


Abstract
Whereas the strong and stable suppression of specific microRNA activity would be essential for the functional analysis of these molecules, and also for the development of therapeutic applications, effective inhibitory methods to achieve this have not yet been fully established. In our current study, we tested various RNA decoys which were designed to efficiently expose indigestible complementary RNAs to a specific miRNA molecule. These inhibitory RNAs were at the same time designed to be expressed in lentiviral vectors and to be transported into the cytoplasm after transcription by RNA polymerase III. We report the optimal conditions that we have established for the design of such RNA decoys (we term these molecules TuD RNAs; tough decoy RNAs). We finally demonstrate that TuD RNAs induce specific and strong biological effects and also show that TuD RNAs achieve the efficient and long-term-suppression of specific miRNAs for over 1 month in mammalian cells.
INTRODUCTION
miRNAs are small (20–24 nt) regulatory noncoding RNAs, which are initially transcribed as pri-miRNAs by RNA polymerase II (1). Pri-miRNAs are cleaved by Drosha in the nucleus to generate pre-miRNAs, which are in turn processed to mature miRNAs after the export to the cytoplasm. The miRNAs then modulate the expression of multiple target genes at the post-transcriptional level as a component of the RNA-induced silencing complex (RISC). In the case of complete complementarity between an miRNA and its target mRNA sequence, the miRNA will induce cleavage of this transcript, thus causing a rapid reduction in its levels. However, most mammalian miRNAs have limited levels of complementarity with their target sequences, located in the 3′ untranslated region (3′-UTR) of the mRNAs, and cause either translational inhibition or rapid deadenylation of these transcripts in cytoplasmic processing bodies (P-bodies). In this regard, it has been reported that miRNAs form many regulatory networks with coding genes (2). A growing body of evidence also now indicates that miRNAs, which are expressed from specific promoters (3), play important roles in differentiation, development, oncogenesis and in the cellular defence response to infection (4–6).
To perform comprehensive functional analysis of a specific miRNA molecule, a methodology that specifically inhibits its activity is essential. There are some available techniques designed to inhibit miRNA function, including the introduction of short, single-stranded oligonucleotides that are chemically modified such as 2′-O-methyl (2′-OMe) RNA (7,8), locked nucleic acid (LNA) molecules and ‘antagomirs’ (9,10). These reagents are chemically synthesized to have complementarity to mature miRNAs, are resistant to cellular nucleases, and probably also function as noncleavable substrates of RISC. However, since they are designed to be introduced into cells by transfection, their inhibitory effects are invariably transient.
DNA vectors that express a ‘microRNA sponge’, a competitive inhibitor of microRNA, have been recently described (11). The transient expression of these microRNA sponge vectors inhibits miRNA function efficiently but for no longer than 1 month. This may be partly due to the relatively low levels of the inhibitory RNAs in the stable transfectant which are expressed via RNA polymerase II. The establishment of a method that would inhibit miRNA for a more prolonged period was thus very desirable and was needed to enable a more detailed analysis of the networks formed between miRNA and coding genes. To enable the long-term suppression of miRNA, we chose a decoy RNA system using lentiviral vectors that harbour RNA expression cassettes driven by RNA polymerase III. We thereby designed a series of decoy RNA molecules that were predicted to be efficiently exportable to the cytoplasm, where the target mature miRNA is present. We have also optimized the entire secondary structures of these decoys and the sequence of miRNA-binding site (MBS) and have thus established a novel and highly potent miRNA inhibitory system which persists for well over 1 month.
MATERIALS AND METHODS
Plasmid construction
Oligonucleotide pairs, listed in Supplementary Table S1, were annealed and cloned into the pMXs-GIN vector (12) digested with NotI and SalI to generate pMXs-GIN-miR140-5pT and pMXs-GIN-miR140-3pT (Supplementary Figure S2A and B), GFP reporter vector to detect the activity of miR-140-5p and miR-140-3p, respectively. The 0.7-kb BamHI–EcoRI fragment of pMXs-GFP (13) was inserted into the BamHI–EcoRI site of pcDNA3.1 (Invitrogen) to generate pcDNA3.1-GFP. The BamHI and EcoRI sites of pcDNA3.1-GFP were blunt ended using Klenow fragment. The 1.7-kb BglII–EcoRV fragment of this plasmid was then inserted into the same sites of pQCXIH (Clontech) to generate pSSCG. The 1.3-kb HindIII–XbaI fragment of pIRES1Hyg (Clontech) was inserted into the equivalent sites of pcDNA3.1 to generate pCMV-Hyg. The 2.1-kb NruI–ApaI fragment of pCMV-Hyg was inserted into the NruI–EcoRV site of pSSCG to generate pSSCH. For the construction of miR-140-5p/140-3p retrovirus expression constructs, we amplified 0.5-kb mouse miR-140-5p/140-3p fragment by PCR from mouse genomic DNA using the primers listed in Supplementary Table S1, followed by cloning into pCR2.1 (Invitrogen). The 0.5-kb BamHI–XhoI fragment of this plasmid was cloned into the pSSCH vector digested with BamHI and SalI to generate pSSCH-miR140-5p/140-3p.
For parental lentivirus vector construct (pLSP), we first amplified 0.3-kb ▵U3/HIV-1 3′LTR fragment by PCR from pLenti6/V5-GW/lacZ (Invitrogen) using the primers listed in Supplementary Table S2 and cloned the PCR product into pCR2.1 to generate pCR2.1-▵U3/HIV-1 3′LTR. We amplified 4.1-kb fragment by PCR from pCR2.1-▵U3/HIV-1 3′LTR using the primers listed in Supplementary Table S2 and digested it with NheI and SalI. An oligo pair, listed in Supplementary Table S2, was annealed and cloned into this digested PCR fragment. The 0.3-kb XbaI–PmeI fragment of this plasmid was cloned between the XbaI–StuI sites of pLenti6/V5-GW/lacZ to generate pLenti6/V5-GW/lacZ-NBES. The 1.0-kb ClaI–XbaI fragment of pSSSP (14) was cloned between the ClaI and XbaI sites of pLenti6/V5-GW/lacZ-NBES to generate pLSP (Supplementary Figure S1D).
An oligo pair, listed in Supplementary Table S3, was annealed and cloned into pmU6 (15) digested with BbsI and EcoRI to generate pmU6-TuD-shuttle. For the construction of TuD RNA expression vectors in a lentiviral background, a series of oligonucleotide pairs were synthesized as listed in Supplementary Table S4. Each of these pairs was annealed and cloned into the pmU6-TuD-shuttle digested with BsmBI. Each mU6-TuD RNA cassette, the 0.4-kb BamHI–EcoRI fragment, was then subcloned into the lentivirus vector pLSP digested with BamHI and EcoRI. These cassettes were also inserted into the BamHI-EcoRI sites of pSL1180 (Pharmacia) to generate TuD RNA expression plasmids. An oligo pair, listed in Supplementary Table S3, was annealed and cloned into the pmU6 digested with BbsI and EcoRI to generate pmU6-protoshuttle. For the construction of Decoy RNA expression plasmids, a series of oligonucleotide pairs were synthesized as listed in Supplementary Table S5. Each oligo pair (Decoy RNA #001–009, 014–017) was annealed and cloned into the pmU6 lentiviral vector digested with BbsI and EcoRI. Oligo pairs for Decoy RNA #010–013 were annealed and cloned into the pmU6-protoshuttle digested with BsmBI. The oligo pair for Decoy RNA #018-022 was annealed and cloned into the pmU6-TuD-shuttle digested with BsmBI. For the construction of mU6-Decoy RNA #023 cassette, the oligo pair listed in Supplementary Table S5 was PCR amplified and cloned into pCR2.1. The 0.2-kb BsmBI fragment of this plasmid was then subcloned into the pmU6-TuD-shuttle digested with BsmBI. Each mU6-Decoy RNA cassette, the 0.4-kb BamHI–EcoRI fragment, was then further subcloned into a lentivirus vector pLSP digested with BamHI and EcoRI.
For the construction of luciferase reporter plasmids, the 0.8-kb KpnI–HindIII fragment of pGL4.74 (Promega) was cloned into pGL4.12 (Promega) digested with KpnI and HindIII to generate pTK4.12-CP+. An oligonucleotide pair, listed in Supplementary Table S6, was annealed and cloned into pTK4.12-CP+ digested with EcoRI and XbaI to generate pTK4.12. Oligonucleotide pairs, listed in Supplementary Table S6, were annealed and cloned into the pGL4.74 digested with XbaI and FseI to generate pGL4.74-T21 and pGL4.74-T16, respectively.
For the construction of the sponge-miR-21 expression vector, the oligo pair listed in Supplementary Table S7 was annealed, self-amplified by PCR and cloned into pCR2.1. The 0.2-kb XhoI–AgeI fragment of this plasmid was subcloned between the XhoI and AgeI sites of pLenti6/V5-GW/lacZ to generate pLenti6/CMV-sponge-miR-21/lacZ. The 0.3-kb XbaI–ApaI fragment of pCS2-Venus (16) was cloned between the XbaI and ApaI sites of pSL1180 to generate pSL1180-polyA. The 3.9-kb HindIII–AgeI fragment of pLenti6/CMV-sponge-miR-21/lacZ was cloned between the HindIII and AgeI sites of pSL1180-polyA to generate pSL1180-CMVsponge-21.
Cell culture and construction of stable cell lines
Cell lines, HeLaS3, PA-1, HCT-116, SW480, HT29, TIG-3/E/TERT and 3Y1 cells were cultured at 37°C in DMEM containing 10% fetal bovine serum (FBS). HeLaS3 cells were seeded at 1 × 105 cells per well in six-well plates and transduced after 24 h with pMXs-GIN, pMXs-GIN-miR140-5pT and pMXs-GINmiR-140-3pT viral stocks (<1 × 104 TU) in the presence of 8 µg/ml of Polybrene and selected with G418 (1 mg/ml) at 24 h after transduction. After 2 weeks of selection, the G418 was removed from the medium. HeLaS3 cells harbouring the miR-140-5p or miR-140-3p reporters were seeded at 1 × 105 cells per well in six-well plates and transduced after 24 h, with the pSSCH-miR140-5p/140-3p virus stock (<1 × 104 TU) in the presence of 8 µg/ml of Polybrene and selected with hygromycin (0.5 mg/ml) from 24 h after the transduction. After 2 weeks of selection, the hygromycin was removed from the medium.
Virus transduction and FACS analysis
HeLaS3 cells harbouring both the miR-140-5p reporter and miR140-5p/140-3p vector and HeLaS3 cells harbouring both miR-140-3p reporter and miR140-5p/140-3p vector were seeded at 1 × 105 cells per well in six-well plates in DMEM containing 10% FBS. After 24 h, the cells were transduced with each TuD RNA virus stock (2 × 105 TU) or Decoy RNA virus stock (2 × 105 TU) in the presence of 8 µg/ml of Polybrene. The medium was then changed to DMEM containing 10% FBS and puromycin (1 µg/ml) after a further 24 h. After 7 days of selection, the puromycin was removed from the medium. The GFP expression levels were measured using FACS Calibur (BD).
Purification of nuclear and cytoplasmic small RNAs
Nuclear and cytoplasmic cellular fractions were isolated from untransduced or TuD RNA tansduced HeLaS3 cells harbouring both miR-140-5p reporter and miR140-5p/140-3p vector 14 days after transduction. Fourteen dishes (10 cm in diameter) for each cell culture were rinsed twice with ice-cold phosphate-buffered saline (PBS), harvested in a further 2 ml ice-cold PBS by scraping, and centrifuged for 5 min at 1500 r.p.m. at 4°C. Cell pellets were suspended with NP-40 lysis buffer [10 mM Tris–HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40], incubated on ice for 10 min, and then centrifuged for 5 min at 1500 r.p.m. at 4°C. The cytoplasmic RNA fraction was prepared from the supernatant by adding ISOGEN (Wako Pure Chemical Industries, Osaka, Japan). Nuclear pellets underwent two additional washes with 4 ml of NP-40 lysis buffer. RNAs smaller than 200 bp were finally purified with mirVana miRNA Isolation Kit (Ambion, Austin, TX) from purified cytoplasmic and nuclear fractions, respectively according to the manufacturer's protocol. Five percent of the total yield of small RNAs from nuclear and cytoplasmic fractions was analysed by northern blotting to allow for a comparison with the same cell numbers.
Northern blotting analysis
Small RNAs were separated on denaturing 18% polyacrylamide gels in 80°C incubator, transferred to Hybond-XL membranes (Amersham BioSciences) and crosslinked with 60 mJ/cm2 UV. The synthesized DNA probe sequences are listed in Supplementary Table S8 and all probes were 5′end-labeled with [γ-32 P] ATP using T4 polynucleotide kinase (TAKARA). Hybridizations were performed using Ultrahyb-Oligo buffer (Ambion) at 37°C according to manufacturer's instructions and membranes were washed using 2× SSC, 0.5% SDS buffer. Radioactive signals were visualized using a FLA-5100 (FUJIFILM).
MiR qRT-PCR
Total RNA from TuD RNA transduced HeLaS3 cells harbouring both miR-140-5p reporter and miR140-5p/140-3p vector was prepared using the mirVana miRNA Isolation Kit (Ambion) 10 days after transduction. From HCT-116 cells that were transfected with TuD RNA expression vectors, total RNAs were also prepared using the mirVana miRNA Isolation Kit at 72 h after transfection. Expression of mature miRNAs was determined by miR-qRT-PCR using miRNA-specific looped RT-primers and TaqMan probes as recommended by the manufacturer (Applied Biosystems, CA). U6 snRNA was used as an internal control. PCR was performed in triplicate using the 7300 Real Time PCR System (Applied Biosystems).
Luciferase assays
PA-1, HCT-116, SW480, HT29, TIG-3/E/TERT and 3Y1 cells were seeded at densities of 1.5 × 105, 1.0 × 105, 3.5 × 105, 3.0 × 105, 7.0 × 104 and 1.2 × 105 cells per well, respectively, in 24-well plates in DMEM containing 10% FBS the day before transfection. The cells were transfected them in triplicate with Lipofectamine 2000 (Invitrogen) and 10 ng of pTK4.12 (Firefly luciferase plasmid), 100 ng of RLuc target reporter plasmid, and 500 ng of TuD RNA expression plasmid or 500 ng of ‘miRNA sponge’ expression plasmid. We cotransfected locked nucleic acid (LNA/DNA) antisense oligonucleotides at 50 nM. LNA/DNA antisense oligonucleotides were synthesized by ThermoElectron and contained locked nucleic acids at eight consecutive centrally located bases (17). The LNA/DNA antisense oligonucleotide sequences are listed in Supplementary Table S9. We performed all assays at 48 h after transfection using the dual luciferase assay (Promega, Madison, WI) on GLOMAX (Promega) for PA-1, SW480, HT29, TIG-3/E/TERT and 3Y1 cells. We assayed at 72 h after transfection for HCT-116 cells.
Western blotting
Total proteins were extracted from cells using 1.5× SDS Buffer and protein concentrations were measured using the Bio-Rad protein assay kit. Protein extracts were separated by 12% SDS–PAGE and transferred onto PVDF-membrane (Millipore). Immunoblotting was performed by incubating the membrane with the antibodies anti-PDCD4 (ab51495, Abcam) and anti-Actin (612656, BD transduction) for 2 h at room temperature. Secondary antibodies conjugated with horseradish peroxidase were incubated with the membranes for 1 h at room temperature after three washes with PBS with Tween-20. Signals were detected using ECL reagent (Amersham).
Assay for cell proliferation and apoptosis
PA-1 cells were seeded at 1 × 104 cells per well in 48-well plates in DMEM containing 10% FBS. After 24 h, the cells were transduced with each TuD RNA expression virus vector stock (1 × 105 TU) in the presence of 8 µg/ml of Polybrene. Medium was changed 24 h after transduction. Metabolic activity of the cells was determined using a luminescent ATP-based assay, CellTiter GLO (Promega), on GLOMAX (Promega) according to the manufacturer's instructions. PA-1 cells were seeded at 3 × 103 cells per well in 48-well plates in DMEM containing 10% FBS. After 18 h, the cells were transduced with each TuD RNA virus stock (6 × 103, 1.5 × 104 and 3 × 104 TU) in the presence of 8 µg/ml of Polybrene. Medium was changed 24 h after transduction. Forty-eight hours after transduction, caspase-3 and caspase-7 activities of the cells were determined using Caspase GLO 3/7 (Promega) on GLOMAX according to the manufacturer's instructions.
RESULTS
Development of sensitive assay system for detecting miR-140-3p activity
We developed a highly sensitive assay system for miR-140-3p activity using HeLaS3 cells which expresses marginal levels of endogenous miR-140-3p and miR-140-5p (18). We constructed a retrovirus-based GFP reporter harboring a 21-bp insert just downstream of the GFP gene that was fully complementary to the mature miR-140-3p (designated the miR-140-3p reporter) (Supplementary Figure S1A). We then transduced this reporter construct into HeLaS3 cells and established mixed cell populations that stably expressed GFP. We next constructed retrovirus vectors expressing RNA molecules containing the entire pre-miRNAs embedding both miR140-5p and miR140-3p (denoted the miR140-5p/140-3p vector; Supplementary Figure S1C). These vectors were transduced into HeLaS3 cells harboring the miR-140-3p reporter and mixed cell populations were obtained. These cells showed <20% of the GFP expression levels found in untransduced HeLaS3 cells harboring the miR-140-3p reporter (Supplementary Figure S2A–C).
Design and potency of prototype decoy RNAs
We designed decoy RNAs with simple secondary structures that would expose RNA sequences with perfect complementarity to mature miR-140-3p. These prototype decoys (Figure 1A) form a stem–loop structure, the loop of which comprises an MBS that is fully complementary to mature miR-140-3p in addition to three linker bases in both ends. To facilitate the stable expression of these decoy RNAs in cells, we utilized lentivirus vectors carrying short RNA expression cassettes driven by the RNA polymerase III promoter (mU6) which is present in both LTRs after integration (Supplementary Figure S1D). Since RNA transcripts generated by RNA polymerase III are terminated with UU or UUUU, the RNA decoys are expected to have 2–4-nt 3′-protruding ends in the stem (Figure 1A).
Figure 1.

Structures of decoy RNAs for miR140-3p. The bold black curved arrow represents the miRNA-binding site (MBS) (5′ → 3′). The red lines represent the linkers in these molecules. (A) Structure of the prototype-decoy RNA (Decoy RNA #001-006). (B) Structures of Decoy RNA #002 and #007-013. I, II, III and IV indicate the stems present in these molecules. The lengths of each stem sequence (I, II, III and IV) for the Decoy RNA panel are shown in Table 2. (C) Structures of Decoy RNA #018–022. The lengths of linker sequence for the Decoy RNA panel are shown in Table 4. (D) Structures of Decoy RNA #013, 020 and 023. The MBS sequence for the Decoy RNA panel are shown in Table 5.
We expected that our decoy RNAs synthesized in the cellular nuclei would be exported to the cytoplasm, and interact with the RISCs containing miR-140-3p. In this regard, the nuclear transport of small RNAs with a stem–loop structure has previously been examined in detail and the length of the stem has been found to be critical for the efficiency of this process via the function of Exp-5 (19). Moreover, since the binding activity of hairpin RNAs to Exp-5 has been shown to be drastically reduced when the length of the stem sequence is <16bp, we lengthened the stem of our prototype RNA decoys from 17 to 24 bp (Decoy RNA #001–#006, Table 1, Supplementary Figure S3). We utilized shRNA molecules that targeted the coding region of the bacteriophage Cre recombinase gene (shCre) as a negative control (NC) for the decoy vectors. Because the prototype RNA decoys with an 18-bp stem showed slightly higher inhibitory effects and also because dsRNAs over 20 bp are potential targets for digestion by Dicer in the cytoplasm, we decided to employ a stem length of 18 bp for all subsequent studies.
Table 1.
Effects of stem length on the inhibitory properties of prototype-decoy RNA molecules
| Decoy RNA | Length of stem | Relative GFP expression (%) |
|---|---|---|
| NCa | – | 16.9 (0.7)b |
| #001 | 17 | 23.5 (1.2) |
| #002 | 18 | 24.4 (0.8) |
| #003 | 19 | 22.2 (1.5) |
| #004 | 20 | 21.1 (1.5) |
| #005 | 21 | 23.8 (3.6) |
| #006 | 24 | 22.4 (2.5) |
aThe shRNA molecule that targeted the coding region of the bacteriophage shCre was used as a NC.
bThe expression levels were normalized to those of HeLaS3 harboring the miR-140-3p reporter alone and are represented by the mean SEM (n = 3).
Optimization of decoy RNA inhibitor potency by altering the secondary structure and the MBS of the prototype molecules
To increase the potency of our decoy RNAs, we altered the design of these molecules so that their secondary structures were more complex than the prototypes (Figure 1B). We again inserted the corresponding expression cassettes into lentivirus vectors and introduced the resulting constructs into the same HeLaS3 cell system. These newly designed decoy RNAs (Decoy RNA #007–#013, Figure 1B, Supplementary Figure S3) were found to be more efficient than the earlier prototype molecule, Decoy RNA #002 (Table 2). Importantly, when a 3-nt linker sequence was inserted at both ends of the MBS of Decoy RNA #009 (to produce Decoy RNA #013) the GFP expression levels in the transduced cells were equivalent to HeLaS3 cells harbouring the miR-140-3p reporter alone (Table 2). This indicates a complete suppression of the miR-140-3p activity conferred by the exogenous miR-140-5p/miR-140-3p vector.
Table 2.
Comparison of the inhibitory effects of decoy RNAs with different stem-structures
| Decoy RNA | Length of the stem (bp)a |
Relative GFP expression (%) | |||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| NC | – | – | – | – | 16.9 (0.7) |
| #002 | 18 | – | – | – | 24.4 (0.8) |
| #007 | 18 | 8 | – | – | 28.2 (2.6) |
| #008 | 18 | – | – | 8 | 26.1 (1.8) |
| #009 | 18 | 8 | – | – | 33.7 (2.7) |
| #010 | 18 | 8 | – | – | 42.4 (2.9) |
| #011 | 18 | 8 | 8 | – | 51.7 (1.1) |
| #012 | 18 | 8 | 8 | – | 55.2 (1.3) |
| #013 | 18 | 8 | – | – | 99.5 (5.7) |
aSten I, II, III and IV indicate stem structures shown in Figure 1B.
We screened several alternative MBS sequences to further optimize the decoy activity. It has been reported that MBS regions which contain four extra nucleotides between positions 8 and 9 from the 3′ end of a perfectly complementary MBS, cause translational attenuation rather than RNA cleavage (20). We thus speculated that an MBS containing extra nucleotides may more strongly inhibit miRNA function than a fully complementary MBS (MBS-pf) as cleavage of the decoy RNA molecules themselves would be avoided. To screen for the most potent MBS, we modified our prototype decoy molecule (see Figure 1A) by inserting 1–4 nt between positions 10 and 11 from 3′ end of the perfectly complementary MBS, where the RISC cleaves the target mRNAs. When we tested these new decoys (Decoy RNA #002, #014–#017, Supplementary Figure S3), we found that the 4-nt insertion (MBS-4ntin) produced the largest recovery of GFP expression among the decoys tested (52%; Table 3).
Table 3.
MBS sequences in the prototype-decoy RNA significantly affect its inhibitory potency
 |
As the insertion of linkers between the MBS and the stem in Decoy RNA #009 greatly increased its inhibitory effects (Figure 1B and Table 2; compare Decoy RNA #009 and #013), we examined the effects of linker length changes of between 1 and 5 nt using Decoy RNA #009 type RNA, the MBS of which is substituted with MBS-4ntin (Decoy RNA #018–022, Figure 1C and Supplementary Figure S3). Insertions of 1–3 nt resulted in strong inhibitory effects, whereas the 4-nt or 5-nt-insertion decoys showed only moderate suppressive activity (Table 4). We thereafter adopted the 3-nt linker length between the MBS and the stem RNA in our analyses.
Table 4.
Comparision of the inhibitory effects of decoy RNAs with different length of linker sequences
| Decoy RNA | Length of linker | Relative GFP expression (%) |
|---|---|---|
| NC | – | 16.9 (0.7) |
| #018 | 1 | 96.4 (12.4) |
| #019 | 2 | 93.5 (11.4) |
| #020 | 3 | 103.9 (20.4) |
| #021 | 4 | 60.6 (9.9) |
| #022 | 5 | 70.1 (13.7) |
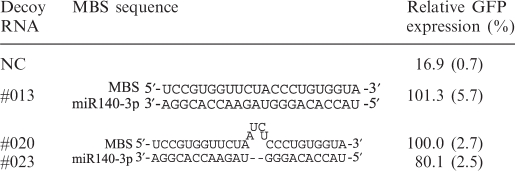
We integrated our optimization results in an attempt to increase the inhibitory effects of the Decoy RNAs #012 (see Figure 1B). We thus inserted 3-nt linkers between the stem sequence and MBS and also substituted MBS-pf with MBS-4ntin to generate Decoy RNAs #023 (Figure 1D and Supplementary Figure S3), respectively. Decoy RNA #023 showed a much improved potency (an increased GFP expression recovery from 55.2% to 80.1%; Tables 2 and 5). We also tested Decoy RNA #020, but there was no improvement in Decoy RNA #020 when compared with Decoy RNA #013. It would be because their inhibitory activities were almost saturated to fully cancel out the exogenously introduced miR-140-3p activity.
Table 5.
Effects of MBS sequences and linkers connecting the MBS and stem regions
 |
From these results, we tentatively concluded that the structures of Decoy RNA #013 and #020 (Figure 2A) have great potential in the design of efficient inhibitors of miRNA function. These decoys contain two MBS regions, which are flanked by two stem structures through 3-nt-linkers. We have tentatively designated this decoy TuD (Tough Decoy) RNA and further analysed its characteristics and specificity as outlined below. The TuD RNA expression cassette can be quite readily constructed as shown in Figure 2B.
Figure 2.

(A) Representative structure of the TuD RNAs. (B) Schematic representation of the generation of TuD RNA expression cassettes driven by mouse U6 promoter. 80–90mer synthetic oligonucleotides pairs are annealed and cloned between the two BsmBI sites present in the BamHI–EcoRI fragment (the original cassette) to generate TuD RNA expression cassettes.
Generality, specificity and duration of the inhibitory effects of TuD RNAs
To test the generality and specificity of the inhibitory effects of TuD RNA, we constructed a reporter cell system for miR-140-5p in which the insertion sequence of the miR-140-3p reporter was substituted with a sequence that was fully complementary to miR-140-5p (Supplementary Figure S1B). The inhibitory effects of TuD-miR140-3p, harbouring an MBS with four extra nucleotides (TuD-miR140-3p-4ntin; Decoy RNA#020) and TuD-miR140-5p-4ntin (Supplementary Figure S4) were determined using both the miR-140-3p and miR-140-5p reporter cell systems. In the miR-140-3p reporter system, TuD-miR140-3p-4ntin introduction resulted in a complete recovery of GFP expression but TuD-miR140-5p-4ntin expression showed only marginal effects (Figure 3A and Supplementary Figure S2D and E). In the miR-140-5p reporter system, the converse result was obtained with these two TuD RNA constructs (Figure 3B). These results indicate that the inhibitory effects of these TuD RNAs are highly efficient and specific to their target miRNAs, lending further support to the wide applicability of these molecules to miRNA suppression.
Figure 3.

Generality, specificity and duration of the inhibitory effects of TuD RNA. (A) Effects of TuD-miR140-5p-4ntin or TuD-miR140-3p-4ntin on miR-140-3p activity detected by the GFP reporter cell system as shown in Table 1, except that the GFP expression levels were determined at 8–12 days post-transduction (Supplementary Figure S3D and E). (B) Effects of TuD-miR140-5p-4ntin or TuD-miR-140-3p-4ntin on miR-140-5p activity. HeLaS3 cells harbouring both the miR-140-5p reporter and the miR140-5p/140-3p vector were transduced with the lentivirus vectors expressing the corresponding TuD RNAs and the GFP expression levels were determined at 8–12 days post-transduction. The expression levels were normalized to those of HeLaS3 cells harbouring miR-140-5p reporter alone and are represented by the mean ± SEM (n = 3). (C) Time course of the inhibitory effects of TuD-miR140-3p-4ntin on miR-140-3p activity. Relative GFP expression levels were determined as shown in Table 1. (D) Levels of mature miR-140-5p in the cells described in (B) determined by quantitative real-time RT-PCR. The miR-140-5p expression levels were normalized to those of HeLaS3 cells harbouring both the miR-140-5p reporter and the miR140-5p/140-3p vector and are represented by the mean ± SEM (n = 3). U6 snRNA was served as an endogenous control. (E) Analysis of the sub-cellular localization of TuD RNAs. The migration positions of Y4 small cytoplasmic RNA (Y4 scRNA, 93 nt) and ACA1 small nucleolar RNA (ACA1 snoRNA, 130 nt) on the same gel are indicated by black and open triangles, respectively. Y4 scRNA and ACA1 snoRNA were served as marker RNAs of cytoplasmic and nuclear fractions, respectively. Un, untransduced cells; 5p, TuD-miR140-5p-transduced cells; 3p, TuD-miR140-3p-transduced cells; N, nucleus; C, cytoplasm.
One important benefit of using lentivirus-based vectors is that the expression of the decoy RNAs is maintained over long periods. We therefore transduced the TuD-miR140-3p-4ntin vector into HeLaS3 cells harbouring both the miR-140-3p reporter and the miR140-5p/140-3p vector and monitored the time course of the GFP expression levels for more than 1 month (Figure 3C). The results showed that TuD-miR140-3p-4ntin, but not TuD-miR140-5p-4ntin, efficiently inhibited the target miR-140-3p in HeLaS3 cells over the duration of this experiment.
We directly determined the miR-140-5p levels using quantitative real time RT-PCR. TuD-miR140-5p-4ntin but not TuD-miR140-3p-4ntin, induced a ∼65% reduction in the apparent expression levels of miR-140-5p (Figure 3D). This suggested that whereas the amount of free miR-140-5p was reduced to about 1/3 of its control levels, it was possible that dsRNA molecules formed between TuD RNA and mature miRNA were too stable for the mature single-stranded miRNA to be detected by real time PCR (21).
Sub-cellular localization of TuD RNAs
We analysed the sub-cellular localization of TuD RNA in HeLaS3 expressing TuD-miR140-3p-4ntin or TuD-miR140-5p-4ntin. Fourteen days after transduction, both nuclear and cytoplasmic RNA fractions were prepared from the cells and analysed by northern blotting analysis after PAGE at high temperature to minimize intra- and inter-molecule base-pairing. Specific probes for TuD-miR140-5p-4ntin and TuD-miR140-3p-4ntin RNA (122 nt and 120 nt, respectively) detected sharp bands of 120 nt and the broad bands around 90 nt (Figure 3E). Since the same RNA samples showed much stronger signals in these lower broad bands when PAGE was performed at room temperature, the full-length TuD–RNA molecules with intra-molecule base pairing formed these bands. TuD-miR140-3p-4ntin and TuD-miR140-5p-4ntin molecules were shown to mainly reside in cytoplasm, whereas a small portion of TuD-miR140-5p-4ntin molecule was also detected in cellular nuclei. These results confirm efficient nuclear export of the synthesized TuD RNA molecules and also suggest that the sequence of the MBS portion in each TuD RNA molecule slightly affects nuclear export efficiencies.
The inhibitory effects of TuD RNA on endogenous miRNA
To examine whether the inhibitory functions of TuD RNA had broader applicability, we next performed several experiments using a different miRNA, endogenous miR-21, as the target and employed different assay systems whereby a comparison with traditional synthetic miRNA inhibitors was possible. We designed two TuD-miR 21s, TuD-miR21-4ntin and TuD-miR21-pf, via the same principles and procedures used to generate TuD-miR 140-3p-4ntin, and TuD-miR140-3p-pf, respectively (Supplementary Figure S4). We next tested whether these decoys would suppress endogenous miR-21 activity in PA-1 cells and transiently cotransfected them using DNA-based vectors together with Dual Luciferase Reporters (DLR) i.e. the Renilla luciferase reporter with or without the insertion of a 22-bp DNA sequence fully complementary to the mature miR-21 into its 3′-UTR as well as the Firefly luciferase reporter as a transfection control (Supplementary Figure S5A–C). As shown in Figure 4A, PA-1 cell populations expressing TuD-miR21-4ntin showed a 25-fold higher expression of miR-21 reporter Renilla luciferase activity, normalized with Firefly luciferase activity, compared with a vector for negative control (TuD-NC) (Supplementary Figure S4). TuD-miR21-pf showed lower inhibitory effects but still induced a significant recovery corresponding to about a 14-fold activation. Importantly, the levels of recovered reporter activity were close to those of the control reporter without the miR-21 target sequence. As expected, this control reporter activity was found to be very constant and independent of the decoy RNAs introduced. Transfection of locked nucleic acid antisense oligonucleotides (LNA/DNA), LNA/DNA-miR21 showed a 2-fold higher expression of the miR-21 target reporter compared with LNA/DNA-scramble transfected PA-1 cells (Figure 4A), showing that TuD RNAs have a much greater inhibitory potency towards miRNA than conventional synthetic reagents under these transfection conditions.
Figure 4.

Inhibitory effects of TuD RNA on endogenous miR-21 activity. TuD RNA expression plasmid vectors or LNA/DNA antisense oligonucleotides were transiently transfected into PA-1 (A) or HCT-116 (B) cells together with the Renilla luciferase miR-21 reporter (miR-21-RL) (open bars) or the untargeted control Renilla luciferase reporter (UT-RL) (black bars) as well as the Firefly luciferase reporter (FL) as a transfection control. After performing a dual luciferase assay, the expression levels were normalized to the ratio of the activity of miR-21-RL to that of FL in TuD-NC vector-transfected PA-1 (A) or HCT-116 (B) cells and are represented by the mean ± SEM (n = 3). (C) Comparison of the inhibitory effects of TuD RNA and a conventional miRNA inhibitory vector. A dual luciferase assay was performed 72 h after transfection in HCT-116 cells. The ratio of miR-21-RL/FL to UT-RL/FL are represented by the mean ± SEM (n = 3) as the expression levels. (D) The levels of mature miR-21 in RNA from HCT-116 cells transfected with TuD-miR21 expression vectors determined by quantitative real time RT-PCR. The miR-21 expression levels were normalized to those of HCT-116 cells transfected with TuD-NC expression vector and are represented by the mean ± SEM (n = 3). U6 snRNA was served as an endogenous control. (E) The levels of pre- and mature miR-21 in the same RNA samples as (D) determined by northern blotting. Y4 scRNA was served as a loading control.
We performed similar experiments using HCT-116 cells, which reportedly have higher levels of endogenous miR-21 than PA-1 cells (18,22). We obtained very similar results in this cell system, except that the inhibitory effects of TuD-miR21-pf were dramatically lower than in PA-1 cells (Figure 4B). We have hypothesized that large amounts of endogenous miR-21 would efficiently cleave the TuD-miR21-pf molecule thus significantly reducing its concentration as compared with TuD-miR21-4ntin. To test this hypothesis, we determined the expression level of TuD-miR21-4ntin and TuD-miR21-pf in PA-1 and HCT-116 cells transfected in similar procedures to those shown in Figure 4A and B, except that the reporter plasmids were not included in the transfection. In PA-1 cells, both TuD-miR21-4ntin and TuD-miR21-pf were detected as strong bands (Figure 5A). In HCT-116 cells, TuD-miR21-4ntin was also highly expressed, whereas the expression level of TuD-miR21-pf was very low (Figure 5B), consistent with our hypothesis that TuD-miR21-4ntin is stable, whereas TuD-miR21-pf is susceptible to digestion, especially when cellular concentrations of miR-21 are high. Considering these results, TuD-miRX-4ntin should be used rather than TuD-miRX-pf, which does not have the expected activity as Tough Decoy.
Figure 5.

Detection of TuD RNA molecules by northern blotting. TuD RNA expression plasmid vectors were transfected into PA-1 (A) or HCT-116 (B) cells and TuD-miR21-4ntin and TuD-miR21-pf were detected by northern blotting. The positions of Y4 scRNA (93 nt) and ACA1 snoRNA (130 nt) are indicated by black and open triangles, respectively. Y4 scRNA was served as a loading control.
We next compared the inhibitory effects of transiently transfected TuD RNA expression vectors with those of another miRNA inhibition vector, ‘miRNA sponge’ using this miR-21 luciferase reporter system in HCT-116 cells (Figure 4C). Transfection of LNA/DNA-miR21 and ‘miRNA sponge’ expression vector showed a 2.3-fold and 2.5-fold higher expression of the miR-21 target reporter compared with LNA/DNA-scramble transfected HCT-116 cells, respectively. On the other hand, TuD-miR21-4ntin and TuD-miR21-pf showed 63-fold and 5.5-fold higher expression. These results show that TuD RNAs have a much greater inhibitory potential towards miRNA than this conventional miRNA inhibition vector under these transfection conditions.
Effect of TuD RNA on the expression levels of the target miRNA
Using real time RT-PCR, the apparent expression levels of miR-140-5p were shown to be reduced by TuD-miR140-5p induction (Figure 3D), and similar results were obtained when RNA samples prepared from similar cultures used in Figure 4B were analysed by real time RT-PCR (Figure 4D). We next analysed the pre-miR-21 and mature miR-21 levels of untransfected or TuD RNAs transfected HCT-116 cells by northern blot analysis after PAGE at high temperature to minimize intra- and inter-molecule base-pairing (Figure 4E). Importantly, this northern blotting analysis indicated that the levels of not only pre-miR-21 but also mature miR-21 were unaffected in the presence of the corresponding TuD RNAs. These apparently contradictory observations would indicate that the absolute amount of mature miR-21 is unaffected by TuD RNA introduction, and that efficient and stable formation of base-pairing between mature miR-21 and TuD RNA molecules would have hampered the detection of mature miR-21 by real-time RT-PCR as has been reported in another RNA inhibitor of miRNA (21).
Efficient suppression of endogenous miR-21 activity by stably transduced TuD-miR21 by lentivirus vector
As for biological activity of miR-21, several reports have revealed that it often stimulates cellular proliferation and plays anti-apoptotic roles in several human tumour cell lines (23,24). To test biological effects of the specific suppression of endogenous miR-21 activity, we have transduced lentivirus vectors expressing TuD-miR21-4ntin and TuD-NC into PA-1 cells at a multiplicity of infection (MOI) of 10, where 10 copies of vector provirus in average are integrated into each cell in the transduced culture. We then analysed cellular proliferation by ATPase-based assay (Figure 6A). Cells transduced with TuD-miR21-4ntin have shown only marginal growth (1.6-fold on 4 days after the transduction), whereas TuD-NC transduced cells have grown to more than 5-fold. When an endogenous miR-21 target, PDCD4 protein (24,25) was monitored in the parallel cultures, the expression of PDCD4 was found to be increased by TuD-miR21-4ntin compared with TuD-NC (Figure 6B). When the enzymatic activities of caspase-3 + 7 per cell were determined in similar cultures 2 days after the transduction, for apoptosis by those in TuD-miR21-4ntin transduced cells was about 7- and 3-fold of those in untransduced cells and TuD-NC transduced cells, respectively (Figure 6C), indicating that induction of apoptosis would at least partly explain the reduction of overall growth rate. These cytostatic phenotypes have become detectable even when lentivirus was transduced at an MOI of 2.
Figure 6.

Biological effects induced by TuD RNA. (A) Inhibition of endogenous miR-21 function induces growth suppression and apoptosis. Cell growth assay was performed after PA-1 cells were transduced with lentivirus vectors expressing either TuD-miR21-4ntin or TuD-NC at an MOI of 10. Cell proliferation was monitored by ATPase based assays and normalized to those of untransduced PA-1 cells on Day 0 and are represented by the mean ± SEM (n = 3). (B) Effects of transduced TuD RNA expression lentivirus vectors upon the expression of endogenous PDCD4 protein, a target of miR-21. PA-1 cells were transduced with the TuD RNA expression lentivirus vectors with an MOI of 10 and total proteins were prepared 20 h after transduction. PDCD4 (top) as well as the β-actin loading control (bottom) were detected by western blotting. (C) Caspase-3 + 7 activation was assayed 48 h after transduction with indicated TuD RNA expression lentivirus with an MOI of 2, 5 and 10. Caspase-3 + 7 activity levels/cell was normalized to that of untransduced PA-1 cells and are represented by the mean ± SEM (n = 3).
The effects of TuD RNA on other family members of the target miRNA
To examine the specificity of miRNA more rigidly, we next tested whether the TuD RNA targeted to a miRNA exhibits inhibitory effects to other family members of the miRNA, which share the same core sequence (2–8 bases from the 5′-end of the miRNA). We have chosen the miR-15a/-15b/-16/-195/-424/-497 family and tested in HCT-116 cells, where expression of miR-16 is predominant (Supplementary Figure S6). So we expect the reporter containing the sequence perfectly complementary to miR-16 is mainly reflecting amounts of functional miR-16. In this setting, we induced high-level expression of TuD-miR195-4ntin and TuD-miR497-4ntin, respectively in HCT-116 cells, in which endogenous miR-195 and miR-497 are expressed only marginally (Supplementary Figure S6). The results of reporter assays (Figure 7) indicated the inhibitory activity of TuD-miR195-4ntin is no less than that of TuD-miR16-4ntin, whereas TuD-miR497-4ntin has slightly reduced the inhibitory effect when compared with TuD-miR16-4ntin. Since the reporter expression is not expected to result from the suppression of endogenous miR-195 or miR-497, we concluded that TuD-RNA do not basically discriminate miRNA members that belong to the same miRNA family.
Figure 7.

Inhibitory effects of TuD RNA on other family members of the target miRNA. (A) RNA sequences of miR-16, 195 and 497 and homology between them. Sequences shown in blue box are the heptameric seed sequence (2–8 from the 5′ end). Black bars indicated homologous nucleotides. (B) The imperfect pairing between miR-16 and MBSs of the corresponding TuD RNAs. Black dots indicate G-U pairs. (C) TuD RNA expression plasmid vectors were transiently transfected into HCT-116 cells together with the Renilla luciferase miR-16 reporter (open bars) or the untargeted control Renilla luciferase reporter (black bars) as well as the Firefly luciferase reporter as a transfection control. After performing a dual luciferase assay, the expression levels were normalized to the ratio of the activity of miR-16-RL to that of FL in TuD-NC vector-transfected cells and are represented by the mean ± SEM (n = 3).
DISCUSSION
We expressed a series of designed RNA molecules with potential RNA decoy activity and screened their ability to suppress specific miRNAs using several highly sensitive human assay cell systems as well as direct quantification of a protein targeted by an endogenous microRNA. We conclude from our analyses that TuD RNAs are efficient and long-term suppressors of miRNAs.
A TuD RNA stem of 18 bp in length (Figure 1B, stem I) promotes efficient transport to the cytoplasm via the function of Exp-5 (Figure 3E). The two stems flanking the MBS (stems I and II) provide some resistance to cellular RNase as well as to degradation by the RISC complex containing the corresponding miRNA. The linker sequences (∼3 nt) between the MBS and the flanking stems markedly increase the MBS accessibility to the corresponding miRNA in a RISC. The combination of these characteristics significantly contributes to the strong inhibitory effects of TuD RNAs.
We designed RNA decoys with more complex structures than the TuD RNAs, and found that they also functioned as efficient RNA decoys (Figure 1B and D, and Tables 2, 5). Our data, however, show that TuD RNA (Decoy RNA #020) inhibits miRNA more efficiently than Decoy RNA #023 (Table 5).
An MBS region that was perfectly complementary to the corresponding mature miRNA (MBS-pf) and an MBS that had an additional 4-nt insert between nucleotides 10 and 11 (MBS-4ntin) were also compared. Whereas MBS-4ntin was basically found to confer more inhibitory efficiency than MBS-pf (Figure 4A and B), both had similar miRNA inhibitory effects under conditions of near saturation (compare Decoy RNA #013 and #020 in Table 5). In the HCT-116-miR-21 reporter system, where endogenous miR-21 is abundant, TuD-miR21-4ntin inhibited miR-21 much more efficiently than TuD-miR21-pf (Figure 4B), whereas in the PA-1-miR-21 reporter system, where endogenous miR21 is less abundant, both of these TuD RNAs showed high levels of inhibition, which were more efficient than the LNA-miR21 inhibitor. To explain these phenomena, we analysed the expression levels of TuD RNAs and found that a high concentration of miR-21 in HCT-116 cells would lead to the efficient cleavage of TuD-MBS-pf itself (Figure 5B), which, in turn, significantly reduces the inhibitory potential of this RNA decoy. We therefore recommend that, as a TuD RNA molecule, TuD-miRX-4ntin should be used rather than TuD-miRX-pf, which do not have the expected full activity as Tough Decoy. As for TuD-miR21-4ntin, its potent inhibitory effects were shown by Dual luciferase reporter assay in a human teratocarcinoma cell line (PA-1) (Figure 4A) and a human colon cancer cell line (HCT-116) (Figure 4B). Whereas we usually transfect 500 ng of this TuD-miR21-4ntin expression plasmid into a well (24-well plate), analysis of dosage-dependency of this plasmid indicated that input plasmid DNA is saturated around 300 ng/well (24-well plates) and that the DNA amount as little as 10 ng/well still show about 1/3 of the full luciferase recovery when tested in HCT-116 cells (Supplementary Figure S7). Importantly, strong inhibitory effects of this TuD RNA were also observed in human colorectal tumor cell lines (SW480 and HT29), an immortalized human diploid embryonic lung fibroblast cell line (TIG-3/E/TERT) (26) and also a rat fibroblast cell line (3Y1) as shown in Supplementary Figure S8.
Importantly, we found from our analyses that TuD RNA achieves stable and long-term suppression of its target miRNA (Figure 3C). We also showed that TuD RNA can specifically and strongly inhibit endogenous miRNA to induce specific biological effects. This tool could therefore be applied to the analysis of regulatory networks which is formed either transiently or stably between miRNA and coding genes. This is also applicable to the construction of a knockdown mouse for specific miRNAs. Since the mouse U6 promoter that is regulated by Cre-loxP has already been well established (27,28), time- and tissue-specific miRNA knockdown in mice should be possible using modified TuD RNA expression systems. As shown in Figure 2B, the TuD RNA expression cassette is relatively easy to construct, it is also possible to prepare a TuD RNA library against the entire human miRNA repertoire to enable comprehensive analyses of these molecules in future studies. Because of these properties, TuD RNAs will be very useful tools for future studies of miRNA biology and potentially also for the design of novel human gene therapies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) (grant number 17016015); a grant from the Japan Society for the Promotion of Science (to T.H); and Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases funded by the contract research fund from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Funding for open access charge: a grant in AID for Scientific Research on Priority Areas from MEXT.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Miyawaki for providing the pCS2-Venus plasmid. We are grateful for M. Ohno for advice concerning detection of short RNA with hairpin structures by northern blotting analysis. We also thank S. Kawaura and A. Kato for assistance in the preparation of this manuscript.
REFERENCES
- 1.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Fujita S, Iba H. Putative promoter regions of miRNA genes involved in evolutionarily conserved regulatory systems among vertebrates. Bioinformatics. 2008;24:303–308. doi: 10.1093/bioinformatics/btm589. [DOI] [PubMed] [Google Scholar]
- 4.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 6.Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. A cellular microRNA mediates antiviral defense in human cells. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 7.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–141. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 11.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haraguchi T, Mizutani T, Yamamichi N, Ito T, Minoguchi S, Iba H. SiRNAs do not induce RNA-dependent transcriptional silencing of retrovirus in human cells. FEBS Lett. 2007;581:4949–4954. doi: 10.1016/j.febslet.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 13.Kitamura T. New experimental approaches in retrovirus-mediated expression screening. Int. J. Hematol. 1998;67:351–359. doi: 10.1016/s0925-5710(98)00025-5. [DOI] [PubMed] [Google Scholar]
- 14.Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, Kimura S, Ito T, Yahagi N, Ichinose M, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471–5481. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
- 15.Yu JY, DeRuiter SL, Turner DL. RNA interference by expression of short-interfering RNAs and hairpin RNAs in mammalian cells. Proc. Natl Acad. Sci. USA. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 17.Naguibneva I, Ameyar-Zazoua M, Nonne N, Polesskaya A, Ait-Si-Ali S, Groisman R, Souidi M, Pritchard LL, Harel-Bellan A. An LNA-based loss-of-function assay for micro-RNAs. Biomed. Pharmacother. 2006;60:633–638. doi: 10.1016/j.biopha.2006.07.078. [DOI] [PubMed] [Google Scholar]
- 18.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng Y, Cullen BR. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res. 2004;32:4776–4785. doi: 10.1093/nar/gkh824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vermeulen A, Robertson B, Dalby AB, Marshall WS, Karpilow J, Leake D, Khvorova A, Baskerville S. Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA. 2007;13:723–730. doi: 10.1261/rna.448107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, Ganser A, Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmittgen TD, Jiang J, Liu Q, Yang L. A high-throughput method to monitor the expression of microRNA precursors. Nucleic Acids Res. 2004;32:e43. doi: 10.1093/nar/gnh040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 24.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J. Biol. Chem. 2008;283:1026–1033. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 25.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 26.Akagi T, Sasai K, Hanafusa H. Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl Acad. Sci. USA. 2003;100:13567–13572. doi: 10.1073/pnas.1834876100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Cre-lox-regulated conditional RNA interference from transgenes. Proc. Natl Acad. Sci. USA. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiscornia G, Tergaonkar V, Galimi F, Verma IM. CRE recombinase-inducible RNA interference mediated by lentiviral vectors. Proc. Natl Acad. Sci. USA. 2004;101:7347–7351. doi: 10.1073/pnas.0402107101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.