

Abstract
The Alternative Reading Frame (ARF) protein suppresses tumorigenesis through p53-dependent and p53-independent pathways. Most of ARF’s anti-proliferative activity is conferred by sequences in its first exon. Previous work showed specific amino acid changes occurred in that region during primate evolution, so we programmed those changes into human p14ARF to assay their functional impact. Two human p14ARF residues (Ala14 and Thr31) were found to destabilize the protein while two others (Val24 and Ala41) promoted more efficient p53 stabilization and activation. Despite those effects, all modified p14ARF forms displayed robust p53-dependent anti-proliferative activity demonstrating there are no significant biological differences in p53-mediated growth suppression associated with simian versus human p14ARF residues. In contrast, p53-independent p14ARF function was considerably altered by several residue changes. Val24 was required for p53-independent growth suppression whereas multiple residues (Val24, Thr31, Ala41 and His60) enabled p14ARF to block or reverse the inherent chromosomal instability of p53-null MEFs. Together, these data pinpoint specific residues outside of established p14ARF functional domains that influence its expression and signaling activities. Most intriguingly, this work reveals a novel and direct role for p14ARF in the p53-independent maintenance of genomic stability.
Keywords: p14ARF, p53, Mdm2, p21, chromosomal instability, primate, cell cycle arrest
Introduction
The mammalian INK4a/ARF locus encodes two unrelated tumor suppressor proteins, p16INK4a and the alternative reading frame protein, ARF [1]. Both gene products share the same nucleotide sequences within a common second exon. ARF is specified by a unique first exon (exon 1β) that utilizes an overlapping reading frame in exon 2 to generate a protein that is structurally and functionally distinct from p16INK4a [2]. Whereas p16INK4a acts in the retinoblastoma (Rb) pathway, ARF protects against aberrant cell growth by activating the p53 tumor suppressor in response to oncogenic stimuli [1]. The significance of the INK4a/ARF locus to human health is illustrated by its high rate of inactivation in human cancer, second in frequency only to p53 [3,4].
The chief function of ARF (p14ARF in humans, p19ARF in mice) is to activate p53, a transcription factor that induces the expression of numerous apoptosis and cell cycle inhibitory genes, including p21WAF1/CIP1 [1]. ARF stabilizes and stimulates p53 activity by binding and inhibiting p53 antagonists, such as Mdm2, which normally ubiquitinates p53 and promotes its degradation [1]. ARF also has p53-independent antiproliferative and tumor suppressive activities [5–9] that are complex and poorly understood at the mechanistic level, but likely result from its association with a diverse array of cellular proteins [10]. ARF primarily resides in nucleoli due to its association with nucleophosmin (NPM) [11–13], although small fractions exist in the nucleoplasm and mitochondria [14–16]. Nucleoplasmic ARF is most effective at binding Mdm2 and activating p53 [11,14,17] whereas nucleolar and mitochondrial localization correlate best with ARF’s p53-independent activities [18–23].
ARF function is tightly conserved across species despite significant divergence in its nucleotide and amino acid sequences. Most of ARF’s growth inhibitory activities are conferred by its N-terminal residues encoded by exon 1β [14,24–27]. Residues 1 to 14 are highly conserved and constitute the minimal region required for Mdm2 binding, p53 activation, and growth suppression, although greater activity is observed for longer peptides [14,25,28,29]. The same N-terminal residues are required for ARF’s ability to inhibit rRNA synthesis and polysome formation [19,30], and they mediate ARF’s binding to most (but not all) of its other partners besides Mdm2. Notable exceptions are CtBP [31] and Foxm1b [32], which functionally associate with distinct N-terminal residues of mouse p19ARF (42–54 for CtBP and 26–44 for Foxm1b). Other residues (82–101 in p14ARF and 26–37 in p19ARF) promote ARF stability and cooperate with amino acids 1–14 to mediate ARF protein-protein associations, nucleolar localization, and the sumoylation of its partners [14,23–25,29,33,34].
We previously examined the sequence conservation of ARF exon 1β among 14 different species of primates including human, great apes, old world monkeys and the more divergent new world monkeys [35]. The sequences are nearly identical with the exception of several discrete amino acid variations that distinguished human ARF from its primate counterparts. The evolutionarily conserved nature of the amino acid changes implied they provide some functional advantage to human ARF. Here we tested the biological significance of those individual variations on human p14ARF function. Our findings identified single p14ARF residues distinct from previously recognized functional domains that regulate its stability and p53-independent activities.
Materials and methods
Cells, constructs and protein expression
Human U20S osteosarcoma cells, human kidney 293T cells, ARF−/− MEFs, and triple knockout (TKO) MEFs lacking ARF, p53, and Mdm2 (both MEFs kindly provided by Martine Roussel and Chuck Sherr [St. Jude Children’s Research Hospital]) were maintained in complete medium (DMEM containing 10% fetal bovine serum, 2 mM glutamine, and 100 μg/ml of penicillin and streptomycin). Complete medium was supplemented with 55 μM 2-mercaptoethanol and 0.1 mM non-essential amino acids for growth of MEFs.
The specific exon 1β amino acid changes representative of the different primates were programmed into human p14ARF by site-directed mutagenesis using a wild-type p14ARF cDNA template. Primer sequences for each construct will be readily provided upon request. Standard polymerase chain (PCR) reaction conditions were used with each set of primers containing the desired mutations, and all products were verified by sequencing in pCRII-Topo (Invitrogen) before subcloning into pEGFP-N1 or pMSCV-IRES-GFP plasmids.
For expression of GFP-tagged ARF proteins, U20S cells were transfected with pEGFP-N1 plasmids containing wild-type or mutated p14ARF cDNAs using FuGENE 6 transfection reagent (ROCHE) or by modified calcium phosphate precipitation [36]. For expression of untagged ARF proteins, bicistronic pMSCV-IRES-GFP plasmids were used to generate retroviruses encoding GFP alone (vector) or each p14ARF mutant plus GFP by calcium phosphate transfection of 293T cells. Sequential infections of low density MEF cultures (1.5 × 105 cells per 100 mm dish) were carried out as described [26], with a few changes. The first round of infection was performed using 4 ml of virus (supplemented with 8 μg per ml polybrene) for 6 hours or overnight, the second round performed with the same volume of virus for 24 hours, and 5 ml complete medium was then added until cells were harvested. Flow cytometry was performed after each infection to assess the percent of GFP-positive cells in the population (infection efficiency) using a Becton Dickinson FACScan.
Subcellular localization assays
Transfected U20S cells (ARF-GFP proteins) or infected MEFs (untagged ARF forms) were plated at 1 × 105 cells per glass coverslip in 6-well dishes. The next day, cells were fixed in 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 10 min. Samples were either immediately processed for nuclear staining (DAPI, 1 μg per ml for 1 min) or subjected to staining for ARF using anti-p14ARF mouse monoclonal antibodies (Sigma, 1 μg per ml) followed by DAPI staining, exactly as described [37]. ARF localization was examined by confocal microscopy (Zeiss) and quantified using GraphPad Prism software.
Proliferation and chromosomal instability
Cell cycle arrest and changes in percent tetraploidy (chromosomal instability) was assessed by flow cytometric analysis of DNA content using a FACScan (BD Biosciences) after staining nuclei with propidium iodide [2]. Flow cytometric profiles were analyzed using ModFit LT Version 2.0 software. Progression into S phase was measured by incorporation of the thymidine analog, bromodeoxyuridine (BrdU), into newly synthesized DNA by immunofluorescent staining of ARF and BrdU, exactly as described [24]. Growth curves of infected MEFs were generated by plating cells at 3 × 104 cells per well (12-well dishes) in duplicate or triplicate and counting cells every 2 days. The statistical significance of data compiled from at least three or more independent experiments was determined by a two-tailed, unpaired equal variance Student’s t-test.
p53 reporter assay
A p53 reporter construct, p53-luc (Stratagene), containing several p53 responsive promoter elements fused to firefly luciferase was used to assess p53 transcriptional activity. U20S cells were co-transfected with each p14ARF plasmid, p53-luc, plus a renilla luciferase construct pRL-SV40 (Promega, 80 ng) to normalize for transfection efficiency, as described [37]. Two days after transfection, cells (duplicate samples) were lysed and measured for luciferase activity using a Dual Luciferase Reporter Assay System (Promega) and SIRIUS Luminometer V3.1 (Berthold Detection Systems).
Protein Analyses
For Western blot analyses, frozen cell pellets were lysed on ice in NP-40 buffer (50 mM Tris, pH 7.5, 120 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.1 mM Na3VO4) supplemented with protease and phosphatase inhibitors. Lysates were incubated on ice for 30 min prior to sonication (5-s pulse) and clarification by microcentrifugation at 15,000 rpm for 10 min at 4°C. Equivalent amounts of total cellular protein (100 μg per sample) were separated by denaturing SDS-PAGE and transferred to PVDF (Millipore) membranes. Proteins were detected by enhanced chemiluminescence (ECL, Amersham) with the following antibodies: anti-GFP (Ab290 rabbit polyclonal, 1:5,000 dilution, Abcam), anti-p14ARF (DCS-240 mouse monoclonal, 2 μg per ml, Novus), anti-Mdm2 (2A10 mouse monoclonal, 1:20, kindly provided by Dr. Gerry Zambetti at St. Jude Children’s Research Hospital), anti-NPM (FC-61991 mouse monoclonal, 1 μg per ml, Zymed Laboratories), anti-p53 (Ab-7 sheep polyclonal, 1:500, Oncogene Research Products or D0-1 mouse monoclonal, 1 μg per ml), anti-p21 (human and mouse-specific antibodies from PharMingen used at suggested conditions), anti-GAPDH (Ab8245 mouse monoclonal, 1:10,000, Abcam) and anti- tubulin (GTU-88 mouse monoclonal, 1:10,000, Sigma).
For half-life studies, transfected U20S or infected ARF−/− MEFs (both p53-positive) were replated onto 6-well dishes, allowed to adhere overnight, and treated with cycloheximide (100 μg per ml) or left untreated (0 hour control) for various time points. Cells were harvested and p14ARF protein levels examined by Western blot analyses of whole cell lysates (100 μg per lane). Band intensities were quantified using NIH ImageJ software to determine half-lives for each protein.
Results
Human p14ARF and other primate forms of the protein are distinguished by just a few unique amino acid differences [35]. The exon 1β specific changes were differentially conserved among the simian subgroups, which consist of great apes, old world monkeys, and the more distantly related new world monkeys and prosimians (Figs. 1A and 1B). The most prominent difference is the change at residue 31 to threonine in human ARF from alanine in all other primates (Fig. 1A). Another major change occurred at residue 60; human and great apes encode a leucine, whereas monkeys (both old world and new world) encode a histidine. In addition to those differences, new world monkeys have several other distinct residues in the N-terminus, one of which (Ser14) lies within human p14ARF’s essential functional domain (residues 1–14).
Figure 1.

Evolutionary changes in the N-terminus of primate p14ARF proteins. A, alignment of amino acid sequences of exon 1β of different primate species. Seven different sequences are aligned from human (HU, Homo sapiens), great apes (GA, both Pan paniscus [pygmy chimpanzee] and Gorilla gorilla [gorilla]), old world monkeys (OM, both Papio ursinus [baboon] and Macaca arctoides [stump-tail macaque]), new world monkey (NM, Ateles belzebuth [long-haired spider monkey]) and prosimian (Pro, Varecia variegate [lemur]). p14ARF residues that are identical in mouse p19ARF are indicated by bolded lines above the human sequence. B, Schematic of the primate evolutionary tree depicting the timeline of divergence between various primates. M, millions of years.
To begin analyzing the effect of simian-specific exon 1β residues on human ARF function, p14ARF mutants bearing those changes were generated and fused at their C-terminal ends to green fluorescent protein (GFP) (Fig. 2A). Mutants are labeled according to the individual change(s) that were introduced relative to wild-type (WT) p14ARF although the multiple changes representative of new world monkeys (A14S;V24E;T31A;A41S;L60H) are abbreviated as “SEASH” for simplicity. The modified ARF proteins were transiently expressed in human U20S osteosarcoma cells (ARF-null), a well-established system used to examine p14ARF localization and function [14,24,25,38]. All of the p14ARF-GFP fusion proteins accumulated predominantly in the nucleoli (Supplementary Fig. S1), showing no effect of primate specific residues on human ARF subcellular distribution.
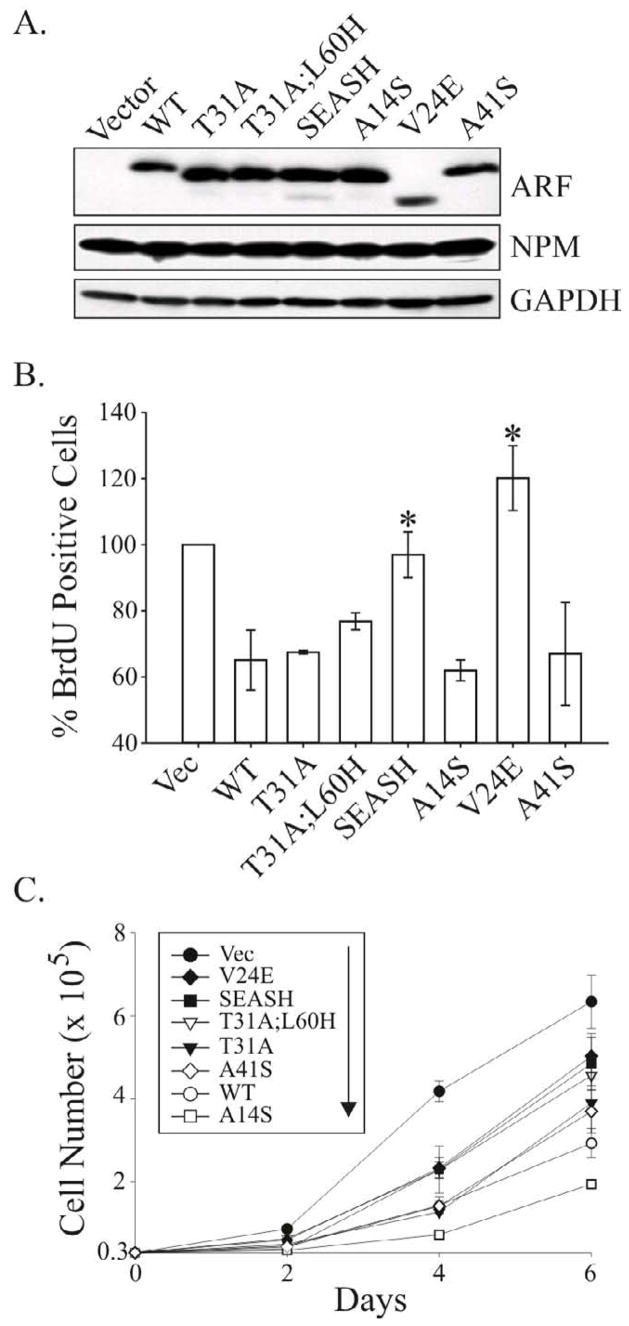
Figure 2.

Expression and activity of GFP-tagged p14ARF mutants in human U20S cells. A, schematic of the C-terminal GFP-tagged forms of p14ARF that were assayed. B, immunoblot analysis of p53, Mdm2, NPM, tubulin (loading control), and GFP or ARF-GFP expression levels in whole cell lysates. C, BrdU incorporation in ARF-positive cells showing SEASH ARF-GFP is statistically impaired in blocking DNA synthesis compared to other p14ARF forms. Data were quantified from 3 or more experiments (error bars representing the standard deviation from the mean). D, half-life analyses showing SEASH residues from new world monkeys stabilize p14ARF. Cells expressing WT or SEASH p14ARF-GFP were treated with cycloheximide (CHX) for the indicated times, and total cell lysates were analyzed by immunoblotting for ARF and GAPDH (loading control).
Western analyses of p53 and its transcriptional target, Mdm2, showed all p14ARF mutants effectively stabilized and activated p53 (Fig. 2B). Similar results were obtained in reporter assays measuring p53 transcriptional activity (Supplementary Fig. S2). In keeping with those data, BrdU incorporation assays revealed each p14ARF protein inhibited DNA synthesis (Fig. 2C), although the SEASH ARF-GFP mutant displayed reduced activity compared to other forms. There was no change in activity with either the T31A and/or L60H variants compared to WT human ARF. The decreased activity of SEASH ARF bearing Ser14, Glu24 and Ser41 suggested those amino acids specifically affect ARF-mediated growth suppression.
Immunoblots of total cell lysates consistently showed increased levels of SEASH ARF-GFP compared to other p14ARF mutants (Fig. 2B) suggesting greater protein stability. To explore that possibility, half-life studies of the human and SEASH ARF proteins were performed using the protein synthesis inhibitor, cycloheximide. Previous analyses of exogenously expressed mouse and human ARF yielded variable results with half-lives ranging between 1–6 hours for untagged ARF compared to roughly 10 hours for GFP-tagged ARF [39–41]. In keeping with the reported stability of GFP-ARF, we found that GFP-tagged WT p14ARF had a half-life of approximately 8 hours (Fig. 2D). In contrast, SEASH ARF-GFP expression levels remained constant for 16 hours and were not reduced until the 24 hour time point, implicating Ser14, Glu24 and Ser41 residues in the control of p14ARF expression.
Based on differences in activity and stability displayed by SEASH ARF-GFP, additional modified forms of ARF were generated to directly evaluate the impact of simian residues Ser14, Glu24 and Ser41 on p14ARF stability and antiproliferative activity (Fig. 3A). To avoid concerns that the GFP tag artificially increases the half-life of ARF, constructs were subcloned into the bicistronic pMSCV-IRES-GFP retroviral plasmid to enable co-expression of untagged p14ARF proteins along with GFP. Retroviruses encoding empty vector or the different ARF forms were transduced into ARF−/− mouse embryo fibroblasts (MEFs). High infection efficiencies of 95 to 99% were achieved for all viruses, as measured by flow cytometric analysis of GFP positivity (not shown). Western blotting verified expression of all modified ARF forms (Fig. 3B). For unknown reasons, V24E migrated at a faster rate on gels whereas WT and A41S p14ARF migrated more slowly (Fig. 3B). The faster migration of V24E (by approximately 1–2 kDa) is not consistent with alternative translation from Met48 (the only other Met in p14ARF), which would encode an 84 amino acid protein (as opposed to the 132 amino acid full-length p14ARF) and migrate at approximately 9 kDa. Sequencing also confirmed no changes in coding outside of the intended mutation. A faster migrating band of similar size to V24E is readily detected in SEASH ARF lysates, although all of the ARF forms exhibit that band depending on the experiment and exposure of the film (data not shown). We therefore speculate that a glutamic acid at position 24 promotes a naturally occurring post-translational modification, such as proteolytic cleavage, or mimics a phosphorylation that results in altered ARF migration.
Figure 3.

Simian-specific residues Ala31 and Ser14 stabilize the p14ARF protein. A, schematic of the untagged forms of p14ARF assayed throughout the remainder of the study. B, Western blot of ARF and GAPDH (loading control) showing differential expression levels of the indicated ARF forms following efficient and equivalent transduction (> 95%) of MSCV-ARF-IRES-GFP retroviruses into ARF-null MEFs. C, half-life analyses of the WT p14ARF and modified forms in human U20S cells. Cells were treated with cycloheximide (CHX) for the indicated times two days after transfection with the indicated ARF construct. Representative Western blots of ARF and GAPDH (loading control) expression in whole cell lysates are shown, with the calculated half-lives denoted in parentheses to the left of each set of blots. Half-lives were determined by quantification of blots using ImageJ software.
Despite equivalent infection efficiencies, WT, V24E and A41S p14ARF proteins were reproducibly expressed at lower levels compared to T31A, T31A;L60H, SEASH and A14S (Fig. 3B). Half-life analyses showed that reduced expression correlated with decreased stabilities of the WT, V24E and A41S proteins (T1/2 ≤ 4 hrs) compared to T31A, SEASH and A14S (T1/2 ≥ 7.5 hr) ARF proteins (Fig. 3C). These data show that the simian-specific residues Ser14 and Ala31 bestow greater stability to the p14ARF protein. The results also verify the implication from previous studies that GFP tagging artificially stabilizes ARF, which was demonstrated by the increased stability of GFP-tagged WT and SEASH ARF proteins compared to their untagged counterparts (compare half-lives of the proteins in Figs. 2D and 3C).
The biological consequences of altered stability and expression of the different ARF forms was then tested. Analyses of GFP expression by flow cytometry (not shown) and immunoblotting (Fig. 4A) revealed equivalent and efficient (≥ 95%) infection of our retroviruses into ARF−/− MEFs. As in Figure 3, the ARF proteins were variably expressed with the lowest expression levels observed for WT and V24E p14ARF proteins (Fig. 4A). The ability of each ARF protein to stabilize p53 and upregulate Mdm2 generally correlated with its level of expression, although V24E and A41S were repeatedly impaired in both regards relative to WT p14ARF. The limited activities of the mutants cannot be attributed to expression level differences since both V24E and A14S were expressed at equivalent or higher levels, respectively, compared to the WT protein. There was no differential effect of the ARF proteins on NPM expression and, interestingly, all forms of ARF effectively induced the p53 target, p21.
Figure 4.

Limited impact of simian ARF residues on p53-dependent cell cycle arrest. (A, B and D), ARF−/− MEFs were infected with MSCV-IRES-GFP vector control or the indicated MSCV-ARF-IRES-GFP retroviruses and analyzed two days later. A, representative Western blots examining expression of ARF, GAPDH (loading control), GFP (indicator of equivalent infection), endogenous NPM and regulators of p53 signaling (p53, Mdm2 and p21). B, representative histograms of the DNA content in successfully infected cells showing similar cell cycle inhibitory activities for the different ARF forms. G1 and G2-M peaks are shaded in gray while the percentage of cells in S phase cells is denoted and highlighted graphically in black. Notably, when results from 3 or more experiments were pooled together, no statistical difference in S phase reduction were seen between any of the modified ARF forms (Fig. S3). C, bar graph depicting the relative ability of each ARF protein to stimulate p53 transcriptional activity in U20S cells co-expressing a p53-luciferase reporter construct. D, bar graph comparing the percent of BrdU incorporation in ARF-positive cells (vector controls normalized to 100%). In both C and D, data were averaged from at least three independent experiments and subjected to student’s t-test analyses. Error bars represent standard deviations from the mean, and asterisks (*) indicate statistically significant differences (p<0.05) for the sample compared to vector control and human ARF.
Despite variations in their abilities to stabilize p53, the robust upregulation of p21 suggested that each ARF form efficiently activated p53. That conclusion is supported by the fact that ARF can activate p53 without stabilizing the protein [24] and by our observation that ARF failed to induce p21 in p53−/− MEFs (data not shown). Moreover, each ARF protein elicited a rapid G1 and G2 phase cell cycle arrest (known to be p53-dependent) and stimulated p53 transcriptional activity in reporter assays (Figs. 4B and 4C). The V24E mutant displayed reduced activation of p53 in reporter assays compared to similarly expressed WT p14ARF although it retained activity compared to vector control (Fig. 4C). BrdU incorporation assays also showed that all p14ARF proteins effectively inhibited DNA synthesis relative to actively proliferating vector control cells (Fig. 4D). While the SEASH mutant tended to inhibit replication less effectively than other ARF forms (similar to SEASH ARF-GFP in Fig. 2C), only the A41S point mutant was statistically less active than WT and other p14ARF proteins (Fig. 4D). Importantly, identical results were obtained in human U20S cells (data not shown). Together, these observations suggest there are no biologically significant differences in p53-mediated growth suppression conferred by the simian ARF residues to p14ARF. However, measurable differences in V24E and A41S activities suggest human ARF residues Val24 and Ala41 improve the efficiency with which p14ARF stabilizes p53, activates p53 and inhibits DNA replication.
We next examined the impact of simian residues on p53-independent functions of p14ARF. The untagged ARF proteins were expressed by retroviral-mediated expression in triple knockout (TKO) MEFs lacking p53, ARF, and Mdm2. Flow cytometric analysis of GFP expression confirmed greater than 98% infection for all viruses (data not shown). Much more equivalent expression of the different ARF forms was observed in TKO cells as opposed to p53-positive MEFs (see Figs. 3 and 4), although levels of WT, V24E and A41S were still slightly lower (Fig. 5A). It is worth noting that these levels are extremely comparable to the high levels of endogenous ARF normally observed in p53-null MEFs and cell lines. Interestingly, while most forms of ARF inhibited DNA replication independent of p53, SEASH p14ARF was inactive and V24E expressors showed modestly higher BrdU incorporation relative to vector (Fig. 5B; note that vector controls were normalized to 100% to allow comparisons of raw data from multiple experiments).
Figure 5.
Differential p53-independent anti-proliferative activities conferred by human versus simian-specific ARF residues. MEFs lacking p53, Mdm2 and ARF were infected with the indicated retroviruses and assays begun 72 to 96 hours post-infection. A, representative Western blots showing relative expression levels of the different ARF forms, NPM and GAPDH (loading control) in the samples. B, bar graph comparing the percent of BrdU incorporation in ARF-positive cells (vector [vec] controls normalized to 100%). Error bars represent the standard deviation from the mean for three or more experiments. Asterisks (*) denote statistically significant differences (p<0.05) exist between the indicated samples and human ARF. C, representative growth curves of successfully infected populations (>95% GFP-positive) expressing vector (Vec) or the indicated p14ARF proteins. Note that the key is aligned to correspond directly to the position of each curve in the plot, with Vec cells at the top (most rapidly proliferating) and A14S expressors (most slowly proliferating) at the bottom.
Growth curves were obtained to directly measure the effect of each ARF protein on proliferation (Fig. 5C). All forms of p14ARF suppressed proliferation to some degree compared to vector expressing cells. In keeping with results from the BrdU assay, V24E and SEASH mutants were the least active despite high levels of expression that were comparable to or increased relative to active inhibitory forms of p14ARF. These data suggest the reduced p53-independent activity of SEASH p14ARF is largely conferred by Glu24, indicating that Val24 normally plays an important role in p14ARF function in cells lacking p53.
Recent work implies a role for ARF in maintaining genomic stability independent of p53 [37,42,43]. In particular, TKO MEFs lacking ARF displayed greater chromosomal instability (CIN) than ARF-positive MEFs lacking just p53 and Mdm2 [37]. Therefore, we tested if re-expression of the p14ARF proteins would prevent the CIN that normally accumulates over time in TKO cells (Fig 6). Infected TKO MEFs were monitored throughout a 6 week time course by flow cytometric analyses of DNA content to assess levels of tetraploidy (a measure of CIN) and GFP expression. The latter showed each population remained 88 to 99 percent GFP-positive by week 6 (data not shown), and Western blots likewise demonstrated sustained expression of GFP and p14ARF at both 3 weeks (Fig. 6A) and 5 weeks post-infection. The percent of tetraploid cells in all populations was similar at week 1 post-infection, and as anticipated, tetraploidy steadily increased over time in the vector control population (Fig. 6B). A similar trend was observed for A41S expressors (Fig. 6B) although significant aneuploidy was apparent by week 6 (Fig. 6C). Strikingly, WT and SEASH p14ARF expressors attained a stable, pseudo-diploid genome with reduced tetraploidy. In contrast, V24E populations became more highly tetraploid (and at an accelerated rate) compared to control cells. The other forms of p14ARF (T31A, T31A;L60H and A14S) initially reduced tetraploidy in the populations, although this was not sustained despite high level expression of each protein. These results, which were reproducible in at least three independent experiments, show that human p14ARF residues Val24, Thr31 and Ala41 are critical for maintaining and/or promoting chromosomal stability.
Figure 6.
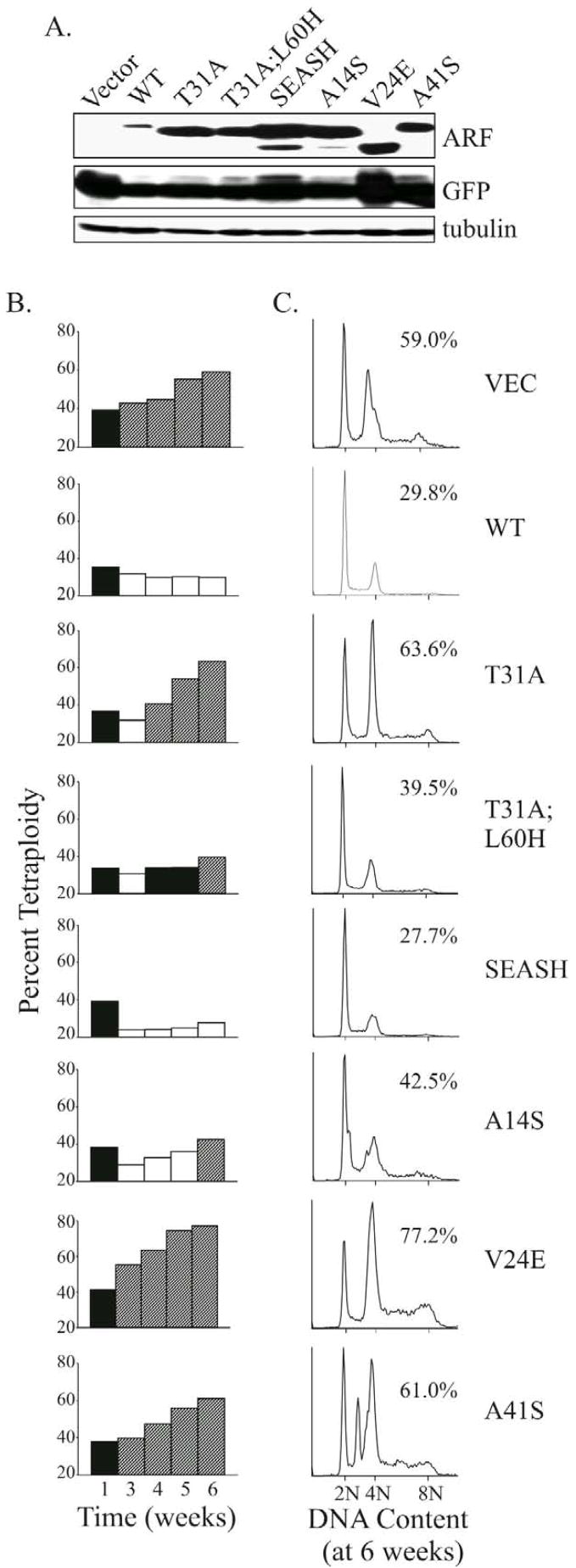
Differential p53-independent effects of human versus simian-specific ARF residues on chromosomal instability. MEFs lacking p53, Mdm2 and ARF were infected with the indicated retroviruses and maintained on a 3T3 protocol for 6 weeks. Representative data from several independent experiments are shown in A–C. A, Western blots confirming sustained expression of the different ARF forms and GFP in the populations at 3 weeks post-infection. Tubulin served as a loading control. B, comparative bar graphs of the percent tetraploidy in each population over time, as measured by flow cytometric analyses of DNA content. Increases (diagonally striped bars) or decreases (open bars) relative to the percent tetraploidy at week 1 after infection (filled bars) are depicted. C, histograms of DNA content for each population at week 6 post-infection are shown, with the percent of tetraploid cells designated. Although not shown, flow cytometric analyses of GFP fluorescence revealed all populations remained 88 to 99 percent GFP positive by week 6.
Discussion
There is now significant evidence that ARF blocks tumor development and metastasis in vivo through p53-independent mechanisms that promote senescence and/or apoptosis [6–9]. This may have important therapeutic ramifications since a large percentage of human tumors lack functional p53 but retain wild-type ARF. At present, the molecular basis of p53-independent ARF signaling is not well understood although numerous potential regulators have been identified [10]. Here, we identify individual p14ARF residues that are essential for its ability to inhibit proliferation and CIN in p53-null cells. Such findings should ultimately prove valuable for defining the biologically relevant, p53-independent mechanisms of ARF action.
Nearly all human cancer cells exhibit aneuploidy, a hallmark of CIN, and several studies recently implied a p53-independent role for ARF in maintaining genomic integrity [37,42,43]. This study provides the first direct evidence that p14ARF controls chromosomal stability in p53-null cells, establishing a new p53-independent function of ARF. Three key residues, Val24, Thr31 and Ala41 (which reside outside the established functional domains of human p14ARF) were critical for maintaining and/or promoting chromosomal stability. There is high conservation of Val/Leu24 in ARF across species (seen in human, mouse, rat, opossum and chicken, e.g.) and Ala41 is identical between mouse and human ARF (Gly in rat and opossum). In contrast, Thr31 is not well conserved (Arg in mouse, Ser in rat, Val/Leu in opossum and chicken). Notably, Thr31 was not sufficient to prevent CIN in the context of other amino acid changes in p14ARF, such as A14S, V24E or A41S. We also discovered that Glu24 not only interfered with ARF’s ability to inhibit chromosomal instability, it enhanced the development of tetraploidy. Such results suggest a dominant-negative activity for V24E ARF, although not in the classical sense because TKO cells lack endogenous ARF. We are not sure why SEASH shows a wild-type phenotype in promoting CIN given the presence of Glu24, but speculate that the particular combination of amino acid changes in the mutant as well as high expression levels may contribute. Finally, comparison of CIN in cells expressing T31A (high levels of tetraploidy) versus T31A;L60H (low levels of tetraploidy) suggests that the monkey-specific His60 plays an important role in maintaining chromosomal stability when residue 31 is an alanine.
There are several possible mechanisms by which ARF could promote chromosomal stability independent of p53. ARF is required for proper mitotic checkpoint control [44] and it activates a p53-independent G2-M phase checkpoint via ATM in response to DNA damage [42]. The link between ARF and ATM is compelling since ATM is a critical suppressor of CIN [45]. Recent work also demonstrates a role for several of ARF’s binding partners (Mdm2, NPM, Mdm4/MdmX and NIAM) in either promoting or inhibiting chromosomal abnormalities [12,37,43,46]. Interestingly, the acquisition of a diploid-like genome over time in WT and SEASH p14ARF expressors suggests they promote the death of tetraploid cells, a possibility we are currently exploring. Given their variable activities in the CIN assay, the different p14ARF forms examined here should facilitate future studies of molecular mechanisms through which ARF promotes genomic stability. With the exception of WT and V24E p14ARF, there was little correlation between ARF’s anti-proliferative activity and inhibition of CIN in p53-null cells. For instance, the SEASH mutant lacked anti-proliferative activity but functioned similar to WT p14ARF in suppressing tetraploidy (see Figs. 5 and 6). Conversely, the T31A and A14S point mutants actively inhibited cell cycling yet were unable to maintain chromosomal stability. Thus, as may be expected, different residues or regions of p14ARF control proliferation and genomic stability in cells lacking p53.
A novel observation from this study was that simian residues Ala31 and Ser14 stabilized the p14ARF protein by decreasing its turnover. The mechanism underlying the decreased stability of WT human p14ARF is currently not known, although the effect was most pronounced in p53-positive cells. Previous studies found that p53 normally inhibits the transcription of ARF as part of a negative feedback loop [38,47,48], but the use of exogenous ARF in our studies suggests a post-transcriptional mechanism. Reduced p14ARF stability could be caused by diminished nucleolar localization and/or NPM binding, which normally protect ARF from proteasomal degradation [11,13,39,40]. However, all forms of ARF showed similar accumulation in nucleoli, and paradoxically, preliminary analyses indicate that NPM binds most efficiently to the least stable protein, WT p14ARF (unpublished data, J.H. and D.E.Q.). It seems likely that other ARF binding partners may preferentially associate with various ARF forms and control their stability. An appealing candidate is the Tat-binding protein 1 (TBP-1), a component of the proteasome that protects ARF from degradation [41,49].
Would it be beneficial if p14ARF was a long-lived protein? Many tumor suppressor proteins, such as p53, are typically short-lived in order to facilitate cell recovery and tissue repair after cell stress or damage. In addition, self-renewal of neuronal and hematopoietic stem cells is reduced by the rise in INK4a/ARF expression that occurs with age, limiting tissue regeneration in older organisms [50]. Our data showed that increased stability of p14ARF point mutants conferred no functional advantage in p53 signaling or p53-independent inhibition of CIN. However, whether or not increased expression would significantly affect the activities of WT p14ARF was not tested. Recent studies of transgenic “super-INK4a/ARF” mice do suggest higher levels of ARF may be advantageous to an organism by reducing oxidative damage and increasing cancer resistance, although the individual contributions of ARF versus p16INK4a in this setting cannot been distinguished [51,52].
The potential involvement of phosphorylation in controlling p14ARF activities is suggested by our findings. Specifically, loss of Thr31 in the T31A mutant severely impaired p14ARF’s ability to maintain chromosomal stability in p53-null cells and markedly enhanced its stability in p53-positive cells. Likewise, the acquisition of serines in the A14S and A41S mutants reduced their inhibition of CIN and A14S heightened ARF stability. The A41S mutant also displayed an inefficient ability to stabilize p53 and inhibit DNA synthesis despite the same or greater expression compared to WT p14ARF. At present, there is no evidence that p14ARF is a phosphoprotein. In fact, our previous analysis of phospho-labeled U20S cells expressing exogenous p14ARF showed the protein is not detectably phosphorylated under normal growth conditions [11]. Whether wild-type p14ARF is phosphorylated in response to particular stresses, such as DNA damage arising from chromosomal instability or chemotherapeutic drugs, remains to be determined. Additional studies would also need to be performed to test if the serine point mutants (A14S or A41S) or the native simian ARF proteins are regulated by phosphorylation.
Two p14ARF residues were found to contribute to efficient regulation of p53. When compared to WT p14ARF, V24E and A41S mutants were modestly but consistently impaired in stabilizing p53 and V24E was compromised in p53 transcriptional activation. Thus, Val24 and Ala41 normally contribute to those processes. Indeed, Val24 resides in a highly conserved region previously shown to control Mdm2 binding [53] and p53 stabilization [24]. Importantly, previous work established that p53 stabilization is not required for ARF signaling [24], which is consistent with the fact that both A41S and V24E mutants potently induced p53-dependent cell cycle arrest despite minimal stabilization of p53. Mutation of Ala41 to Ser also caused a modest reduction in ARF’s ability to block BrdU incorporation even though the cells exhibited a similar G1 and G2 arrest profile to other ARF expressors. A comparable phenotype was observed in ARF-arrested p21-null MEFs in which ARF induced a later G1 checkpoint (at G1-S as opposed to mid-G1), where it was suggested that replication had been initiated but elongation blocked [54]. Ala41 is well conserved across ARF species and lies in a region (residues 26–44) that mediates mouse p19ARF association with the oncogenic Foxm1b transcription factor [32]. Thus, A41S may be impaired in its ability to bind and inhibit Foxm1b, which would result in lowered expression of the p21-related CDK inhibitor, p27Kip1.
It is tempting to speculate about the evolutionary implications of our findings. The equivalent growth inhibitory activity of WT and mutant p14ARF proteins in p53-positive cells might suggest that ARF-p53 signaling was not significantly modified during primate evolution. By comparison, p53-independent regulation of proliferation and CIN by p14ARF was markedly altered by simian residues with wild-type human ARF generally exhibiting superior activity. A key exception was the similar activity of the more distantly related SEASH and wild-type p14ARF proteins in regulating chromosomal stability. Those results make it difficult to draw conclusions about primate evolution of p14ARF from these studies, as does the fact that exon 2-encoded residues which distinguish the species were not concomitantly modified in our mutants. One substitution within exon 2 of p14ARF has been identified in great apes (S73R) whereas four variations occur in old world monkeys (S73R, G83R, R115W and A121T) (unpublished data, A.T. and V.L.). While most studies have shown that ARF’s activities are governed by exon 1β domains, a number of exon 2 encoded residues were recently found to be important for ARF association with a mitochondrial protein, p32, and for mediating ARF-induced apoptosis [16]. The simian residues listed above have not yet been tested in that regard.
In sum, our study identifies individual p14ARF residues that govern its expression and certain signaling activities, most notably p53-independent proliferation and maintenance of genomic stability. Further studies exploring the specific pathways that regulate each of these events will provide valuable insight into p14ARF function. This is relevant to our fundamental understanding of cancer given the major importance of ARF and chromosomal stability to tumor suppression, and growing recognition that ARF’s p53-independent functions have significant anticancer effects.
Acknowledgments
This work was supported by grants from the Philip Foundation (A.T.), the CNRS – UMR 6187 (A.T.), the Ecole Doctorale ICBG of Poitiers and the Region Poitou-Charentes (A.T.), and the National Institutes of Health (D.E.Q.). We thank Anne Cantereau (Service commun d’Imagerie Confocale, CNRS – UMR 6187) and the Central Microscopy Research Facility at the University of Iowa for technical assistance with confocal microscopy, as well as James Habrioux (Service commun d’Informatique, CNRS – UMR 6187) and Tarik Smith (University of Iowa) for their assistance.
The abbreviations used are
- ARF
alternative reading frame protein
- Mdm2
mouse double minute-2
- CIN
chromosomal instability
- MEFs
mouse embryo fibroblasts
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Op Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 2.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 3.Hainaut P, Soussi T, Shomer B, Hollstein M, Greenblatt M, Hovig E, Harris CC, Montesano R. Database of p53 gene somatic mutations in human tumors and cell lines: updated compilation and future prospects. Nucleic Acids Res. 1997;25:151–157. doi: 10.1093/nar/25.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–77. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 5.Carnero A, Hudson JD, Price CM, Beach DH. p16(INK4A) and p19(ARF) act in overlapping pathways in cellular immortalization. Nat Cell Biol. 2000;2:148–155. doi: 10.1038/35004020. [DOI] [PubMed] [Google Scholar]
- 6.Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19ARF tumor suppressor. Genes Dev. 2000;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eymin B, Leduc C, Coll JL, Brambilla E, Gazzeri S. P14(ARF) induces G(2) arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene. 2003;22:1822–1835. doi: 10.1038/sj.onc.1206303. [DOI] [PubMed] [Google Scholar]
- 8.Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19(Arf) suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. Plos Biol. 2004;2:1138–1149. doi: 10.1371/journal.pbio.0020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ha L, Ichikawa T, Anver M, Dickins R, Lowe S, Sharpless NE, Krimpenfort P, DePinho RA, Bennett DC, Sviderskaya EV, Merlino G. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci USA. 2007;104:10968–10973. doi: 10.1073/pnas.0611638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- 11.Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE. Nucleophosmin (B23) Targets ARF to Nucleoli and Inhibits Its Function. Mol Cell Biol. 2005;25:1258–1271. doi: 10.1128/MCB.25.4.1258-1271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 13.Colombo E, Bonetti P, Lazzerini Denchi E, Martinelli P, Zamponi R, Marine JC, Helin K, Falini B, Pelicci PG. Nucleophosmin Is Required for DNA Integrity and p19Arf Protein Stability. Mol Cell Biol. 2005;25:8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llanos S, Clark PA, Rowe J, Peters G. Stabilisation of p53 by p14ARF without relocalization of Mdm2 to the nucleolus. Nat Cell Biol. 2001;3:445–452. doi: 10.1038/35074506. [DOI] [PubMed] [Google Scholar]
- 15.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, Kimchi A. A Short Mitochondrial Form of p19ARF Induces Autophagy and Caspase-Independent Cell Death. Mol Cell. 2006;22:463–475. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 16.Itahana K, Zhang Y. Mitochondrial p32 Is a Critical Mediator of ARF-Induced Apoptosis. Cancer Cell. 2008;13:542–553. doi: 10.1016/j.ccr.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C, Smith BA, Bandyopadhyay K, Gjerset RA. DNA Damage Disrupts the p14ARF-B23(Nucleophosmin) Interaction and Triggers a Transient Subnuclear Redistribution of p14ARF. Cancer Res. 2005;65:9834–9842. doi: 10.1158/0008-5472.CAN-05-1759. [DOI] [PubMed] [Google Scholar]
- 18.Itahana K, Bhat KP, Jin AW, Itahana Y, Hawke D, Kobayashi R, Zhang YP. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 19.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424. doi: 10.1016/s1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 20.Datta A, Nag A, Raychaudhuri P. Differential regulation of E2F1, DP1, and the E2F1/DP1 complex by ARF. Mol Cell Biol. 2002;22:8398–8408. doi: 10.1128/MCB.22.24.8398-8408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P. Myc-ARF (Alternate Reading Frame) Interaction Inhibits the Functions of Myc. J Biol Chem. 2004;279:36698–36707. doi: 10.1074/jbc.M312305200. [DOI] [PubMed] [Google Scholar]
- 22.Brady SN, Yu Y, Maggi LB, Jr, Weber JD. ARF Impedes NPM/B23 Shuttling in an Mdm2-Sensitive Tumor Suppressor Pathway. Mol Cell Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tago K, Chiocca S, Sherr CJ. Sumoylation induced by the Arf tumor suppressor: A p53-independent function. Proc Natl Acad Sci USA. 2005;102:7689–7694. doi: 10.1073/pnas.0502978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korgaonkar C, Zhao L, Modestou M, Quelle DE. ARF function does not require p53 stabilization or Mdm2 relocalization. Mol Cell Biol. 2002;22:196–206. doi: 10.1128/MCB.22.1.196-206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber JD, Kuo ML, Bothner B, DiGiammarino EL, Kriwacki RW, Roussel MF, Sherr CJ. Cooperative signals governing ARF-Mdm2 interaction and nucleolar localization of the complex. Mol Cell Biol. 2000;20:2517–2528. doi: 10.1128/mcb.20.7.2517-2528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quelle DE, Cheng MG, Ashmun RA, Sherr CJ. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16(INK4A) but not by the alternative reading frame protein p19(ARF) Proc Natl Acad Sci USA. 1997;94:669–673. doi: 10.1073/pnas.94.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 28.Midgley CA, Desterro JMP, Saville MK, Howard S, Sparks A, Hay RT, Lane DP. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene. 2000;19:2312–2323. doi: 10.1038/sj.onc.1203593. [DOI] [PubMed] [Google Scholar]
- 29.Lohrum MA, Ashcroft M, Kubbutat MH, Vousden KH. Contribution of two independent MDM2-binding domains in p14(ARF) to p53 stabilization. Curr Biol. 2000;10:539–542. doi: 10.1016/s0960-9822(00)00472-3. [DOI] [PubMed] [Google Scholar]
- 30.Rizos H, McKenzie HA, Ayub AL, Woodruff S, Becker TM, Scurr LL, Stahl J, Kefford RF. Physical and Functional Interaction of the p14ARF Tumor Suppressor with Ribosomes. J Biol Chem. 2006;281:38080–38088. doi: 10.1074/jbc.M609405200. [DOI] [PubMed] [Google Scholar]
- 31.Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Targeting of C-Terminal Binding Protein (CtBP) by ARF Results in p53-Independent Apoptosis. Mol Cell Biol. 2006;26:2360–2372. doi: 10.1128/MCB.26.6.2360-2372.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, Costa RH. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–850. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rizos H, Darmanian AP, Mann GJ, Kefford RF. Two arginine rich domains in the p14ARF tumour suppressor mediate nucleolar localization. Oncogene. 2000;19:2978–2985. doi: 10.1038/sj.onc.1203629. [DOI] [PubMed] [Google Scholar]
- 34.Tompkins V, Hagen J, Zediak VP, Quelle DE. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle. 2006;5:641–646. [PubMed] [Google Scholar]
- 35.di Tommaso A, Soler C, Roos C, Kitzis A, Ladeveze V. The ink4a/arf Locus Evolution in Primates: Characterization of Three ARF Sequences. DNA Cell Biol. 2004;23:167–173. doi: 10.1089/104454904322964760. [DOI] [PubMed] [Google Scholar]
- 36.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tompkins VS, Hagen J, Frazier AA, Lushnikova T, Fitzgerald MP, di Tommaso A, Ladeveze V, Domann FE, Eischen CM, Quelle DE. A Novel Nuclear Interactor of ARF and MDM2 (NIAM) That Maintains Chromosomal Stability. J Biol Chem. 2007;282:1322–1333. doi: 10.1074/jbc.M609612200. [DOI] [PubMed] [Google Scholar]
- 38.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuo ML, den Besten W, Bertwistle D, Roussel MF, Sherr CJ. N-terminal polyubiquitination and degradation of the Arf tumor suppressor. Genes Dev. 2004;18:1862–1874. doi: 10.1101/gad.1213904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodway H, Llanos S, Rowe J, Peters G. Stability of nucleolar versus non-nucleolar forms of human p14(ARF) Oncogene. 2004;23:6186–6192. doi: 10.1038/sj.onc.1207854. [DOI] [PubMed] [Google Scholar]
- 41.Pollice A, Sepe M, Villella VR, Tolino F, Vivo M, Calabro V, La Mantia G. TBP-1 protects the human oncosuppressor p14ARF from proteasomal degradation. Oncogene. 2007 doi: 10.1038/sj.onc.1210313. [DOI] [PubMed] [Google Scholar]
- 42.Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, Khochbin S, Gazzeri S. p14(ARF) activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol. 2006;26:4339–4350. doi: 10.1128/MCB.02240-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang P, Greiner TC, Lushnikova T, Eischen CM. Decreased Mdm2 expression inhibits tumor development induced by loss of ARF. Oncogene. 2006;25:3708–3718. doi: 10.1038/sj.onc.1209411. [DOI] [PubMed] [Google Scholar]
- 44.Khan SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci USA. 2000;97:3266–3271. doi: 10.1073/pnas.050560997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shiloh Y. ATM And Related Protein Kinases: Safeguarding Genome Integrity. Nat Rev Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 46.Matijasevic Z, Steinman HA, Hoover K, Jones SN. MdmX Promotes Bipolar Mitosis To Suppress Transformation and Tumorigenesis in p53-Deficient Cells and Mice. Mol Cell Biol. 2008;28:1265–1273. doi: 10.1128/MCB.01108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and mdm2. Proc Natl Acad Sci USA. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robertson KD, Jones PA. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol Cell Biol. 1998;18:6457–6473. doi: 10.1128/mcb.18.11.6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pollice A, Nasti V, Ronca R, Vivo M, Iacono ML, Calogero R, Calabro V, La Mantia G. Functional and Physical Interaction of the Human ARF Tumor Suppressor with Tat-binding Protein-1. J Biol Chem. 2004;279:6345–6353. doi: 10.1074/jbc.M310957200. [DOI] [PubMed] [Google Scholar]
- 50.Kim WY, Sharpless NE. The Regulation of INK4/ARF in Cancer and Aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Matheu A, Pantoja C, Efeyan A, Criado LM, Martin-Caballero J, Flores JM, Klatt P, Serrano M. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004;18:2736–2746. doi: 10.1101/gad.310304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Vina J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 53.Bothner B, Lewis WS, DiGiammarino EL, Weber JD, Bothner SJ, Kriwacki RW. Defining the molecular basis of Arf and Hdm2 interactions. J Mol Biol. 2001;314:263–277. doi: 10.1006/jmbi.2001.5110. [DOI] [PubMed] [Google Scholar]
- 54.Modestou M, Puig-Antich V, Korgaonkar C, Eapen A, Quelle DE. The ARF tumor suppressor inhibits growth through p21-dependent and p21-independent pathways. Cancer Res. 2001;61:3145–3150. [PubMed] [Google Scholar]