK inhibition regardless of NRAS or BRAF mutation status. Interestingly, treating this cell line panel with doses of CI-1040 that resulted in only modest reductions in ERK phosphorylation yielded no cell death even in the most sensitive cell lines (data not shown). Additionally, removal of CI-1040 treatment after 24 hours allows all cell lines to completely recover and avert cell death. Recent evidence indicates that Bim is quickly phosphorylated and degraded in response to MAPK reactivation (17). Collectively, it is essential to completely block MEK/ERK signaling for a significant amount of time (e.g., 48 to 96 hours) in order to elicit efficient cell death. However, the most resistant cell lines (e.g., C8161 and SK-MEL-28) remain viable even after 144 hours of continuous MEK inhibition (data not shown).
A recent study using the MEK inhibitors U0126 and CI-1040 found that while phosphorylated ERK may be a valid marker for assessing MEK inhibition, it did not correlate with inhibition of melanoma cell growth or BRAF mutational status (19). A separate study using anthrax lethal toxin induced MEK inhibition reported that melanoma cells harboring mutant BRAF are more sensitive compared with those containing mutant NRAS. However, there were exceptions to this finding as two mutant BRAF cell lines, SK-MEL-5 and A2058, had similar levels of sensitivity as the NRAS mutant cells and a cell line wild-type for BRAF, SK-MEL-31, was highly sensitive to MEK inhibition (25). In addition, SK-MEL-5 and SK-MEL-28 are highly resistant to apoptosis induced by anthrax lethal toxin (6), which is similar to what we observed with CI-1040.
Previous studies have suggested that inactivation of the BH3-only protein Bad by MAPK signaling promotes survival in melanoma cells and that interfering with this signaling may represent a tumor specific target (5). Our findings indicate that Bim and Bmf, but not Bad or Bid, are functionally required to induce apoptosis following MEK inhibition. It has previously been reported that Bim, Bid, and Puma bind all of the pro-survival proteins, while the remaining BH3-only proteins are more restricted. While Bad and Bmf were reported to only interact with Bcl-2, Bcl-xL, and Bcl-w (32), recent data suggests that Bmf can also interact with Mcl-1 and Bfl-1 albeit with different affinities (9, 33). Furthermore, Bmf was first identified in a screen for binding partners to Mcl-1 and the BH3 domain in Bmf is most similar to that in Bim (30).
Several studies have described a role for Bim and Bmf in cell death mediated by specific processes. For example, Bmf has been reported to play a role in apoptosis mediated by histone deacetylase (HDAC) inhibitors (34) and both Bim and Bmf are implicated in the cell death response to TGF-β (35) and arsenic treatment (36). In addition, Bim and Bmf play key roles in mammary epithelial anoikis and morphogenesis (16). Here we demonstrate that Bim and Bmf mediate cell death in response to MEK inhibition in melanoma. Interestingly, RNAi targeting either Bim or Bmf is sufficient to rescue cells from cell death in some models while reduction in both proteins is required to significantly suppress apoptosis in other systems. These discrepancies are likely due to differences in cell types and/or expression of pro-survival Bcl-2 family members.
Expression of the p53-responsive BH3-only protein Puma has been reported to be reduced during melanoma progression and low Puma expression correlates with poor prognosis (37). While no significant difference in Puma expression was observed between cell lines, loss or reduced expression occurred over the CI-1040 treatment time course (Figures 2 and 3). Additionally, expression of another p53-responsive BH3-only protein Noxa is dramatically reduced or lost upon CI-1040 treatment. Taken together, this data suggests that these proteins play little or no role in sensitivity or resistance to MEK inhibition as their expression is reduced upon MEK inhibition. However, it is probable that these active BH3 only proteins are responsible for apoptosis induced by ABT-737.
Two distinct models of how BH3-only proteins induce apoptosis have been proposed (38, 39). While it is accepted that Bax or Bak are required for cell death, it is unclear whether BH3-only proteins activate Bax or Bak directly or indirectly. Our results are consistent with the direct activation model that requires a functional “activator” (Bim) and “sensitizer” (Bmf) BH3-only pair to induce apoptosis. Bim activation occurs rapidly and robustly upon MEK inhibition in all cell lines examined regardless of the level of apoptosis induced (Figure 3 and 4), but cell death occurs only when an additional BH3-only member (Bmf) is concurrently active. Bim and Bmf RNAi data are supportive of this model since reducing the expression of either one dramatically blocks cell death (Figure 5A). Bim binds to and antagonizes all Bcl-2 pro-survival members, while Bmf binds to a smaller subset with different affinity (9). Further investigation may distinguish which model, if either, is correct.
Our results suggest that a MEK-independent mechanism of retaining Bmf in the cytoskeletal compartment (e.g., DLC2) is lacking in the sensitive melanoma cell lines and is a major contributing factor to apoptosis that is directed by MEK inhibition. Several kinases, including JNK1 (MAPK8), have been implicated in the regulation and sequestration of Bmf to DLC2 (40). Combined targeting of these kinases or upstream components with MEK inhibition may expand the utility of MAPK as a therapeutic target. Single agent MEK inhibition must be nearly complete in order to initiate apoptosis in even the most sensitive cell lines. Results from early phase clinical trials have demonstrated that tolerable doses of current generation MEK inhibitors PD 0325901 and AZD6244 reduce phospho-ERK levels ∼84% and ∼79%, respectively (41, 42). Additionally, the greatest tumor reduction (70%) in a melanoma patient, whose tumor contained an NRAS mutation, had undetectable phospho-ERK levels in the tumor following sustained daily AZD6244 treatment (41). Complete and sustained MEK inhibition is unlikely to be achieved unilaterally in vivo due to an increase in frequency and severity of side effects upon dose escalation and bioavailability of these inhibitors in organs in which melanoma commonly metastasizes (e.g., brain and liver). Therefore, by understanding the mechanism driving cell death in response to MEK inhibition in melanoma, logical combinatorial therapies can be developed. Consistent with this idea, a novel BH3 mimetic, TW-37, which binds Mcl-1, Bcl-xL, and Bcl-2, in combination with CI-1040 demonstrated synergy in a MEK inhibitor resistant melanoma cell line in vivo (23). Despite its inability to antagonize Mcl-1, which all of the melanoma cell lines examined express (Figure 3), ABT-737 had relatively high single agent activity in this panel of melanoma cell lines and when combined with a less potent MEK inhibitor (PD98059) cell death was enhanced (Figure 2). Our results provide new insights into how MEK inhibition induces apoptosis in melanoma cells. Future studies will determine the mechanism by which Bmf remains sequestered in melanoma cells resistant to MEK inhibition.
Supplementary Material
Acknowledgements
We thank Dr. Maureen Murphy for providing CACL cells. We also thank Dr. Christin Tse and Dr. Judith Sebolt-Leopold for helpful discussions. Grant Support: The National Institutes of Health, the Melanoma Research Foundation, the James A. Schlipmann Melanoma Cancer Foundation, and the Nevada Cancer Institute.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Charlie Guild Melanoma Foundation R, CA
- 3.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Cheung M, Sharma A, Madhunapantula SV, Robertson GP. Akt3 and mutant V600E BRaf cooperate to promote early melanoma development. Cancer Res. 2008;68:3429–39. doi: 10.1158/0008-5472.CAN-07-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenmann KM, VanBrocklin MW, Staffend NA, Kitchen SM, Koo HM. Mitogen-activated protein kinase pathway-dependent tumor-specific survival signaling in melanoma cells through inactivation of the proapoptotic protein bad. Cancer Res. 2003;63:8330–7. [PubMed] [Google Scholar]
- 6.Koo HM, VanBrocklin M, McWilliams MJ, Leppla SH, Duesbery NS, Woude GF. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc Natl Acad Sci U S A. 2002;99:3052–7. doi: 10.1073/pnas.052707699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang YF, Jiang CC, Kiejda KA, Gillespie S, Zhang XD, Hersey P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin Cancer Res. 2007;13:4934–42. doi: 10.1158/1078-0432.CCR-07-0665. [DOI] [PubMed] [Google Scholar]
- 8.Karst AM, Li G. BH3-only proteins in tumorigenesis and malignant melanoma. Cell Mol Life Sci. 2007;64:318–30. doi: 10.1007/s00018-006-6364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 10.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 11.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 12.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A. 2001;98:9666–70. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:R883. doi: 10.1016/s0960-9822(00)00843-5. [DOI] [PubMed] [Google Scholar]
- 14.Yu C, Minemoto Y, Zhang J, et al. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol Cell. 2004;13:329–40. doi: 10.1016/s1097-2765(04)00028-0. [DOI] [PubMed] [Google Scholar]
- 15.Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–69. [PubMed] [Google Scholar]
- 16.Schmelzle T, Mailleux AA, Overholtzer M, et al. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A. 2007;104:3787–92. doi: 10.1073/pnas.0700115104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cartlidge RA, Thomas GR, Cagnol S, et al. Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 2008 doi: 10.1111/j.1755-148X.2008.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–6. doi: 10.1074/jbc.M301010200. [DOI] [PubMed] [Google Scholar]
- 19.Smalley KS, Contractor R, Haass NK, et al. Ki67 expression levels are a better marker of reduced melanoma growth following MEK inhibitor treatment than phospho-ERK levels. Br J Cancer. 2007;96:445–9. doi: 10.1038/sj.bjc.6603596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeh AH, Bohula EA, Macaulay VM. Human melanoma cells expressing V600E B-RAF are susceptible to IGF1R targeting by small interfering RNAs. Oncogene. 2006;25:6574–81. doi: 10.1038/sj.onc.1209674. [DOI] [PubMed] [Google Scholar]
- 21. http://www.sanger.ac.uk/genetics/CGP/Studies/ [Google Scholar]
- 22.Chen M, Granger AJ, Vanbrocklin MW, et al. Inhibition of avian leukosis virus replication by vector-based RNA interference. Virology. 2007;365:464–72. doi: 10.1016/j.virol.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Verhaegen M, Bauer JA, Martin de la Vega C, et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006;66:11348–59. doi: 10.1158/0008-5472.CAN-06-1748. [DOI] [PubMed] [Google Scholar]
- 24.Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–72. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 25.Abi-Habib RJ, Urieto JO, Liu S, Leppla SH, Duesbery NS, Frankel AE. BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol Cancer Ther. 2005;4:1303–10. doi: 10.1158/1535-7163.MCT-05-0145. [DOI] [PubMed] [Google Scholar]
- 26.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–93. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 28.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 29.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 30.Puthalakath H, Villunger A, O'Reilly LA, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- 31.Day CL, Puthalakath H, Skea G, et al. Localization of dynein light chains 1 and 2 and their pro-apoptotic ligands. Biochem J. 2004;377:597–605. doi: 10.1042/BJ20031251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Smits C, Czabotar PE, Hinds MG, Day CL. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure. 2008;16:818–29. doi: 10.1016/j.str.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006;13:129–40. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 35.Ramjaun AR, Tomlinson S, Eddaoudi A, Downward J. Upregulation of two BH3-only proteins, Bmf and Bim, during TGF beta-induced apoptosis. Oncogene. 2007;26:970–81. doi: 10.1038/sj.onc.1209852. [DOI] [PubMed] [Google Scholar]
- 36.Morales AA, Gutman D, Lee KP, Boise LH. BH3-only proteins Noxa, Bmf, and Bim are necessary for arsenic trioxide-induced cell death in myeloma. Blood. 2008;111:5152–62. doi: 10.1182/blood-2007-10-116889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karst AM, Dai DL, Martinka M, Li G. PUMA expression is significantly reduced in human cutaneous melanomas. Oncogene. 2005;24:1111–6. doi: 10.1038/sj.onc.1208374. [DOI] [PubMed] [Google Scholar]
- 38.Letai A. Restoring cancer's death sentence. Cancer Cell. 2006;10:343–5. doi: 10.1016/j.ccr.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–25. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Show MD, Hill CM, Anway MD, Wright WW, Zirkin BR. Phosphorylation of mitogen-activated protein kinase 8 (MAPK8) is associated with germ cell apoptosis and redistribution of the Bcl2-modifying factor (BMF). J Androl. 2008;29:338–44. doi: 10.2164/jandrol.107.003558. [DOI] [PubMed] [Google Scholar]
- 41.Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–46. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menon SS, Whitfield LR, Sadis S, et al. Pharmacokinetics (PK) and pharmacodynamics (PD) of PD 032901, a second generation MEK inhibitor after multiple oral doses of PD 032901 to advanced cancer patients.. 2005 ASCO Annual Meeting; Chicago, IL. 2005 June 1.2005. [Google Scholar]; Journal of Clinical Oncology; ASCO Annual Meeting Proceedings.; Jun 1, 2005. 2005. p. 3066. Part I of II. 2005. p. 3066. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.