

Abstract
Reduced penetrance in genetic disorders may be either dependent or independent of the genetic background of gene carriers. Hirschsprung disease (HSCR) demonstrates a complex pattern of inheritance with ≈50% of familial cases being heterozygous for mutations in the receptor tyrosine kinase RET. Even when identified, the penetrance of RET mutations is only 50–70%, gender-dependent, and varies with the extent of aganglionosis. We searched for additional susceptibility genes which, in conjunction with RET, lead to phenotypic expression by studying 12 multiplex HSCR families. Haplotype analysis and extensive mutation screening demonstrated three types of families: six families harboring severe RET mutations (group I); and the six remaining families, five of which are RET-linked families with no sequence alterations and one RET-unlinked family (group II). Although the presence of RET mutations in group I families is sufficient to explain HSCR inheritance, a genome scan reveals a new susceptibility locus on 9q31 exclusively in group II families. As such, the gene at 9q31 is a modifier of HSCR penetrance. These observations imply that identification of new susceptibility factors in a complex disease may depend on classification of families by mutational type at known susceptibility genes.
A major hallmark of phenotypes displaying a complex pattern of inheritance is the segregation of multiple factors. In principle, linkage analysis on a collection of families can identify multiple loci, although an individual family may segregate only a few of the relevant genes. Multilocus segregation is compatible with two distinct hypotheses. Thus, a phenotype could be multigenic or multifactorial, resulting from the cumulative effects of mutations in multiple genes; disease expression and penetrance may then be sensitive to an individual's genetic background. This hypothesis is in contrast to the simple association of a disorder with multiple genes, genetic (locus) heterogeneity, in which mutations in any one of several genes are sufficient for phenotypic expression and are independent of genetic background. In either scenario, linkage heterogeneity exists, but, for the multigenic hypothesis to prevail, we need to demonstrate multiple gene segregation in the same families (1). Although unequivocal evidence for multigenic inheritance exists in the many rodent models of human disease, and is strongly suspected in complex human disorders, it remains largely unproven. However, in the human, in rare instances, retinitis pigmentosa can arise from digenic inheritance (2); segregation of insulin-dependent diabetes mellitus depends on interactions between both HLA and an X-linked gene (3).
Hirschsprung disease (HSCR), or congenital aganglionosis, is a model complex genetic disorder in which we can test these hypotheses. HSCR is associated with the lack of intrinsic ganglion cells in the myenteric and submucosal plexuses of the distal gastrointestinal tract. The phenotype has ≈100% heritability (4) but shows variable presentation and reduced penetrance, even in multiplex kindreds, with affected individuals differing in the length of aganglionosis, association with pigmentary anomalies and sensorineural deafness, and severity of intestinal blockage. Segregation analyses (4) of HSCR have established two genetic hallmarks: (i) recurrence risks are ≈ 3–25%, with a non-Mendelian pattern of inheritance; and (ii) recurrence risk to relatives varies with length of aganglionosis and gender of the proband. Molecular genetic analyses have identified five functionally dependent HSCR susceptibility genes in pathways crucial for the normal development of the enteric nervous system (5–18).
The major HSCR gene is the receptor tyrosine kinase RET, in which coding sequence mutations can be identified in 50% of familial and 35% of sporadic cases (19–21). The functional ligand for RET is the type β transforming growth factor-like glial cell line-derived neurotrophic factor (GDNF), which activates RET through a three-component complex by using the glycosylphosphatidylinositol-linked anchor protein GFRA1 (glial cell line-derived neurotrophic factor-family receptor) (8, 9). Mutations in GDNF are rare in HSCR (10–13); no mutations in GFRA1 have been revealed on extensive testing (14). A second biochemical pathway compromised in HSCR is encoded by the G-protein coupled endothelin-B receptor EDNRB (15) and its ligand endothelin-3 (EDN3); ≈5% of familial and sporadic HSCR cases are explained by mutations in these genes (16). Finally, mutations in the gene encoding the transcription factor SOX10, which regulates EDNRB expression (17), have also been identified in multiple patients with HSCR and pigmentary anomalies (Shah-Waardenburg syndrome) (17, 18).
In addition to finding mutations in single genes, we and others have presented preliminary evidence of multigenic inheritance in HSCR. In a large inbred Mennonite kindred segregating HSCR, we identified a hypomorphic change (W276C) in EDNRB on chromosome 13q22 and demonstrated significant nonrandom association between HSCR and polymorphisms within the RET gene (15, 16). In addition, the penetrance of HSCR in this kindred depends on genetic interaction between RET and EDNRB because parent-to-offspring transmission of W276C is significantly associated with transmission of multiple polymorphisms within RET (ref. 16; M. Carrasquillo and A.C., unpublished data). Extensive sequence analysis of the RET coding sequence has failed to identify a mutation in the Mennonites. Nevertheless, the strong association between HSCR and RET, and association between the joint transmission of RET and EDNRB to patients, emphasizes that EDNRB in conjunction with RET determines HSCR susceptibility in this kindred and that the RET mutation is located in the non-coding regions. Additionally, in some outbred families, GDNF mutations do not appear to act alone but co-occur with mutations in RET or trisomy 21 (10, 11). These data underscore the complex inheritance and provide the first clues that multiple genes may be required to modulate the expression of HSCR.
To test whether HSCR results from multigene segregation in outbred families, we have focused attention on 12 families showing vertical transmission. In such families, few segregating loci are expected a priori because they largely, not exclusively, segregate long-segment HSCR and have higher penetrance than short-segment HSCR (4, 21). We chose families with three or more affecteds across two or more generations with phenotypic characteristics typical of other reported large families. The variable expressivity and reduced penetrance in these families may be explained by environmental effects, stochastic developmental events, or multigenic inheritance. To distinguish between these hypotheses, we conducted allele-sharing tests and linkage analysis to identify multiple segregating genomic regions, followed by mutation analysis of known genes. We present data that demonstrate the segregation of RET in the majority (11/12) of families and additional segregation of a new locus at 9q31. Mutation analysis identified severe RET mutations in only 50% of families. Importantly, 9q31 segregation was limited to those six families lacking coding sequence missense or nonsense RET mutations. Thus, the nature of the RET mutation in HSCR patients is a strong predictor of the presence (segregation) of additional genetic changes, supporting multigenic inheritance. A corollary of these observations is that HSCR patients heterozygous for multiple mutations have weak (hypomorphic) changes at RET and, thus, explains the reduced RET penetrance.
Materials and Methods
Family Ascertainment.
Multiplex families segregating HSCR were ascertained under informed consent and were classified into those with affected siblings only and those with three or more affected individuals in two or more generations. The latter families show vertical transmission of HSCR and have higher penetrance. We identified 12 families for this study, of which families 1–6 and 8–10 have been reported previously (21, 22); the Mennonite families (15) are not a part of this study. Genomic DNA was extracted and lymphoblastoid cell lines were established from participating members as described (22).
Linkage Analysis.
Polymorphic microsatellite markers were genotyped by using standard methods (22), and allele sizes were determined by using Centre d'Etude du Polymorphisme Humain (CEPH) individual 1347-02 as a genotyping standard. Parametric linkage analysis is powerful, but the major hindrance is the lack of an accurate inheritance model (23) and concomitant reduction of power of detecting linkage when parameters are specified incorrectly. One cannot a priori predict the number of different loci, the mutant allele frequency at each locus, and the penetrance associated with each HSCR susceptibility allele. We, therefore, relied on model-free tests of allele-sharing at marker loci among all affecteds within each family and searched for regions in which allele-sharing exceeds Mendelian expectations. All multipoint parametric and nonparametric linkage analyses were performed by using the computer program genehunter 1.1 (24). The genetic markers used for linkage analysis had intermarker map distances as published (25); allele frequencies from CEPH families were used for these markers and were not different from those estimated from founders in the 12 families. For lod score analysis, we specified an autosomal-dominant mode of inheritance, a mutant allele frequency of 1%, and penetrance of 58% in males and 29% in females; these estimates are based on our previous segregation analysis (22). Linkage analyses were also performed allowing for heterogeneity, by using the “Total Heterogeneity” command in genehunter; we calculated both a heterogeneity lod score and estimated the proportion of linked families (α). To compute the posterior probability of linkage on chromosome 10, we placed the disease locus at the RET intron 5 marker and used Eq. 9.11 in ref. 23; linkage was declared whenever the posterior probability exceeded the estimated α. Nonparametric analysis used the nonparametric linkage (NPL) score and the “sharing all” option.
Mutation Detection.
Mutation analysis was performed on all 21 exons of RET comprising 3,474 bp and 1,700 bp of adjacent intronic sequence by using published human cDNA and partial genomic sequences (26–29) as wild-type standards. Single-strand conformational polymorphism, denaturing gradient gel electrophoresis, and automated nucleotide sequencing using dye terminator chemistry were performed by standard methods (6, 14, 21).
RNA Isolation and Reverse Transcription (RT)–PCR.
Total RNA was extracted from lymphoblastoid cell lines by using RNAzolB (Cinna/Biotecx Laboratories, Friendswood, TX); extractions were performed twice to help reduce DNA contamination. RNA (5 μg) was reverse transcribed by using SuperscriptII (GIBCO/BRL) with random hexamer primers, and 3 μl of the RT reaction was used for subsequent 50-μl PCR amplifications. Primers for specific amplification of RET exon 11 were designed such that forward and reverse oligonucleotides were in exon 10 and 12, respectively, so that the product would span introns and control for genomic DNA contamination. The primers used were RETX10F: 5′-TTCCCTGAGGAGGAGAAGTGC-3′ and RETX12R: 5′-CCACTTTTCCAAATTCGCCTT-3′; these were used in amplification reactions as follows: 30 s at 94°C; 30 s at 59°C; and 30 s at 72°C for 35 cycles.
Results
RET Is the Major Gene for HSCR.
Human chromosome 10 is the site of two HSCR genes, RET at 10q11 (19, 20) and GFRA1 at 10q25 (14). We searched for linkage in 12 multiplex families segregating HSCR (Fig. 1) by using 22 polymorphic microsatellite markers spanning the entire genetic length of chromosome 10 and at a resolution of 8 centimorgans (cM) (Fig. 2a). We performed both parametric and nonparametric multipoint linkage analysis using the computer program genehunter (24). We detected significant linkage with the RET locus with a nonparametric linkage (NPL) score of 11.3 (P = 7.7 × 10−8) and a lod score of 9.4 (Table 2; Fig. 2a). No linkage was evident at the GFRA1 locus consistent with our previous observations (14). Haplotype analysis using markers flanking and within RET demonstrated that all families except 12 were linked to RET (Table 1). Consequently, RET is the major segregating factor in multigenerational HSCR families, but additional genes must also exist. We demonstrated significant linkage heterogeneity (χ2 = 5.1, P = 0.023) and obtained a multipoint heterogeneity lod score of 10.5 at RET with α = 91% of families linked (23). Calculations of the posterior probability of linkage for each family showed that 11 of the 12 families have a >97% probability of being linked to RET (Table 1). There was no evidence for a common RET haplotype in the linked families.
Figure 1.

HSCR families used in linkage analysis. Individuals with confirmed HSCR and clinical diagnosis of chronic severe constipation are indicated by filled and shaded symbols, respectively. Asterisks indicate individuals used in genotyping.
Figure 2.
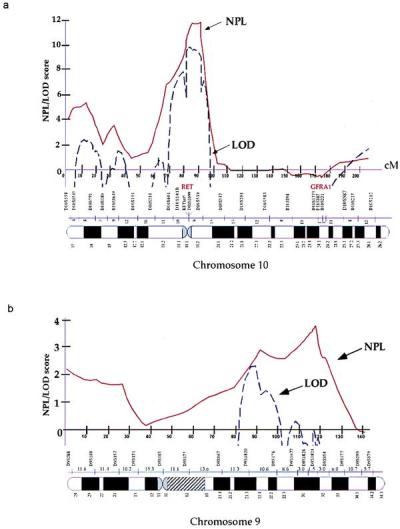
Linkage analysis of HSCR. (a) Chromosome 10. (b) Chromosome 9. The x axis indicates the genetic map of the indicated chromosome with intermarker distances in centimorgans (cM), and the y axis indicates the linkage score. (Solid line, NPL score; dashed line, parametric lod score. Although depicted on the same graph, NPL and lod scores have distinct properties and are not synonymous; thus, they should not be numerically compared against each other.)
Table 2.
Linkage analysis of chromosome 10 and 9 genetic markers and family classification by RET mutational type
| Family type | RET
|
9q31 locus
|
||
|---|---|---|---|---|
| NPL (P value) | lod score (α) | NPL (P value) | lod score (α) | |
| Missense/nonsense mutations, | 9.1 | 7.7 | 0.4 | 0.0 |
| families 1–6 | (6.2 × 10−5) | (1.00) | (0.5) | (0) |
| Splice variants/no detected | 5.3 | 3.9 | 5.3 | 4.8 |
| changes, families 7–12 | (4.5 × 10−5) | (0.86) | (6.8 × 10−5) | (1.0) |
| All families | 11.3 | 10.5 | 3.8 | 2.3 |
| (7.7 × 10−8) | (0.91) | (0.002) | (0.46) | |
Families were classified according to presence (families 1–6) or absence (families 7–12) of missense or nonsense mutation. Nonparametric analysis provided the NPL score and P value; parametric analysis provided the heterogeneity lod score and the estimated proportion of linked families (α) (see Materials and Methods).
Table 1.
Segregation of RET and a chromosome 9q31 locus in HSCR families
| Family | Posterior probability of RET linkage* | RET†
|
9q31 haplotype shared‡ | ||
|---|---|---|---|---|---|
| Mutation | Type | Evolutionary conservation | |||
| 1 | 1.00 | N394K | n | m, c, z | no |
| 2 | 0.99 | R360W | nc | m | no |
| 3 | 0.99 | R77C | nc | m, c | no |
| 4 | 1.00 | S32L+ | nc | m | no |
| 5 | 0.99 | W942X+ | — | m, c, z, d | no |
| 6 | 0.99 | E921X+ | — | m, c, z, d | no |
| 7 | 0.99 | S649S | — | — | yes |
| 8 | 0.98 | IVS17 + 5G > A | — | — | yes |
| 9 | 0.97 | ND | — | — | yes |
| 10 | 1.00 | ND | — | — | yes |
| 11 | 0.99 | ND | — | — | yes |
| 12 | 0.005 | ND | — | — | yes |
Posterior probability of RET linkage (see Materials and Methods): haplotype sharing among all affecteds within each family, based on markers D10S141-RETint5-D10S1099, was consistent with posterior probability estimates (shared in families 1–11; not shared in family 12).
† RET mutations in exons [+, reported previously (7); ND, none detected]; listed are mutational change, residue change classified as nonconservative (nc) or neutral (n), and evolutionary conservation of human residue (m, c, z, and d denoting conservation in mouse, chicken, zebrafish, and Drosophila, respectively).
‡ Haplotype sharing within each family based on markers D9S176-D9S1832-D9S1677-D9S1828.
Diverse RET Mutations in HSCR.
To determine the nature of the mutations in RET, we conducted extensive mutation analysis of all probands in the linked families (Table 1). We identified eight putative mutations distributed across the RET gene in the 11 linked families relative to reference sequence data (26–29) and studies of control individuals: four probands had missense changes (N394K, R360W, R77C, and S32L in families 1–4, respectively), two probands had nonsense mutations (W942X in family 5 and E921X in family 6), and two families harbored sequence alterations that could affect normal splicing (S649S in family 7 and IVS17 + 5G → A in family 8). The synonymous Serine change at residue 649 (TCG → TCA) in exon 11 of the RET gene has previously been observed (11) in one patient. With the observation of this second mutation, we sought to determine whether this was a polymorphism. The G-to-A nucleotide change created a DdeI restriction site and allowed a rapid screen of 60 CEPH control individuals and 100 HSCR probands; we did not encounter this again in 320 chromosomes. The second splicing mutation occurs in intron 17 in the consensus splice donor site, altering the sequence of the splice donor site from (exon/intron) AG/GTGAGT to AG/GTGAAT. The remaining three RET-linked families (families 9–11) have been extensively screened, but no causative sequence changes could be identified.
To assess which changes might be functional mutations, and which benign rare variants, we also compared the nature of the residue altered (conservative, nonconservative, or neutral) and the evolutionary conservation of that amino acid by protein sequence alignment of human, mouse, chicken, zebrafish, and Drosophila RET. As shown in Table 1, the changes in families 1–4 are either nonconservative residue changes or alterations at evolutionarily conserved residues and likely lead to diminished protein function. The mutations in families 5 and 6 are nonsense changes at very highly conserved sites. Thus, we conclude that the sequence alterations in families 1–6 are RET mutations that lead to HSCR. These mutations are likely loss-of-function variants.
The RET S649S Synonymous Change Is a Variant Splicing Mutation.
The synonymous substitution S649S is a priori an unlikely candidate mutation. We hypothesized that the underlying nucleotide change (TCG → TCA) causes aberrant splicing of RET exon 11 because the substitution creates a sequence [CTTCATCGTCTCAGT] that closely resembles the consensus splice acceptor site [(ct)10ncagG] (Fig. 3a). If this novel site within exon 11 were used in splicing, then this would result in the deletion of the first 23 amino acids of exon 11. Given that exon 11 encodes the transmembrane domain of RET, it is likely that this deletion would not allow membrane anchoring and would be a loss-of-function allele.
Figure 3.

Functional consequences of the S649S variant in RET splicing. (a) Normal and mutant splicing events of RET exon 11 and its resulting translation. The site of the G → A transition observed in HSCR family 7 does not alter the Serine649 residue but does create a sequence closely resembling the consensus splice acceptor site [(ct)10ncagG]. (b) RT-PCR on RNA isolated from a lymphoblastoid cell line. One band of the expected size (383 bp) on normal splicing and a smaller band (314 bp) resulting from aberrant exon 11 splicing are amplified in affected carriers of the Serine variant (lanes 2–6). The unaffected grandmother (lane 1) who does not carry the S649S variant has only one RT-PCR product of the expected size (383 bp) on normal splicing. (c) BanI restriction digest of RT-PCR products. An expressed polymorphism in RET, which alters a BanI restriction site in exon 11, was used to analyze RT-PCR products. The wild-type band in lane 3 (labeled as 3+) and the mutant band in lane 3 (3−), as well as the band in lane 1 (1+), were gel-extracted. Two bands (239 bp and 144 bp) are observed after the digestion of lane 1, indicating homozygosity for the BanI site. Lane 3+ contains three bands: one undigested band (383 bp) and two bands resulting from the BanI digest (239 bp and 144 bp), indicating that the wild-type band observed in b has been amplified from both the wild-type and mutant alleles. The novel acceptor splice site is thus only partially used when present in an affected individual.
We used RT-PCR analysis (Fig. 3b) to show functionally that the mutation base at residue 649 creates a novel splice acceptor site within RET exon 11 that is used in splicing in lymphoblastoid cells. However, these data do not clarify whether the new splice acceptor site is used exclusively or not. To determine whether the RT-PCR band of wild-type molecular weight was a single molecular species or not, we used an expressed BanI polymorphism, G691S, in exon 11 for which carriers of the S649S variant were also heterozygous. As shown in Fig. 3c, the wild-type band consisted of amplification products from two sources: the transcript carrying the wild-type sequence and the transcript carrying the S649S mutation. Therefore, the novel splice site is only partially used. On this basis, we conclude that the S649S mutation is a hypomorphic allele and that carriers will express an intermediate level of RET. We were unable to assay the effect on splicing caused by IVS17 + 5G → A because of unavailability of cell lines from family 8.
A New HSCR Locus at Chromosome 9q31 Modifies RET Penetrance.
To search for additional susceptibility factors, we have begun a genome-scan in the 12 pedigrees, initially concentrating on chromosome 9 based on a two-point lod score of 1.51, in one family, with a microsatellite marker within the Hexabrachion locus at 9q32–34. We genotyped and performed linkage analysis using 16 polymorphic microsatellite markers spanning the entire genetic length of chromosome 9 at a map resolution of 12 cM (Fig. 2b). We obtained weak evidence of linkage with a peak NPL score of 3.8 (P = 0.002) between markers D9S1677 and D9S1828 (Table 2). Parametric analysis provided a peak lod score ≈20-cM proximal to the peak NPL value, probably because of incorrect specification of parameters in the analysis. Further, we obtained a peak heterogeneity lod score of 2.3 with α = 46% of families linked. These results are consistent with haplotype analysis (Table 1; Fig. 2b), which shows segregation of the 9q31 locus in families 7–12 only. There was no evidence for a common 9q31 haplotype in the linked families.
The results of mutation analysis at RET and linkage analysis on chromosome 9 suggested classification of the 12 families into two groups: RET-linked families 1–6 (group I), which carried severe nonsense and missense coding sequence RET mutations but no segregation at 9q31; and those without RET missense or nonsense mutations (families 7–12, group II) but segregation at 9q31. This association between RET mutation type and 9q31 segregation is highly significant (χ2 = 12, 2 df, P = 0.0025) and demonstrates that segregation of the additional 9q31 locus is restricted to families that do not have missense or nonsense RET mutations. We reanalyzed the linkage data by the type of RET mutation (Fig. 4) and show significant linkage in the group II families with a NPL score of 5.3 (P = 6.8 × 10−5) between markers D9S1677 and D9S1828; as expected, no evidence of linkage was obtained in the group I families. Analogous parametric analysis showed a peak lod score of 4.8 in group II families at the same map location defined by the peak NPL score; no significant linkage to 9q31 was evident in group I families.
Figure 4.
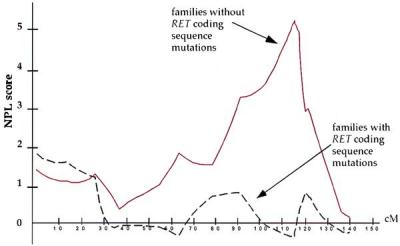
Nonparametric linkage (NPL) analysis of chromosome 9 markers in HSCR on subdividing families by RET mutation status. Dashed line, those with RET coding sequence mutations (families 1–6); solid line, those without RET coding sequence mutations (families 7–12).
To distinguish between multigenic inheritance and locus heterogeneity, we next reanalyzed RET linkage data by family type. As shown in Table 2, RET linkage is significant in both group I and II families with NPL scores of 9.1 (P = 6.2 × 10−5) and 5.3 (P = 4. 5 × 10−5), respectively. Under parametric analysis, significant lod scores were also observed in both groups (lod = 7.7, α = 100% in group I; lod = 3.9, α = 86% in group II). In group I families, RET is the sole contributory factor because there is no evidence of segregation of the 9q31 locus. In the remaining group II families, there is overall significant evidence of linkage to both RET and 9q31 using NPL and lod score analysis. One family (family 12) in this latter group, analogous to those in group I, might harbor a severe mutation in the 9q31 gene only. These data clearly demonstrate the existence of at least two types of HSCR families: families with missense or nonsense RET mutations sufficient to lead to HSCR and those with weak alleles in which additional genetic changes are required for disease expression. We suspect that RET-linked families without recognized mutations (families 9–11) either harbor common polymorphic variants (excluded from being considered as mutations) or noncoding sequence changes that make partial contributions to HSCR risk. The demonstration of 9q31 segregation in these latter families suggest both multigenic inheritance and that HSCR results from the cumulative genetic effects at RET and the yet unknown 9q31 locus in some families.
Discussion
Segregation of Hirschsprung disease is decidedly non-Mendelian; in most families, the phenotype is variable and the mutant genotype displays reduced penetrance (4). The central genetic question is, therefore, whether the reduced penetrance is random and dependent on environmental or stochastic developmental factors, nonrandom and dependent on an individual's genetic background, or both. We have presented significant evidence that the latter is true because HSCR shows multigenic inheritance with RET-mediated susceptibility being the major theme. Moreover, additional genes with large effect, such as the one we have mapped to 9q31, must also exist.
Significant evidence argues that RET is the major gene for HSCR and is involved in ≈80% of families (6, 19–21, 33). Nevertheless, investigators have failed to find coding sequence mutations in >50% of familial cases (6–8, 21). This inability to detect RET coding sequence mutations in linked families is not attributable to mutation detection technology but is a persistent finding that requires alternative explanations, such as: (i) RET linkage is spurious in small families; (ii) a gene closely linked to RET harbors susceptibility mutations; (iii) mutations other than those in the RET coding sequence, including introns and the promoter regions, compromise RET function; and (iv) RET is under epigenetic regulation that can affect its normal expression and function.
Scenario i is undoubtedly true for some families but cannot be the major explanation because the sum total of families without identified RET mutations still demonstrates linkage to chromosome 10q11 (Table 2). Indeed, family 12 reported here is the first description of a multiplex family in which RET-mediated susceptibility can be definitively excluded. Scenario ii, in which a gene different from, but tightly linked to, RET contributes to susceptibility, cannot be currently ruled out. However, our preliminary analysis of ≈500 kilobases of nucleotide sequence proximal to RET (GenBank accession no. AL022344) has not identified any specific transcripts that might be implicated. Rigorous identification and testing of all genes in this region is required and is currently underway. We conclude that the related scenarios iii and iv are the most likely explanations, both of which suggest misregulation of RET in HSCR.
In this study, we have demonstrated the diverse nature of RET mutations in HSCR (Table 1). Although functional tests have not been performed on each mutation, the genetic data strongly argue for loss-of-function mutations in group I families. The two nonsense mutations we observed, E921X and W942X, occur in the tyrosine kinase domain and are expected to abolish kinase activity as suggested by in vitro functional studies of similar mutants (34). Additional studies imply that the four missense mutations, S32L, R77C, R360W, and N394K, occur in the extracellular domain at residues that are either highly conserved and/or lead to radical substitutions, are expected to alter protein folding, and inhibit transport of RET protein to the cell surface (35–37). These observations, together with microdeletions of chromosome 10q11.1 including RET in rare patients, have supported the view that HSCR results from haplo-insufficiency because 50% RET cannot support normal enteric development in the human. In group I families, these severe mutations may be sufficient to lead to HSCR.
The group II families hold the key to further understanding of HSCR. Although these families show significant linkage to RET, only two splice variants, IVS17 + 5G → A and S649S, of which S649S is a weak RET allele (Fig. 3), have been identified. Such weak alleles compromise but do not abrogate RET activity and thus require additional genetic changes for HSCR expression. The segregation of 9q31 in group II families adds weight to this conclusion. In three of the remaining five (group II) families, despite a >97% probability of RET linkage, we have failed to identify any coding sequence RET mutations. We conclude that mutations in group II families are hypomorphs and harbor mutations in the noncoding regulatory regions. In addition, polymorphisms within the gene, which have been excluded as disease-causing, may contribute to disease risk as well.
Genetic disorders with variable expressivity and incomplete penetrance can arise from either structural or regulatory hypomorphic mutations. In familial retinoblastoma, structural mutations that result in only partial loss of protein function, rather than complete abrogation of normal gene function, are associated with incompletely penetrant forms of the disease (38, 39). In addition, splicing mutations with variable efficiency in usage of novel splice sites have been proposed as the molecular basis for reduced penetrance in cystic fibrosis (40). Epigenetic (regulatory) modification of a gene, such as through position effects, is another type of mutation that may be responsible for reduced penetrance. First, position effect mutations can occur hundreds of kilobases away from the target gene, explaining both linkage and failure to observe coding mutations (41). Second, position effect changes have phenotypes similar to loss-of-function mutations; several examples are known in the human and mouse (41). The best-studied is the receptor tyrosine kinase c-kit, known to be subject to position effect variegation as a result of specific mutations affecting chromosomal rearrangement and, as a consequence, control elements of the gene. In this case, gene function is not completely abolished, but the effect of the mutation represses transcription in a tissue-specific manner (42). Similar factors may be operating in HSCR.
We postulate that the degree of RET expression is critical for HSCR expression: severe alleles lead to HSCR directly; weak alleles also require the effects of a 9q31 gene. A corollary is that the 9q31 gene is a modifier of HSCR penetrance. Consequently, the 9q31 gene could be a member of a signaling pathway that affects RET expression, and a “hit” at multiple locations within the pathway may be the cause of HSCR in families with weak RET mutations. Proving this hypothesis will require identification of this gene; unfortunately, the current linkage location is still too broad (95% support limits are ≈10 cM) to allow positional cloning. It is possible that this 9q31 gene is the same as that which is mutant in Riley-Day syndrome [Mendelian Inheritance in Man (MIM) 223900]. The latter, also known as familial dysautonomia, is a hereditary neuropathy that results from a loss of subsets of autonomic neurons and is associated with lack of tearing, emotional lability, paroxysmal hypertension, increased sweating, cold hands and feet, corneal anesthesia, and blotching of the skin. Riley-Day syndrome, common in Ashkenazi Jews, has been shown to co-occur with HSCR and has been mapped to an ≈150-kilobase segment in 9q31 (43). This gene, when identified, will be an excellent candidate gene for HSCR.
Acknowledgments
We are indebted to the HSCR families for participation and the American Pseudo-obstruction & Hirschsprung Disease Society, Inc., for cooperation. We gratefully acknowledge the work of Jennifer Scott for family studies, James Gabriel for advice with RNA studies, Kimberly Bentley and Carl Kashuk for excellent technical assistance, and Drs. Hunt Willard and Evan Eichler for comments on the manuscript. This work was supported by a grant from the National Institutes of Health (HD28088) to A.C.
Abbreviations
- HSCR
Hirschsprung disease
- NPL
nonparametric linkage
- RT
reverse transcription
- cM
centimorgans
References
- 1.Chakravarti A, Lander E S. In: Genetics and Biology of Alcoholism. Cloninger C R, Begleiter H, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. pp. 307–315. [Google Scholar]
- 2.Kajiwara K, Berson E L, Dryja T P. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 3.Cucca F, Goy J V, Kawaguchi Y, Esposito L, Merriman M E, Wilson A J, Cordell H J, Bain S C, Todd J A. Nat Genet. 1998;19:301–302. doi: 10.1038/995. [DOI] [PubMed] [Google Scholar]
- 4.Badner J A, Sieber W K, Garver K L, Chakravarti A. Am J Hum Genet. 1990;46:568–580. [PMC free article] [PubMed] [Google Scholar]
- 5.Edery P, Pelet A, Mulligan L M, Abel L, Attie T, Dow E, Bonneau D, David A, Flintoff W, Jan D, et al. J Med Genet. 1994;31:602–606. doi: 10.1136/jmg.31.8.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angrist M, Bolk S, Thiel B, Puffenberger E G, Hofstra R M, Buys C H C M, Cass D T, Chakravarti A. Hum Mol Genet. 1995;4:821–830. doi: 10.1093/hmg/4.5.821. [DOI] [PubMed] [Google Scholar]
- 7.Attie T, Till M, Pelet A, Amiel J, Edery P, Boutrand L, Munnich A, Lyonnet S. Hum Mol Genet. 1995;4:2407–2409. doi: 10.1093/hmg/4.12.2407. [DOI] [PubMed] [Google Scholar]
- 8.Treanor J J S, Goodman L, De Sauvage F, Stone D M, Poulsen K T, Beck C D, Gray C, Armanini M P, Pollock R A, Hefti F, et al. Nature (London) 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 9.Trupp M, Arenas E, Fainzilber M, Nilsson A-S, Sieber B-A, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumae U, et al. Nature (London) 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 10.Angrist M, Bolk S, Halushka M, Lapchak P L, Chakravarti A. Nat Genet. 1996;14:341–344. doi: 10.1038/ng1196-341. [DOI] [PubMed] [Google Scholar]
- 11.Salomon R, Attie T, Pelet A, Bidaud C, Eng C, Amiel J, Sarnacki S, Goulet O, Ricour C, Nihoul-Fekete C, et al. Nat Genet. 1996;14:345–347. doi: 10.1038/ng1196-345. [DOI] [PubMed] [Google Scholar]
- 12.Hofstra R M, Osinga J, Buys C H. Eur J Hum Genet. 1997;5:180–185. [PubMed] [Google Scholar]
- 13.Ivanchuk S M, Myers S M, Eng C, Mulligan L M. Hum Mol Genet. 1996;5:2023–2026. doi: 10.1093/hmg/5.12.2023. [DOI] [PubMed] [Google Scholar]
- 14.Angrist M, Jing S, Bolk S, Bentley K, Nallasamy S, Halushka M, Fox G, Chakravarti A. Genomics. 1998;48:354–362. doi: 10.1006/geno.1997.5191. [DOI] [PubMed] [Google Scholar]
- 15.Puffenberger E G, Hosoda K, Washington S S, Nakao K, deWitt D, Yanagisawa M, Chakravarti A. Cell. 1994;79:1257–1266. doi: 10.1016/0092-8674(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 16.Chakravarti A. Hum Mol Genet. 1996;5:303–307. [PubMed] [Google Scholar]
- 17.Southard-Smith E M, Angrist M, Ellison J, Agarwala R, Baxevanis A, Chakravarti A, Pavan W J. Genome Res. 1999;9:215–225. [PubMed] [Google Scholar]
- 18.Pingault V, Bondurnad N, Kuhlbrodt K, Goerish D E, Prehu M, Puliti A. Nat Genet. 1998;18:171–173. doi: 10.1038/ng0298-171. [DOI] [PubMed] [Google Scholar]
- 19.Romeo G, Ronchetto P, Luo Y, Barone V, Seri M, Ceccherini I, Pasini B, Bocciardi R, Lerone M, Kaarianen H, Martucciello G. Nature (London) 1994;367:377–378. doi: 10.1038/367377a0. [DOI] [PubMed] [Google Scholar]
- 20.Edery P, Lyonnet S, Mulligan L M, Pelet A, Dow E, Abel L, Holder S, Nihoul Fekete C, Ponder B A, Munnich A. Nature (London) 1994;367:378–380. doi: 10.1038/367378a0. [DOI] [PubMed] [Google Scholar]
- 21.Attie T, Pelet A, Edery P, Eng C, Mulligan L M, Amiel J, Boutrand L, Beldjord C, Nihoul Fekete C, Munnich A, et al. Hum Mol Genet. 1995;4:1381–1386. doi: 10.1093/hmg/4.8.1381. [DOI] [PubMed] [Google Scholar]
- 22.Angrist M, Kauffman E, Slaugenhaupt S A, Matise T C, Puffenberger E G, Bolk S, Washington S S, Lipson A, Cass D T, Reyna T, et al. Nat Genet. 1993;4:351–356. doi: 10.1038/ng0893-351. [DOI] [PubMed] [Google Scholar]
- 23.Ott J. Analysis of Human Genetic Linkage. Baltimore: Johns Hopkins Univ. Press; 1991. [Google Scholar]
- 24.Kruglyak L, Daly M J, Reeve-Daly M P, Lander E S. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 25.Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, et al. Nature (London) 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 26.Ceccherini I, Bocciardi R, Luo Y, Pasini B, Hofstra R, Takahashi M, Romeo G. Biochem Biophys Res Commun. 1993;196:1288–1295. doi: 10.1006/bbrc.1993.2392. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H. Oncogene. 1988;3:571–578. [PubMed] [Google Scholar]
- 28.Takahashi M, Buma Y, Hiai H. Oncogene. 1989;4:805–806. [PubMed] [Google Scholar]
- 29.Myers S M, Eng C, Ponder B A, Mulligan L M. Oncogene. 1995;11:2039–2045. [PubMed] [Google Scholar]
- 30.Kusafuka T, Wang Y, Puri P. J Pediatr Surg. 1997;32:501–504. doi: 10.1016/s0022-3468(97)90616-3. [DOI] [PubMed] [Google Scholar]
- 31.Hofstra R M W, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg E-J, Stulp R P, van Ravenswaaij-Arts C, Majoor-Krakauer D, Angrist M, et al. Nat Genet. 1996;12:445–447. doi: 10.1038/ng0496-445. [DOI] [PubMed] [Google Scholar]
- 32.Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra R M, Martelli H, Bidaud C, Munnich A, Lyonnet S. Nat Genet. 1996;12:442–444. doi: 10.1038/ng0496-442. [DOI] [PubMed] [Google Scholar]
- 33.Seri M, Yin L, Barone V, Bolino A, Celli I, Bocciardi R, Pasini B, Ceccherini I, Lerone M, Kristoffersson U, et al. Hum Mutat. 1997;9:243–249. doi: 10.1002/(SICI)1098-1004(1997)9:3<243::AID-HUMU5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 34.Pasini B, Borrello M G, Greco A, Bongarzone I, Luo Y, Mondellini P, Alberti L, Miranda C, Arighi E, Bocciardi R, et al. Nat Genet. 1995;10:35–40. doi: 10.1038/ng0595-35. [DOI] [PubMed] [Google Scholar]
- 35.Iwashita T, Murakami H, Asai N, Takahashi M. Hum Mol Genet. 1996;5:1577–1580. doi: 10.1093/hmg/5.10.1577. [DOI] [PubMed] [Google Scholar]
- 36.Carlomagno F, De Vita G, Berlingieri M T, de Franciscis V, Melillo R M, Colantuoni V, Kraus M H, Di Fiore P P, Fusco A, Santoro M. EMBO J. 1996;15:2717–2725. [PMC free article] [PubMed] [Google Scholar]
- 37.Pelet A, Geneste O, Edery P, Pasini A, Chappuls S, Attie T, Munnich A, Lenoir G, Lyonnet S, Billaud M. J Clin Invest. 1998;101:1415–1423. doi: 10.1172/JCI375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sakai T, Ohtani N, McGee T L, Robbins P D, Dryja T P. Nature (London) 1991;353:83–86. doi: 10.1038/353083a0. [DOI] [PubMed] [Google Scholar]
- 39.Otterson G A, Chen W, Coxon A B, Khleif S N, Kaye F J. Proc Natl Acad Sci USA. 1997;94:12036–12040. doi: 10.1073/pnas.94.22.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rarve-Harel N, Kerem E, Nissim-Rafinia M, Madjar I, Goshen R, Augarten A, Rahat A, Hurwitz A, Darvasi A, Kerem B. Am J Hum Genet. 1997;60:87–94. [PMC free article] [PubMed] [Google Scholar]
- 41.Bedell M A, Jenkins N A, Copeland N G. Nat Genet. 1996;12:229–232. doi: 10.1038/ng0396-229. [DOI] [PubMed] [Google Scholar]
- 42.Duttlinger R, Manova K, Chu T Y, Gyssler C, Zelenetz A D, Bachvarova R F, Besmer P. Development (Cambridge, UK) 1993;118:705–717. doi: 10.1242/dev.118.3.705. [DOI] [PubMed] [Google Scholar]
- 43.Slaugenhaupt S, Blumenfeld A, Liebert C B, Mull J, MacCormack K, LeBel A, O'Leary K, Breakefield X O, Maayan C, McCormick M K, et al. Am J Hum Genet. 1996;59:55. (abstr.). [Google Scholar]