


Abstract
Primary ribosomal protein S4 is essential for 30S ribosome biogenesis in eubacteria, because it nucleates subunit assembly and helps coordinate assembly with the synthesis of its rRNA and protein components. S4 binds a five-helix junction (5WJ) that bridges the 5′ and 3′ ends of the 16S 5′ domain. To delineate which nucleotides contribute to S4 recognition, sequential deletions of the 16S 5′ domain were tested in competitive S4-binding assays based on electrophoretic mobility shifts. S4 binds the minimal 5WJ RNA containing just the five-helix junction as well or better than with affinity comparable to or better than the 5′ domain or native 16S rRNA. Internal deletions and point mutations demonstrated that helices 3, 4, 16 and residues at the helix junctions are necessary for S4 binding, while the conserved helix 18 pseudoknot is dispensable. Hydroxyl radical footprinting and chemical base modification showed that S4 makes the same interactions with minimal rRNA substrates as with the native 16S rRNA, but the minimal substrates are more pre-organized for binding S4. Together, these results suggest that favorable interactions with S4 offset the energetic penalty for folding the 16S rRNA.
INTRODUCTION
Ribosomal protein S4 plays a central role in the biogenesis of the small ribosomal subunit. It regulates its own translation and that of other ribosomal proteins encoded by the alpha-operon (1,2), by recognizing a pseudoknot structure in the leader sequence (3–5). S4 also regulates rRNA expression levels by acting as a general transcription antiterminator, similar to NusA (6). Additionally, S4 nucleates assembly of the 30S subunit along with S7 (7). Thus, S4 helps balance the levels of rRNA, ribosomal proteins and assembled ribosomes.
The role of many ribosomal proteins in assembly is to stabilize the folded conformation of the rRNA (8). Binding of S4 to the 16S rRNA is necessary for addition of protein S16 and other proteins within the 5′ and central domains of the 30S ribosome (9), and S4 was one of two proteins required for initiation of 30S assembly in in vitro reconstitution experiments (7). Base modification and hydroxyl radical footprinting studies indicated that S4 induces structural changes in the 5′ and 3′ domains of the 16S rRNA, that could lead to the further steps in 30S assembly (10,11).
The binding site for ribosomal protein S4 consists of the five-way junction (5WJ) between helices (H) 3, 4, 16, 17 and 18, which flank the 5′ and 3′ ends of the 16S 5′ domain (10,12,13) (Figure 1a and b). The stable C-terminal domains of S4, which include a winged-helix motif (14), directly contacts the center of the 5WJ in the rRNA (15). Outside of a few base-specific contacts in the 5WJ, S4 predominantly interacts with the rRNA backbone.
Figure 1.

Escherichia coli 16S rRNA fragments tested for S4 binding. (a) 16S rRNA; (b) 5′domain; (c) RNA(▵H6-14); (d) RNA(▵H5-14); (e) 5WJ RNA; (f) 5WJ_nt6; (g) 5WJ:BstH17; (h) 5WJ:TthH17; (i) 5WJ▵H3; (j) 5WJ▵H4; (k) 5WJ:H16trunc; (l) 5WJ:H17trunc; and (m) 5WJ:H18trunc. Secondary structures of ▵H6-14 and 5WJ were confirmed by RNase T1 digestion (Figures S1 and S2).
The N-terminal region of free S4 is disordered, but forms a structured loop and short α helix when bound to 16S rRNA (16–19). The N-terminal region of S4 has been implicated in tight, specific binding to the 16S rRNA (Gerstner,R.B. and Draper,D.E., unpublished data). In Thermus thermophilus, the N-terminal domain is further stabilized by coordination of a Zn2+ ion (18). Evidence for co-folding of the S4 N-terminal domain and the 16S rRNA comes from specific changes in the chemical reactivity of the bases in the 16S–S4 complex above 30°C (11).
Given that the S4-binding site includes base-paired nucleotides that are 530 nt apart in the 16S sequence, and that the 16S rRNA is not stably folded in the absence of proteins (10), an unanswered question is which features of the S4 binding site are most important for recognition, and whether interactions in other parts of the 5′ domain contribute to the stability of the complex. Minimal rRNA substrates have been previously identified for primary ribosomal proteins S7 (20), S8 (21,22) and S15 (23). We used the known contacts between the protein and RNA in the 30S ribosome (15) to design minimal rRNA substrates for S4.
Deletions and chemical probing of the 16S rRNA have been previously used to define the binding site for S4. Ungewickell and co-workers (24,25) identified discontinuous fragments of the 5′ domain that were able to bind S4. Draper and co-workers (26,27) used nitrocellulose binding assays to measure the affinity of rRNA fragments for Escherichia coli S4. The smallest continuous fragment with high affinity for S4 at 0°C contained nucleotides 39–500 of the 5′ domain (26). Although small segments could be deleted from this RNA without large effects on binding (27), removing the lower half of the domain (nucleotides 53–359) reduced affinity 5-fold (26). As these residues do not contact S4 directly (11,15,28), the reduced binding presumably arose from alterations in the rRNA structure.
Here, we show that the helix 18 pseudoknot and helices outside the 5WJ are dispensable for tight binding, while specific base contacts at the centre of the 5WJ contribute significantly to the stability of the S4 complex. Precise deletions of other helices in the junction also result in almost total loss of specific binding. Hydroxyl radical footprinting and dimethylsulfate (DMS) base modification of the rRNA alone or in the presence of S4 confirm that mutations compromising the stability of the complex also remove specific interactions within the S4 complex. Thus, these studies delineate the factors required for specific S4 recognition more precisely and form the starting point for further studies of the mechanism of S4–RNA binding and 30S assembly initiation.
MATERIALS AND METHODS
RNA preparation
RNA containing the 5′ domain of E. coli 16S rRNA (nt 21–562) was transcribed from plasmid pRNA1 with T7 RNA polymerase as previously described (29). Deletions and mutations were made in the 5′ domain RNA by standard PCR amplification of pRNA1 (Table S1). 16S rRNA was extracted from native 30S subunits as previously described (30). Radiolabeled transcripts were transcribed from 1 μg EcoRI-linearized plasmid in 40 μl reactions containing 50 μCi α-[32P]-ATP (Perkin Elmer), for 30 min at 37°C. Transcripts were passed over spin columns (TE-100 or TE-400, Clontech). The RNA concentration was estimated from the UV absorption of products from identical reactions lacking α-[32P]-ATP.
S4 purification
Geobacillus (formerly Bacillus) stearothermophilus (Bst) S4 was purified as described by (31), except that the ion-exchange column used was UNO S1 (BioRad). Pooled S4 fractions were dialyzed into reconstitution buffer with 1 M KCl (32), and concentrated as described previously (31). Purified S4 was stored at –80°C in 10% glycerol. Protein concentrations were measured from absorption at 280 nm (ε280 = 18 490 M−1 cm−1) (31). The final preparation was ≥85% full length, as judged by SDS–PAGE, and ≥30–50% active as judged by binding competition. All assays using S4 were done in low-retention microcentrifuge tubes (Fisher Scientific).
Protein-binding assays
For equilibrium binding studies with Bst S4, 0.5 nM 32P-labeled 5′ domain RNA was preincubated 10–20 min at 42°C in 20 µl 80 mM K-HEPES, 330 mM KCl, 4 mM MgCl2 (HKM4) and 20 ng/μl poly(dI–dC) (Sigma). Poly(dI–dC) improved the resolution of S4 complexes without detectably competing for S4 binding (up to 100 ng/µl). Bst S4 (0–400 nM) was added to the RNA and incubated another 10–20 min at 42°C. A 5 μl aliquot was removed from each sample and mixed with 1 μl of native gel loading dye (final 10% glycerol, 0.25% xylene cyanol). Of this mixture, 2–3 μl was loaded on a native 1× TBE 8% polyacrylamide gel. Gels were run at 12–15 W for 5–6 h at ∼10°C. The counts in the bound and free species were quantified using ImageQuant (Molecular Dynamics). Counts above the free RNA in lanes with no protein were defined as background and subtracted from the bound RNA in each lane. The fraction of bound RNA (fB) was fit to the Langmuir binding isotherm to obtain the equilibrium dissociation binding constant (Kd). Nitrocellulose filter binding assays were carried out with 5′ domain RNA and Bst S4 as previously described (31).
Competitive-binding assays
Competitive binding assays were carried out as above, except that 0.5–1.6 nM radiolabeled 5′ domain RNA and 0–400 nM of unlabeled 5′ domain RNA were mixed and preincubated 10–20 min at 42°C, before addition of 32–50 nM Bst S4. For other unlabeled competitor RNAs, the concentration range was 0–1 µM. The fraction of labeled RNA bound to S4 (ƒB) was determined as above, and fit to Equation (1) as described previously (23,33).
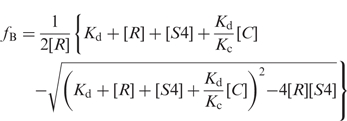 |
1 |
Kd and KC are the dissociation constants of the labeled RNA and unlabeled competitor, respectively, from which Krel = KC/Kd. [R], [S4] and [C] are the concentrations of labeled RNA, S4 protein and competitor RNA, respectively. Competitive binding assays with 0–200 ng/μl poly(A), poly(U) and poly(dI–dC) (Sigma) were done as described above, except that 20 ng/µl poly(dI–dC) was omitted from the reaction buffer.
The Kd of the 5WJ–S4 complex was determined by direct titration of Bst S4 or by competition as described above for the 5′ domain RNA, except that samples were loaded on a 6% TKM2 (30 mM Tris, 100 mM potassium acetate, 2 MgCl2) gel. Competitive binding reactions against the 5WJ RNA used 0.5–1 nM 32P-labeled 5WJ RNA and 22.5 nM S4.
Dual-label binding assays
In a reaction volume of 20 μl, 32P-labeled 5′ domain and 32P-labeled minimal competitor RNA were preincubated 15–20 min at 42°C in HKM4. Unlabeled RNA was added when necessary to ensure that both S4 complexes were detectable. Bst S4 was added to the RNA mixture and incubated another 15–20 min at 42°C. The ratio of total RNA:S4 was varied from 1:1 to 5:1 (34). Loading dye (5 μl) was added to each reaction, and quickly mixed by pipetting. From each reaction, one aliquot was loaded on an 8% TBE gel to resolve the 5′ domain RNA and the other on a 6% TKM2 native gel to resolve the minimal RNA complexes. Gels were run at 12–15 W for 5–6 h at 10°C.
The fractions of bound and free 5′ domain RNA were obtained from TBE gels, while those of smaller competitor RNA were obtained from the TKM2 gel. These fractions were used to calculate Krel:
| 2 |
RNase T1 protection
5′-32P-labeled RNA(ΔH6-14), RNA(ΔH5-14) and 5WJ RNAs were incubated with 25 mM K-HEPES (pH 7.5) plus varying MgCl2 for 20 min at 42°C. In some experiments, 5WJ RNA or its variants were instead incubated with 80 mM K-HEPES (pH 7.5), 330 mM KCl, HKM4, HKM10 or HKM20. The RNA was treated with RNase T1 as previously described (29).
Fe–EDTA hydroxyl radical footprinting
5′ domain RNA (1–3 pmol) was incubated with buffer and S4 as described above (14 μl total volume). Bst S4 was added in 2-fold molar excess over the RNA; Eco S4 was added in 4-fold excess. After incubation at 42°C, reactions were kept in an ice/water bath for 10 min before treatment with Fe–EDTA reagents as previously described (29). 5WJ RNA (0.75–2 pmol) was incubated under the same conditions, and treated with 1 μl 10 mM Fe(II)-20 mM EDTA and 2 μl each of 10 mM sodium ascorbate and 0.3% H2O2 for 1 min on ice. Reactions were quenched with 1 μl 0.1 M thiourea. The RNA was analyzed by primer extension as previously described (29). Primers annealed 3′ of nt 547, 448 and 161.
Chemical base modification
Base modification reactions with dimethyl sulfate (DMS; Sigma) or kethoxal (Research Organics) were carried out as previously described (35), with the following changes. 5′ domain, 5WJ RNA or 5WJ RNA variants (2 pmol) were incubated with buffer and S4 as for Fe–EDTA reactions, except that the total volume was 25 μl. The 5′ domain was treated with 1 μl of 1:4 DMS:95% ethanol for 10 min on ice or 3 μl 111 mg/ml kethoxal in 20% ethanol for 4 h at 4°C. 5WJ RNA was treated with 1 μl 1:3 DMS:95% ethanol or 1 μl 285 mg/ml kethoxal for 4 h at 4°C.
RESULTS
Competitive-binding assays measure the Kd of rRNA–S4 complexes
To define the minimal requirements for S4 RNA binding, competitive binding experiments were used to compare the affinity of protein S4 for various fragments of the E. coli 16S rRNA. We used the Bst S4 protein for these studies, because it is more stable than the E. coli (Eco) protein in solution. The Bst protein has previously been shown to specifically bind both Bst and E. coli 16S 5′ domain rRNAs at 42°C with a dissociation constant of 0.7 nM, while forming nonspecific complexes with a binding constant of 100 nM (31). The E. coli rRNA was used for these studies to take advantage of crystallographic and biochemical information available for E. coli ribosomes.
32P-labeled E. coli 5′ domain RNA (nts 21–562 of 16S rRNA) was incubated with 0–400 nM Bst S4 at 42°C, and the S4–rRNA complex was detected by a native gel mobility shift (see ‘Materials and methods’ section). The apparent binding constant from direct titrations was 20 ± 4 nM, similar to the value obtained from nitrocellulose filter binding assays (Kd = 12 ± 5 nM; data not shown). When unlabeled 5′ domain RNA was added as a competitor, the Kd for the 5′ domain was determined to be 5.5 ± 2.6 nM (Figure 2). This is higher than the previously reported value of 0.7 nM for the specific complex, but much lower than the Kd for nonspecific binding (31). Kinetic dissociation experiments show that the electrophoretically retarded RNA likely contains a mixture of high- and low-affinity complexes that both contribute to the measured Kd (Bellur,D.L. and Woodson,S.A., in preparation). This may account for the discrepancy in Kd values and the small deviation of the binding data in Figure 2b from a two-state model. We consider the relative binding affinities obtained by competition more reliable than the absolute Kd values obtained by direct titration.
Figure 2.

Competitive binding of Bst S4 to the 16S 5′ domain RNA. (a) S4 binding measured by gel mobility shift. 0.5 nM 32P-labeled and 0–400 nM unlabeled 5′domain RNAs were incubated with 31.5 nM Bst S4 in HKM4 buffer at 42°C. Gel is 8% polyacrylamide in TBE. NP, 32P-5′domain RNA only. (b) The fraction of complexed 32P-labeled 5′domain RNA versus competitor RNA was fit to Equation (1). Filled circles, 5′ domain (Kd, 5′domain = 5.5 ± 2.6 nM); filled squares, 16S rRNA (Krel = 1.6 ± 0.8).
S4 bound the native 16S rRNA with similar affinity as the 5′ domain in competition experiments, with a relative dissociation constant Krel = 1.6 (Figure 2). In contrast, nonspecific substrates poly(A), poly(U) and poly(dI–dC) did not compete against the labeled 5′ domain RNA, demonstrating that the complexes were sequence-specific (data not shown). Consequently, the competitive gel-shift assay could be used to compare S4 binding to different substrates.
Designing small target rRNAs
The S4-binding site in the 16S rRNA corresponds to helices H3, H4, H16–18 in the 5′ domain (Figure 1) (10). To test whether the rest of the 5′ domain was therefore dispensable for S4 binding, rRNA substrates missing helix 1 and helices in the lower half of the 5′ domain were prepared. RNA(ΔH6-14) is missing H1 and H6-H14 (Figure 1c), RNA(ΔH5-14) additionally lacked H5 (Figure 1d), while 5WJ RNA contained just the five helices that make up the immediate S4-binding site (H3, H4, H16, H17 and H18; Figure 1e). To minimize disruptions to the rRNA secondary structure, helices flanking large deletions were capped with stable tetraloops. The program Mfold (36) was used to predict the lowest free-energy secondary structure for each substrate, and only RNAs that were predicted to form the correct secondary structure were tested for S4 binding (Figure 1 and Supplementary Table S1).
The secondary structures of the three RNA substrates were probed experimentally by partial digestion with RNase T1, which cleaves after single stranded guanosines. The nuclease reactions were done under conditions similar to our binding assays, at 42°C in 25 mM K-HEPES (pH 7.5) and 0–20 mM MgCl2 (data not shown). All three RNAs formed their native secondary structures in <4 mM Mg2+, the Mg2+ concentration in our binding reactions. As illustrated for RNA(ΔH6-14) in Figure S1, the RNase T1 cleavage patterns were consistent with the secondary structure of the 16S rRNA, except for the tertiary base pair G450-C483 in H17, and G521, which is adjacent to the pseudoknot in H18. Thus, these rRNA fragments were good candidates for binding protein S4.
A minimal rRNA substrate for S4
The relative binding affinities of the rRNA fragments for Bst S4 were measured by competition with 32P-labeled 5′ domain RNA as described above (Figure 1 and Table 1). A relative free energy ΔΔG < 0 indicates the competitor RNA binds protein S4 more weakly than the 16S 5′ domain (i.e. dissociation is more favorable). Among the initial substrates tested, the differences in the dissociation free energy were on the order of ±0.5 kcal/mol, comparable to the precision of the assay (Table 1). Remarkably, the smallest RNA containing just the five-helix junction (‘5WJ’, Figure 1e) bound protein S4 about 2-fold better than the 5′ domain (ΔΔG = 0.7 ± 0.2 kcal/mol) and 3-fold better than the 16S rRNA. Therefore, RNA interactions in the lower half of the 5′ domain do not contribute significantly to the stability of S4 complexes. A slightly different deletion of H6-14 that eliminated part of helix 5 reduced S4 affinity 5-fold in previous studies (26,27), most likely due to structural changes in the RNA.
Table 1.
Relative dissociation constants and free energies for S4 binding measured by competition against 5′ domain RNA
| Competition |
Dual-label |
|||
|---|---|---|---|---|
| RNAa | Krel | ΔΔG (kcal/mol) | Krel | ΔΔG (kcal/mol) |
| 16S rRNA | 1.6 ± 0.8 | –0.3 ± 0.4 | N.D.b | |
| RNA(ΔH6-14) | 1.1 ± 0.4 | –0.1 ± 0.2 | 0.5 ± 0.1 | 0.5 ± 0.1 |
| RNA(ΔH5-14) | 2.3 ± 0.2c | –0.5 ± 0.1c | 0.8 ± 0.3 | 0.2 ± 0.3 |
| 5WJ_nt6 | 0.8 ± 0.6 | 0.1 (−0.4, +0.9) | 0.4 ± 0.1c | 0.6 ± 0.1c |
| 5WJ | 0.4 ± 0.1 | 0.7 ± 0.2 | 0.6 ± 0.1 | 0.5 ± 0.2 |
| 5WJ:H18trunc | N.D.d | 0.9 ± 0.1 | 0.1 ± 0.1 | |
| 5WJ:BstH17 | N.D.d | 0.8 ± 0.4 | 0.2 (–0.3,+0.5) | |
| 5WJ:TthH17 | N.D.d | 5.8 ± 1.8 | –1.1 ± 0.2 | |
| 5WJ:C526A | N.D.d | 0.5 ± 0.2 | 0.4 ± 0.3 | |
aBinding affinity of S4 for competitor RNA was measured relative to 32P-labeled 5′domain RNA at 42°C in HKM4 buffer, as described in ‘Materials and methods’ section. Data were fit to Equation (1) (‘Competition’) or Equation (2) (‘Dual-label’). Krel = Kd Competitor/Kd 5′domain, where Kd 5′domain = 5.5 ± 2.6 nM, with free energies calculated from ΔΔG = –RTlnKrel at 315.15 K. The relative affinities and dissociation free energy values were averaged over three or more experiments for ‘Competition’, and from three to seven reactions for ‘Dual-label’ competitions, unless stated otherwise.
b16S rRNA and 16S–S4 complexes were poorly resolved.
cValues reported from two experiments.
dThese RNAs were tested against 5′ domain RNA only through dual-label competition.
Direct measurement of specific binding by competition
To evaluate whether our results were strongly biased by nonspecific binding of S4 (31), we also performed competitive binding experiments where both RNAs were 32P-labeled (Figure 3a). The advantage of this method is that the protein concentration is lower than the total RNA concentration, minimizing contributions from nonspecific interactions (34). To detect S4 binding to two labeled RNA substrates, an aliquot of each binding reaction was loaded on an 8% TBE gel to resolve the 5′ domain complexes, while another aliquot was loaded on a 6% TKM2 gel to resolve the smaller RNA complexes (Figure 3b). The results of the dual-label competition experiments were in good agreement with the competition assays done with only labeled 5′ domain and excess S4 (Table 1). Thus, we concluded that both sets of competition experiments were likely reporting the relative stabilities of specific S4 complexes.
Figure 3.

Dual-label competitive binding assay. (a) Scheme of the competitive binding experiments with two labeled RNAs. Complexes of the 5′ domain were separated on 8% polyacrylamide gels in TBE, while complexes of the 5WJ variants were separated on 6% polyacrylamide gels in TKM2, as described in ‘Material and Methods’ sections. (b) Distribution of S4 between 5′ domain and 5WJ RNAs. The counts in each complex and in free RNA were quantified and used to calculate Krel [Equation (2)]. For the 5WJ RNA, average Krel = 0.6 ± 0.1. The amount of labeled 5WJ complex decreases as unlabeled 5WJ RNA is added because its specific activity decreases. See Table 1 for further data.
The specificity of the complexes was further tested by exchanging the Eco H17 sequence in the 5WJ RNA for H17 from Bst (Figure 1g) or T. thermophilus (Tth) (Figure 1h). Of the helices that contact S4 directly, H17 varies most in length and sequence among bacterial rRNAs (37). While the sequences of the Eco and Bst 5WJ are 77% identical, H17 shares 42% sequence identity. Not surprisingly, the Bst H17 substitution was more favourable for binding of Bst S4 protein (Krel = 0.78) than the Tth H17 substitution (Krel = 5.8). Therefore, the sequence of H17 is important for S4 recognition.
S4 binds minimal 5WJ RNA very tightly
Further competitive binding experiments against the 5WJ RNA demonstrated that the minimal RNA could form very stable complexes with S4 (Kd ∼ 1 nM). The bound and free forms of the 5WJ RNA were more easily separated on native gels because this RNA is only 200 nt long (Figure S2b). In direct titrations of 5WJ with S4 protein, the average Kd was 13 ± 6 nM, similar to that for the 5′ domain•S4 complex (data not shown). However, competitive binding assays with labeled and unlabeled 5WJ RNAs results in a Kd of 0.72 ± 0.20 nM (Figure 4, closed diamonds), identical to the value of 0.7 nM obtained in nitrocellulose filter binding assays (31).
Figure 4.

Competitive binding of Bst S4 to 5WJ RNA. Representative plot of competition between 32P-labeled 5WJ RNA and unlabeled RNAs with 22.5 nM Bst S4 in HKM4 buffer. Diamonds, WT 5WJ (Kd = 0.72 ± 0.20 nM); circles, 5WJ:H17trunc (Krel = 4.1 ± 3.3); squares, 5WJΔH3 (Krel = 360 ± 130).
The relative affinities of the 16S rRNA and 5′ domain in competition with labeled 5WJ RNAs were Krel = 1.1 ± 0.4 and 2.3 ± 0.4, respectively (Table S2). Therefore, using the Kd for the 5WJ•S4 (0.72 nM) from the self-competition experiments as a reference, we obtain Kd (16S) = 0.7 nM and Kd (5′ dom) = 1.7 nM, again very similar to previous results. S4 is expected to have fewer opportunities to form nonspecific (or non-native) complexes with the smaller 5WJ RNA. This may explain why titrations against the smaller 5WJ RNA yield lower estimates for the S4 dissociation constants. Direct titrations of the 5WJ RNA also yielded a higher Kd than competitive binding experiments, because the former may include contributions from intermediate complexes. These contribute less significantly to competitive binding because the protein:RNA ratio is lower than in direct titrations.
Defining S4-binding site requirements
Since the 5WJ RNA bound S4 strongly and the complex was well resolved on nondenaturing gels, competitive binding assays against labeled 5WJ RNA were used to define the requirements for S4 binding in more detail. Variations in the 5′ and 3′ ends of the RNA substrates established that an RNA beginning at 16S nt 8 (H1) and ending at nt 556 bound S4 2-fold better than 5WJ RNA; others variants, including those containing H1, were 5–50-fold worse (Table S2).
Next, a series of deletions were made to the 5WJ RNA to see if any of the five helices in the S4 recognition site could be eliminated or truncated (Figure 1i–m). Truncating H17 between nts 446 and 487 or removing the 530-loop pseudoknot from H18 (Figures 4 and 5a) were well tolerated, presumably because the deleted residues do not contact protein S4 directly (15). By contrast, truncating H16 or deleting H3 or H4 from the 5WJ RNA had disastrous effects on binding, with ΔΔG = –3.5 kcal/mol reflecting the minimum free energy cost of the deletion to the stability of the complex (Figure 5a). In general, helices in direct contact with protein S4 at the center of the helical junction were necessary to form a stable complex, while nucleotides removed from the junction were dispensable for strong S4 binding (Figure 5b).
Figure 5.

Sequence requirements for S4 binding. Energetic costs of deletions and point mutations in the 5WJ were obtained from competitive S4-binding assays against 32P-labeled 5WJ RNA at 42°C in HKM4 buffer. See Table S2 for further information. (a,b) Deletions in the 5WJ, and differences in dissociation free energy (ΔΔG), relative to the parental 5WJ RNA. (c,d) Base substitutions, as in (a). (b) and (d) Tertiary structure of 5WJ•S4 showing the deletions and mutations, colored as in (a) and (c), respectively, with S4 in cyan. Ribbons made with PDB 2AVY (41) using PyMOL (DeLano Scientific, http://www.pymol.org).
Defining role of individual bases in S4 binding
To further define the sequence requirements for binding, nucleotides that are universally conserved in prokaryotes or in direct contact with S4 were mutated (Figure 5c). In the crystal structure of the T. thermophilus 30S subunit, S4 directly contacts the base and backbone of nts 404, 406 and 437, and the base of nt 405 (15). Single base substitutions at these positions in H16 and H17 reduced binding to the threshold of detection in our competition assays (ΔΔG ≤ –3.6 kcal/mol), reflecting the essential roles played by these nucleotides in S4 binding (Figure 5c). A509 and A510 are universally conserved in prokaryotes (37) and adjacent to the 530-loop pseudoknot in H18. Double mutation of these As to C also abrogated S4 binding, presumably because they disrupt interactions with the N-terminal domain of S4 (Figure 5d). Partial digestion with RNase T1 showed that the A509C, A510C double mutation destabilized the H18 pseudoknot, which could contribute to the loss of binding, while the mutation at U405C did not change the secondary structure of the rRNA (data not shown). The destabilizing effects of these point mutations were independent of whether they were made in the 5WJ or 5′ domain RNA (Table S2).
The H18 pseudoknot is universally conserved among 16S genes and required for efficient translation (38). To test whether the H18 pseudoknot is important for S4 recognition, base pairing of the pseudoknot was disrupted by mutating G524 to C and C526 to A (Figure 5c). While C526A made little difference to the stability of the S4 complex with the 5WJ RNA (ΔΔG = –0.4 kcal/mol), G524C had a more unfavorable effect (ΔΔG = −2.3 kcal/mol). This difference in S4 affinity likely relates to the ability of these mutated residues to base pair with nts 505 and 507, as hydrogen bonding between A526 and G505 is expected to be more favorable than hydrogen bonding between C524 and C507. Since deletion of nts 516–535 in H18 did not affect S4 binding (Figure 5a), however, the H18 pseudoknot cannot be a prerequisite for S4 recognition. Rather, its stability is important only if the potential to form the pseudoknot exists. These results suggest that the 530-loop can form an alternative structure in the absence of S4 that is promoted by mutations in H18, and that S4 binding offsets the energetic cost to correctly folding the wild-type 530-loop.
Tertiary structure of S4 complexes probed by hydroxyl radical footprinting
Hydroxyl radical footprinting of the rRNA backbone was used to determine whether the 5′ domain and 5WJ RNAs form the expected tertiary interactions with Bst and Eco S4 at 42°C (Figure 6). The results are summarized on the secondary structure of the 5WJ in Figure 7. In the presence of Bst or Eco S4, the hydroxyl radical protection pattern was very similar to that of the Eco 16S–S4 complex at 37°C (28). Thus, the 5WJ and 5′ domain RNAs are capable of forming the correct 3D structure in complex with S4 (Figure 7b and c). The few protections that were inconsistent with the predicted solvent accessibility of the 16S rRNA (Figure 7b) could be explained by limited diffusion of hydroxyl radicals 3′ of protected regions (nt 514–516 and 537–538) or an alternate conformation of an internal loop in H17 that is far from the S4-binding site (nts 446–448).
Figure 6.

Hydroxyl radical footprinting on 5′ domain and 5WJ RNAs with S4. Representative footprinting gels covering nucleotides 480–544. The RNAs were probed under different buffer conditions at 42°C (lanes 4–6, HKM4; lanes 7–9, HKM20), with Bst (lanes 5 and 8) and Eco (lanes 6 and 9) S4. UCGA, dideoxy sequence ladders; P, reverse transcriptase pausing control; NT, untreated RNA. The vertical bars next to the lanes represent regions of protection from hydroxyl radical cleavage. Black bars, RNA–RNA interactions, black circles, S4–RNA interactions.
Figure 7.

Structure probing of 5′domain and 5WJ RNAs with S4. (a) Representative sequencing gel showing DMS modification of 5WJ•S4 complex. The gel is labeled as in Figure 6, and symbols are as described in the key. (b,c) Summary of hydroxyl radical footprinting and chemical base modification on (b) 5′domain and (c) 5WJ RNA. Colored circles indicate backbone protections in HKM4. Colored circles with black outlines indicate riboses that are in direct contact with S4 [1j5e, (18); 2i2p, (42)]. Gray symbols, undetermined. Moderate-to-strong protection (squares) or enhanced base modification (triangles) is represented by closed red symbols. Open red symbols indicate Gs weakly or inconsistently modified by kethoxal.
The free 5′ domain and 5WJ RNAs were almost completely folded in 20 mM MgCl2 (HKM20) (Figure 6, lane 7 in both panels), as seen earlier for the 5′ domain RNA (29). Under these conditions, addition of S4 did not increase the number of residues protected by RNA–RNA interactions, although residues buried only by direct contact with the protein were protected more strongly in the presence of S4 (Figure 7b and c). In 4 mM MgCl2 (HKM4, Figure 6, lane 4 in both panels), only some of the RNA interactions were stable in the absence of S4. Under these conditions, many more residues became protected when the protein was added. These results demonstrate that S4-binding reactions in HKM4 involve binding and folding of the RNA. Importantly, the 5WJ RNA alone was more preorganized in 4 mM MgCl2 than the 5′ domain RNA (compare blue and pink circles in Figure 7b and c). The smaller RNA may take on fewer alternative conformations, explaining why the 5WJ RNA binds S4 slightly better than the longer 5′ domain.
Specific base interactions in 5′ domain and 5WJ complexes
The E. coli S4–16S rRNA complex undergoes a conformational change which changes the accessibility of bases in H18 to chemical modification (11). To further determine whether minimal RNA substrates form the correct structure, Eco and Bst S4 complexes were treated with DMS and kethoxal. Figure 7a shows the region 490–535 in 5WJ•S4 complexes, which contains most of the nucleotides modified by DMS when S4 is present.
Both the 5WJ and the 5′ domain RNA showed the expected base modifications in the presence of Bst S4 (4 mM MgCl2) or Eco S4 (20 mM MgCl2), including the ‘530-loop’ pseudoknot in H18 (Figure 7). The DMS modification pattern also confirmed that the secondary structures were correctly formed, in agreement with our RNase T1 results (data not shown). The reactivity of Gs toward kethoxal was largely consistent with previous results, although some products were difficult to assign due to very weak levels of kethoxal modification (Figure 7b and c).
We next tested whether the change in S4-binding affinity was directly linked to the loss of specific interactions between H18 and S4, by probing complexes containing mutations in the pseudoknot or truncation of H18. The C526A mutation or truncation of H18, which decreased relative S4 affinity very little (Table S2), resulted in the same modification pattern as the parental 5WJ RNA (Table S3). Conversely, the mutation G524C lowered the binding affinity 40-fold relative to intact 5WJ RNA (Table S2). In the G524C RNA, residues C524–526 that participate in the pseudoknot were modified in the presence of S4, in stark contrast to the results for every other RNA examined (Table S4). Thus, the reduction in affinity arising from modifications to the H18 pseudoknot are linked to loss of specific interactions with S4.
DISCUSSION
Protein S4 plays an important role in the early stages of 30S ribosome assembly, coordinating assembly of the subunit with production of its RNA and protein components (39). Here, we show that an RNA containing just the five helices within the S4-binding site (5WJ) are sufficient for specific RNA binding. Remarkably, the 200 nt 5WJ RNA binds S4 tightly and with comparable sequence-specificity as larger rRNA fragments, making it useful for further studies on the mechanism of S4–RNA recognition.
Mutagenesis and deletions of bases in H16 and H17 show that these residues make especially important contributions to the thermodynamic stability of the complex. Deletion of nt 410–432 from helix 16 reduced S4 binding more than 380-fold, while mutagenesis of nts 405 and 406 made binding undetectable. Sapag et al. (27) found that deletion of nts 415–432 reduced S4 affinity to 66% of wild type. This difference may be due to S4 interactions with A412 and backbone contacts to G410 and A411 (11,15) which were eliminated in our H16 truncation. Both H16 truncations ((27); this study) remove multiple backbone contacts made by S4 to nts 425–430 (15). Taken together, these results imply that sequence-specific interactions between S4 and bases within the rRNA are important for stable recognition.
A surprising result was that formation of the conserved H18 pseudoknot was not necessary for S4 binding, as deletion of this region had little effect on S4 affinity. On the other hand, destabilizing the pseudoknot, by mutating G524 to C for example, destabilized S4 binding about 40-fold, perhaps by altering the conformation of nts 508–512 that make direct contact with S4 (15). The double mutation in nucleotides 509 and 510 in the 530-loop also severely disrupted S4 binding. This may be due to an altered secondary structure in this region (data not shown).
Interestingly, interactions in helix 16 and 17 that are critical for the stability of the S4 complex form rapidly, as judged by time-resolved hydroxyl radical footprinting (40). By contrast, contacts between S4 and helix 18 saturate more slowly. Thus, interactions that stabilize the specific complex to the greatest extent correlate with those that saturate in the shortest time, supporting the notion that stable interactions at the center of the binding surface contribute to initial encounter complexes as well as the final complex.
Consistent with the idea that the structure of the rRNA is very important for the stability of the S4 complexes, hydroxyl radical footprinting reactions show that the 5WJ RNA is more structured in 4 mM MgCl2 than the same region in the 5′ domain RNA. This advantage may be due to the absence of helices 6–12, which can misfold in the absence of S4 and indirectly affect the conformational stability of the upper 5WJ (29). A more homogenous and stable conformation of the free RNA may not only increase the stability of the S4 complex, but also increase the probability of forming specific RNA–protein contacts. Thus, greater preorganization of the 5WJ RNA (and a higher yield of specific complex) may explain why competition experiments against this RNA yield lower estimates for the binding constant (0.7 nM) than experiments with the 5′ domain (5.5 nM). The kinetics of S4 dissociation from the 5′ domain RNA is consistent with the presence of different types of S4 complexes (Bellur,D.L. and Woodson,S.A., in preparation).
In summary, our results provide a new understanding of how a key ribosomal protein recognizes its rRNA target. We show that all of the five helices that contact S4 in the ribosome are necessary and sufficient for its specific binding to the rRNA. Although S4 makes only few direct contacts with bases in the 16S rRNA, these interactions, which lie near the center of the protein–RNA interface, contribute significantly to specific binding. Our results suggest that the preorganized five-helix junction is the likely target for S4 binding during the initial phase of 30S subunit assembly, and that S4 stabilizes the junction under physiological conditions. Although the H18 pseudoknot is not required for S4 binding, S4 also induces a conformational change in the H18 (‘530-loop’) pseudoknot (11).
These conformational changes, which are stabilized or induced by S4, promote the binding of further proteins during 30S assembly. S16 interacts with helices 15 and 17 that are part of the five-helix junction, while S12 binds the opposite face of helix 18. An interesting question that remains to be addressed is whether S4 must capture the 16S rRNA in a conformation that can assemble further, or whether, as suggested by kinetic footprinting (11,40), the S4–16S rRNA complex reaches its mature structure via induced fit, that in turn leads to subsequent assembly steps.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Institutes of Health (GM60809). Funding for open access charge: National Institutes of Health (GM60809).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank D. Draper for the gift of S4 overexpression plasmids, P. Ramaswamy, A. Cukras and R. Green for E. coli S4 protein, and T. Adilakshmi for 16S rRNA. We also thank D.D., T.A. and P. Fleming for helpful advice, and M. Koppenol, S. Jackson and R. Moss for technical assistance.
REFERENCES
- 1.Olsson MO, Isaksson LA. Analysis of rpsD mutations in Escherichia coli. III. Effects of rpsD mutations on expression of some ribosomal protein genes. Mol. Gen. Genet. 1979;169:271–278. doi: 10.1007/BF00382273. [DOI] [PubMed] [Google Scholar]
- 2.Yates JL, Arfsten AE, Nomura M. In vitro expression of Escherichia coli ribosomal protein genes: autogenous inhibition of translation. Proc. Natl Acad. Sci. USA. 1980;77:1837–1841. doi: 10.1073/pnas.77.4.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deckman IC, Draper DE. Specific interaction between ribosomal protein S4 and the alpha operon messenger RNA. Biochemistry. 1985;24:7860–7865. doi: 10.1021/bi00348a002. [DOI] [PubMed] [Google Scholar]
- 4.Tang CK, Draper DE. Evidence for allosteric coupling between the ribosome and repressor binding sites of a translationally regulated mRNA. Biochemistry. 1990;29:4434–4439. doi: 10.1021/bi00470a025. [DOI] [PubMed] [Google Scholar]
- 5.Spedding G, Draper DE. Allosteric mechanism for translational repression in the Escherichia coli alpha operon. Proc. Natl Acad. Sci. USA. 1993;90:4399–4403. doi: 10.1073/pnas.90.10.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torres M, Condon C, Balada JM, Squires C, Squires CL. Ribosomal protein S4 is a transcription factor with properties remarkably similar to NusA, a protein involved in both non-ribosomal and ribosomal RNA antitermination. EMBO J. 2001;20:3811–3820. doi: 10.1093/emboj/20.14.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nowotny V, Nierhaus KH. Assembly of the 30S subunit from Escherichia coli ribosomes occurs via two assembly domains which are initiated by S4 and S7. Biochemistry. 1988;27:7051–7055. doi: 10.1021/bi00418a057. [DOI] [PubMed] [Google Scholar]
- 8.Draper DE. Protein-RNA recognition. Annu. Rev. Biochem. 1995;64:593–620. doi: 10.1146/annurev.bi.64.070195.003113. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima S, Nomura M. Assembly mapping of 30S ribosomal proteins from E. coli. Nature. 1970;226:1214. doi: 10.1038/2261214a0. [DOI] [PubMed] [Google Scholar]
- 10.Stern S, Wilson RC, Noller HF. Localization of the binding site for protein S4 on 16 S ribosomal RNA by chemical and enzymatic probing and primer extension. J. Mol. Biol. 1986;192:101–110. doi: 10.1016/0022-2836(86)90467-5. [DOI] [PubMed] [Google Scholar]
- 11.Powers T, Noller HF. A temperature-dependent conformational rearrangement in the ribosomal protein S4.16 S rRNA complex. J. Biol. Chem. 1995;270:1238–1242. doi: 10.1074/jbc.270.3.1238. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann RA, Muto A, Fellner P, Ehresmann C, Branlant C. Location of ribosomal protein binding sites on 16S ribosomal RNA. Proc. Natl Acad. Sci. USA. 1972;69:1282–1286. doi: 10.1073/pnas.69.5.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greuer B, Osswald M, Brimacombe R, Stoffler G. RNA-protein cross-linking in Escherichia coli 30S ribosomal subunits; determination of sites on 16S RNA that are cross-linked to proteins S3, S4, S7, S9, S10, S11, S17, S18 and S21 by treatment with bis-(2-chloroethyl)-methylamine. Nucleic Acids Res. 1987;15:3241–3255. doi: 10.1093/nar/15.8.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies C, Gerstner RB, Draper DE, Ramakrishnan V, White SW. The crystal structure of ribosomal protein S4 reveals a two-domain molecule with an extensive RNA-binding surface: one domain shows structural homology to the ETS DNA-binding motif. EMBO J. 1998;17:4545–4558. doi: 10.1093/emboj/17.16.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brodersen DE, Clemons WM, Jr., Carter AP, Wimberly BT, Ramakrishnan V. Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16 S RNA. J. Mol. Biol. 2002;316:725–768. doi: 10.1006/jmbi.2001.5359. [DOI] [PubMed] [Google Scholar]
- 16.Markus MA, Gerstner RB, Draper DE, Torchia DA. The solution structure of ribosomal protein S4 delta41 reveals two subdomains and a positively charged surface that may interact with RNA. EMBO J. 1998;17:4559–4571. doi: 10.1093/emboj/17.16.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sayers EW, Gerstner RB, Draper DE, Torchia DA. Structural preordering in the N-terminal region of ribosomal protein S4 revealed by heteronuclear NMR spectroscopy. Biochemistry. 2000;39:13602–13613. doi: 10.1021/bi0013391. [DOI] [PubMed] [Google Scholar]
- 18.Wimberly BT, Brodersen DE, Clemons WM, Jr., Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 19.Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. Structure of functionally activated small ribosomal subunit at 3.3 angstroms resolution. Cell. 2000;102:615–623. doi: 10.1016/s0092-8674(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 20.Dragon F, Payant C, Brakier-Gingras L. Mutational and structural analysis of the RNA binding site for Escherichia coli ribosomal protein S7. J. Mol. Biol. 1994;244:74–85. doi: 10.1006/jmbi.1994.1705. [DOI] [PubMed] [Google Scholar]
- 21.Mougel M, Allmang C, Eyermann F, Cachia C, Ehresmann B, Ehresmann C. Minimal 16S rRNA binding site and role of conserved nucleotides in Escherichia coli ribosomal protein S8 recognition. Eur. J. Biochem. 1993;215:787–792. doi: 10.1111/j.1432-1033.1993.tb18093.x. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Jiang L, Zimmermann RA. The binding site for ribosomal protein S8 in 16S rRNA and spc mRNA from Escherichia coli: minimum structural requirements and the effects of single bulged bases on S8-RNA interaction. Nucleic Acids Res. 1994;22:1687–1695. doi: 10.1093/nar/22.9.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batey RT, Williamson JR. Interaction of the Bacillus stearothermophilus ribosomal protein S15 with 16 S rRNA: I. Defining the minimal RNA site. J. Mol. Biol. 1996;261:536–549. doi: 10.1006/jmbi.1996.0481. [DOI] [PubMed] [Google Scholar]
- 24.Ungewickell E, Garrett R, Ehresmann C, Stiegler P, Fellner P. An investigation of the 16-S RNA binding sites of ribosomal proteins S4, S8, S15, and S20 FROM Escherichia coli. Eur. J. Biochem. 1975;51:165–180. doi: 10.1111/j.1432-1033.1975.tb03917.x. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann RA, Mackie GA, Muto A, Garrett RA, Ungewickell E, Ehresmann C, Stiegler P, Ebel JP, Fellner P. Location and characteristics of ribosomal protein binding sites in the 16S RNA of Escherichia coli. Nucleic Acids Res. 1975;2:279–302. doi: 10.1093/nar/2.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vartikar JV, Draper DE. S4-16 S ribosomal RNA complex. Binding constant measurements and specific recognition of a 460-nucleotide region. J. Mol. Biol. 1989;209:221–234. doi: 10.1016/0022-2836(89)90274-x. [DOI] [PubMed] [Google Scholar]
- 27.Sapag A, Vartikar JV, Draper DE. Dissection of the 16S rRNA binding site for ribosomal protein S4. Biochim. Biophys. Acta. 1990;1050:34–37. doi: 10.1016/0167-4781(90)90137-q. [DOI] [PubMed] [Google Scholar]
- 28.Powers T, Noller HF. Hydroxyl radical footprinting of ribosomal proteins on 16S rRNA. RNA. 1995;1:194–209. [PMC free article] [PubMed] [Google Scholar]
- 29.Adilakshmi T, Ramaswamy P, Woodson SA. Protein-independent folding pathway of the 16S rRNA 5′ domain. J. Mol. Biol. 2005;351:508–519. doi: 10.1016/j.jmb.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 30.Nierhaus KH, Dohme F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc. Natl Acad. Sci. USA. 1974;71:4713–4717. doi: 10.1073/pnas.71.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerstner RB, Pak Y, Draper DE. Recognition of 16S rRNA by ribosomal protein S4 from Bacillus stearothermophilus. Biochemistry. 2001;40:7165–7173. doi: 10.1021/bi010026i. [DOI] [PubMed] [Google Scholar]
- 32.Culver GM, Noller HF. Efficient reconstitution of functional Escherichia coli 30S ribosomal subunits from a complete set of recombinant small subunit ribosomal proteins. RNA. 1999;5:832–843. doi: 10.1017/s1355838299990714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weeks KM, Crothers DM. RNA recognition by Tat-derived peptides: interaction in the major groove? Cell. 1991;66:577–588. doi: 10.1016/0092-8674(81)90020-9. [DOI] [PubMed] [Google Scholar]
- 34.Fields DS, Stormo GD. Quantitative DNA sequencing to determine the relative protein-DNA binding constants to multiple DNA sequences. Anal. Biochem. 1994;219:230–239. doi: 10.1006/abio.1994.1262. [DOI] [PubMed] [Google Scholar]
- 35.Moazed D, Stern S, Noller HF. Rapid chemical probing of conformation in 16 S ribosomal RNA and 30 S ribosomal subunits using primer extension. J. Mol. Biol. 1986;187:399–416. doi: 10.1016/0022-2836(86)90441-9. [DOI] [PubMed] [Google Scholar]
- 36.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 37.Gutell RR. Comparative sequence analysis and the structure of 16S and 23S rRNA. In: Zimmerman RA, Dahlberg AE, editors. Ribosomal RNA: Structure, Evolution, Processing, and Function in Protein Biosynthesis. Boca Raton, FL: CRC Press; 1996. pp. 111–128. [Google Scholar]
- 38.Powers T, Noller HF. A functional pseudoknot in 16S ribosomal RNA. EMBO J. 1991;10:2203–2214. doi: 10.1002/j.1460-2075.1991.tb07756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nierhaus KH. The assembly of prokaryotic ribosomes. Biochimie. 1991;73:739–755. doi: 10.1016/0300-9084(91)90054-5. [DOI] [PubMed] [Google Scholar]
- 40.Adilakshmi T, Bellur DL, Woodson SA. Concurrent nucleation of 16S folding and induced fit in 30S ribosome assembly. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 42.Berk V, Zhang W, Pai RD, Cate JH. Structural basis for mRNA and tRNA positioning on the ribosome. Proc. Natl Acad. Sci. USA. 2006;103:15830–15834. doi: 10.1073/pnas.0607541103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.