

Abstract
Neuroaxonal dystrophy (NAD), a distinctive axonopathy characterized by marked enlargement of distal axons, is the hallmark pathologic alteration in aged and diabetic human prevertebral sympathetic ganglia and in corresponding rodent models. NAD is thought to represent the abnormal outcome of cycles of synaptic degeneration and regeneration; a systematic study of identified axon terminals in aged and diabetic prevertebral ganglia, however, has not previously been performed. We examined the initial changes that develop in pre- and postsynaptic elements in sympathetic ganglia of aged and diabetic mice and found numerous synaptic changes involving both presynaptic and postsynaptic elements. Early alterations in presynaptic axon terminal size, vesicle content and morphology culminate in the development of anastomosing membranous tubulovesicular aggregates, accumulation of autophagosomes and amorphous debris that form a continuum with progressively larger classically dystrophic swellings. Dendritic changes consist of the development of swellings composed of delicate tubulovesicular elements and mitochondriopathy characterized by increased numbers of small mitochondria and, exclusively in aged ganglia, megamitochondria. These results support the hypothesis that NAD results from progressive changes in presynaptic axon terminals that likely involve membrane dynamics and which are accompanied by distinctive changes in postsynaptic dendritic elements.
Keywords: Autonomic neuropathy, Aging, Dendritic dystrophy, Diabetic neuropathy, Mitochondriopathy, Neuroaxonal dystrophy
INTRODUCTION
Autonomic neuropathy is an increasingly recognized problem in human aging and diabetes; it may result in a constellation of cardiovascular, genitourinary, sudomotor and alimentary complaints or result in subclinical disease. Autonomic neuropathy may also complicate therapeutic intervention or decrease the safety margin of neurotransmission; in combination with additional superimposed insults, this may produce clinical symptoms. Studies of pre- and paravertebral sympathetic ganglia in autopsied human subjects do not demonstrate substantial progressive loss of sympathetic neurons with increasing age (1–3) or duration of diabetes (3). Moreover, previous studies have not shown consistent structural changes in sympathetic neuronal cell bodies such as chromatolysis or apoptosis; nor has an intraganglionic inflammatory infiltrate in excess of age-matched controls been identified. Instead, the hallmark pathologic alteration reported in aged and diabetic human prevertebral sympathetic ganglia is neuroaxonal dystrophy (NAD), an axonopathy characterized by marked enlargement of distal axons containing a distinctive admixture of cytoskeletal, vesicular and membranous elements (3–7). Immunohistochemical studies of diabetic and aged human sympathetic ganglia suggest that the majority of dystrophic axonal terminals arise from sympathetic neurons (8), possibly secondary to intraganglionic sprouting. This pattern of damage to nerve terminals is likely due to dis- or misconnected ganglionic neurons and, particularly in prevertebral ganglia innervating the viscera, contributes to the loss of integrated reflexes. The findings in animal models of the aging and diabetic sympathetic nervous systems show close correspondence with those in humans, i.e. the development of dystrophic axons in prevertebral ganglia in the absence of significant neuronal loss (3, 9, 10). The fidelity of the neuropathologic findings in animal models to those of aged and diabetic humans suggests that similar pathogenetic mechanisms may be involved and that therapeutic approaches found to be efficacious in the models will predict efficacy in humans.
Although dystrophic changes in intraganglionic terminal axons are robust, unequivocal and consistent neuropathologic findings in aging and diabetes in prevertebral sympathetic ganglia, their pathogenesis and pathophysiological significance are uncertain. Based on their terminal axonal location and resemblance of dystrophic axons to axonal growth cones (the motile tips of regenerating axons), we and others have proposed that NAD represents the abnormal outcome of normal cycles of synaptic degeneration and regeneration (11–13). This process might interfere with synaptic plasticity relevant to synaptic function in general and learning and memory in particular. Dystrophic changes do occur on the terminal portions of axons but only rarely form the identifiable presynaptic elements of synapses (i.e. containing active zones terminating on dendritic elements); this may simply reflect the infrequency of encountering such minute specializations on the surface of individual dystrophic swellings that may reach 5 to 30 microns in diameter. A systematic ultrastructural study of synaptic axon terminals in aged and diabetic prevertebral ganglia has not been previously performed. We propose that structural alterations may initially develop in delicate (i.e. less than 1 μm in diameter) presynaptic axonal terminals; these alterations then progress to form large dystrophic swellings with distinctive collections of subcellular organelles. To test this hypothesis, we examined aged mice and several diabetic mouse models, focusing on the subtle ultrastructural and morphometric changes that develop in identified presynaptic elements. Elucidation of such changes in relatively simple autonomic ganglia may provide insights into the pathogenesis of a large number of human, veterinary and experimental diseases in which dystrophic axons are characteristic findings (14, 15) and in which dysfunction parallels their distribution.
In this study, we demonstrate that there is a continuum of changes in identified presynaptic axon terminals of aging and diabetic mouse sympathetic ganglia beginning with early alterations in synaptic vesicle content and morphology that culminates in the development of membranous aggregates in preterminal axons and formation of multivesicular autophagic bodies. In addition, we identified robust changes in postsynaptic dendritic elements in aged and diabetic mouse ganglia that may also contribute to synaptic dysfunction. We refer to dystrophic swellings involving both axons and dendrites collectively as “neuritic” dystrophy. Unexpectedly, we found that a number of neuronal cell bodies and dendrites in aged but not diabetic animals develop previously unappreciated, distinctive megamitochondria.
MATERIALS AND METHODS
Animals
The animals used in the present study formed the basis for two prior publications on the quantitative determination of the frequency of large, dystrophic swellings in aged and diabetic mice (16, 17). The previous studies did not address the more subtle synaptic alterations that are the current focus.
All animals were housed and cared for in accordance with the guidelines of the Washington University Committee for the Humane Care of Laboratory Animals and with National Institutes of Health guidelines on laboratory animal welfare. All mice were allowed standard rat chow and water ad libitum and maintained on a 12/12-hour light/dark cycle.
Diabetic Mice
The diabetic mice used in this study were the focus of a prior published study in which the frequency of markedly dilated dystrophic neurites, their ganglionic distribution, time course of development and quantitative comparison with age- and strain-matched controls were analyzed (17). Plasma glucose values, body weights and other details of the mouse groups examined are in that reference and are not repeated here. In the current study we analyzed common pathologic features of several groups of type 1 diabetic mouse models as follows: 1) female non-obese diabetic (NOD) mice that were diabetic for 3 to 5 weeks (Taconic Laboratories, Germantown, NY); 2) male streptozotocin (STZ)-induced diabetic non-obese diabetic-severe combined immune deficient (NOD-SCID) mice (18) that were diabetic for 2 to 4 weeks (Jackson Laboratory, Bar Harbor, ME); and 3) male STZ-induced C57BL6 and B6D2F1 mice that were diabetic for 3 to 4 months (Jackson Laboratory, Bar Harbor, ME).
The different models and strains of mice analyzed reflect practical issues such as synchronizing the development of onset of diabetes in NOD-SCID mice and the importance of STZ-induction of diabetes in a variety of strains of mice in anticipation of the use of a variety of knockout mice. We focused on the common pathologic features found in all of these type 1 diabetic models thereby providing evidence that they reflect critical reproducible diabetes-related pathology and not findings that are confined to a single strain. All 3 models developed comparable pathologic findings and are presented together.
Aged Mice
Male CFW strain outbred albino young adult (4 months) and aged (22–24 months) mice were obtained from the NIA colony maintained by Charles River Laboratories, Wilmington, MA.
Tissue Preparation
Animals were anesthetized with ketamine/xylazine and perfused with 50 ml of heparinized saline followed by 100 to 200 ml of 3% glutaraldehyde in 0.1M phosphate buffer, pH 7.3, containing 0.45 mM Ca+2. The superior mesenteric-celiac ganglia (SMG-CG) were dissected as a single block, cleaned of extraneous tissue while maintaining the superior mesenteric artery with the ganglionic block, and fixation was continued overnight at 4°C in the same buffer. Some ganglia were fixed by immersion in the same glutaraldehyde fixative overnight. Superior cervical ganglia were also dissected following perfusion fixation. Tissue samples were postfixed in phosphate-buffered 2% OsO4, dehydrated in graded concentrations of ethanol and embedded in EMbed-812 (Electron Microscopy Sciences, Hatfield, PA) with propylene oxide as an intermediary solvent. One-μm-thick plastic sections were examined by light microscopy after staining with toluidine blue. Ultrathin sections of individual SMG-CG or SCG were cut onto formvar-coated slot grids, which permit visualization of entire ganglionic cross sections. Tissues were subsequently stained with uranyl acetate and lead citrate, examined with a JEOL 1200 electron microscope and images captured with an AMT digital camera (Advanced Microscopy Techniques, Chazy, NY).
Morphometric Studies
Determination of the Cross Sectional Area of Presynaptic Axon Terminals
Sections of entire SMG-CG from various groups of aged and diabetic mice or SCG from NOD mice were examined by an observer blinded to the identity of individual animals. Ganglia were systematically scanned at 20,000× magnification beginning at one tissue margin scanning vertical columns from left to right until the entire ganglionic cross section had been examined. We identified individual active zones (defined by the presence of pre- and postsynaptic densities with immediately adjacent collections of synaptic vesicles) as synapses irrespective of the size or the nature of subcellular organelles composing their associated presynaptic axon terminal. It was not possible to examine all of the synapses composing the entire ganglion because they had been sectioned and used in our previous studies. Therefore, we reasoned that identifying active zones, which did not differ in size in ganglia of aged and young mice, or diabetic and age-matched control mice, the use of classical nonbiased counting strategies would not bias counts by over-representing large synapses. Images of the entire axonal terminal associated with individual active zones were captured at 40,000× magnification irrespective of their contents, i.e. including both dystrophic and normal subcellular elements. The cross sectional areas of presynaptic axonal terminals were determined using imageJ software and expressed as μm2. Approximately 50 to 130 synapses were captured from each ganglionic cross section.
Quantitation of Subpopulations of Nerve Terminals
Images of synapses acquired as described in the previous paragraph were also examined at 40,000× magnification and the numbers of terminals determined as follows: i) those with heterogeneous synaptic vesicles (defined as synapses with ≥ 4 non-dense core vesicles/per synapse that are more than 3 times the diameter of agranular synaptic vesicles); ii) those containing tubulovesicular elements; iii) those containing any number of autophagosomes; and iv) those containing amorphous granular material free in the presynaptic terminal. The results were expressed as percent of axon terminals with individual pathological alterations/total number of axon terminals examined.
Determination of Numbers of Cell Bodies with Megamitochondria
Numbers of cell bodies with megamitochondria (i.e. mitochondria ≥ 2 microns in diameter) were determined by scanning at 7,500× magnification and expressed as percent of neurons with megamitochondria/total number of nucleated cell bodies for each mouse.
Digital Images Manipulation
Images were uniformly increased in contrast and brightness adjusted.
Statistical Analysis
Statistical analysis was performed using a two-tailed t-test.
RESULTS
Young, Non-Diabetic Mice
Synapses in young (2- to 4-month old) non-diabetic mice of all strains were predominantly found on small and large dendrites and dendritic spines as well as (albeit much less frequently) on spines arising directly from perikarya; these results are similar to those of previous studies (19, 20). Large numbers of synapses were surrounded by Schwann and satellite cell processes and held closely within the satellite cell capsule that surrounds individual perikarya (Fig. 1A), an area of which (arrow, Fig. 1A) is shown at higher magnification in Figure 1B. Other synapses were scattered within the ganglionic neuropil outside of the perikaryal satellite cell capsule (i.e. more distant from neuronal cell bodies) and were surrounded by Schwann cell processes.
Figure 1.

Normal young mouse SMG-CG ultrastructure. (A, B) A principal sympathetic neuron is surrounded by presynaptic axon terminals, dendrites (asterisk, B) and dendritic spines composing the neuropil. One area (arrow, A) is shown at higher magnification in B and consists of several presynaptic axon terminals (arrows, B) enclosed in satellite cell processes. Magnification bars: A, 2 μm; B, 500 nm. (C) A typical synapse shows a presynaptic active zone (arrow) with clusters of agranular synaptic vesicles, a postsynaptic density in a small dendrite (*) and rare dense core vesicles. Magnification bar: 500 nm. (D) A typical small dendrite with lucent cytoplasm (*) is immediately postsynaptic to a nerve terminal. Magnification bar: 500 nm.
The presynaptic axon terminals ranged in size from 0.30 to 1.0 μm in diameter and 0.3 to 3.0 μm2 in cross sectional area in young animals with 50% of synapses less than 0.8 μm2 (Table 1; Fig. 2). They had well-defined active zones (arrow, Fig. 1C) and occasionally formed perforated synapses. Presynaptic axon terminals contained collections of small agranular vesicles and a few larger vesicles with dense cores. In typical synapses of young animals, agranular synaptic vesicles, but not those containing large dense cores, were gathered to dense active zones (arrow, Fig. 1C) as well as forming a likely reserve population more distant from active zones. Occasional individual clathrin-coated pits and vesicles were seen within the presynaptic axon terminal but did not form complex aggregates with other coated vesicles or synaptic vesicles. Infrequent synapses with heterogeneous lucent vesicles (1.4% of axon terminals; Table 2), very rare autophagosomes (0.2% of axon terminals; Table 2), and collections of tubulovesicular elements (0.2% of axon terminals; Table 2) were contained within the presynaptic axonal nerve terminal. Collections of amorphous material free within the presynaptic nerve terminals were never seen (Table 2) in non-diabetic young mice.
Table 1.
Effect of Age on Presynaptic Axon Terminal Cross Sectional Area of Sympathetic Ganglia
| Experimental Group | Age | Synapses (#) | Axon Terminal Cross Sectional Area (square microns) |
|---|---|---|---|
| Aged Mouse Exp | |||
| Young (n = 4) | 4 months | 90 ± 12 | 0.86 ± 0.06 |
| Aged (n = 7) | 22 –24 months | 92 ± 10 | 1.22 ± 0.06* |
Values represent the means ± SEM of the mean cross-sectional area of 50 to 125 synapses sampled from the SMG-CG of each of n mice. Statistical comparison:
p ≤ 0.01;
p ≤ 0.05.
Figure 2.

Aged and young mouse SMG-CG presynaptic axon terminal cross sectional area size-frequency histogram.
Table 2.
Distribution of Synaptic Pathology in Aged Mouse Sympathetic Ganglia
| Experiment | Synapses (#/mouse) | Heterogeneous vesicles (% total) | Tubulovesicles (% total) | Autophagosomes (% total) | Amorphous debris (% total) |
|---|---|---|---|---|---|
| Aged Mouse | |||||
| Young (4 months, n = 4) | 85 ± 12 | 1.42 ± 0.2 | 0.2 ± 0.2 | 0.2 ± 0.2 | 0 + 0 |
| Aged (22 24 months, n = 7) | 92 ± 10 | 8.0 ± 1.4* | 6.5 ± 1.5* | 6.2 ± 1.0* | 0.8 + 0.4 |
Synapses were classified into subgroups on the basis of ultrastructural criteria described in the text and expressed as the percentage of the total number of synapses examined. The number of synapses represents the mean number ± SEM of synapses examined in each of n mice. Statistical comparison:
p ≤ 0.01;
p ≤ 0.05 vs. respective young controls.
The postsynaptic elements (arrow, Fig. 1D), chiefly dendrites and dendritic spines, typically contained a postsynaptic density, mitochondria, occasional coated pits and vesicles, variable numbers of ribosomes, endoplasmic reticulum and more electron lucent cytoplasm than presynaptic elements. Large collections of mitochondria or dendritic processes containing large numbers of lucent tubulovesicular elements were not seen in young mouse ganglia. Cell bodies of principal sympathetic neurons did not show chromatolysis, apoptosis or megamitochondria.
AGED MICE
Presynaptic Nerve Terminals
Synaptic alterations in aged mice were distributed comparably to controls, forming the majority of synapses on dendrites, dendritic spines and somal spines.
Morphometric Studies of Presynaptic Nerve Terminals
Our previous studies have shown that presynaptic axonal dystrophy develops reproducibly in the SMG-CG of aged mice but marked dystrophic swellings, heretofore our single focus of analysis, involve only a small percentage of intraganglionic presynaptic axons. Currently, focusing on all presynaptic axon terminals, we found that average cross sectional area is significantly (41%) and reproducibly increased in the SMG-CG of aged mice (Table 1). A size-frequency histogram shows a general shift from a predominance of small axon terminals to larger forms in aged mouse SMG-CG compared to young controls (Fig. 2).
Ultrastructural Characteristics of Presynaptic Nerve Terminals
The overall increase in size of presynaptic axon terminals in aged mouse SMG-CG is a consequence of subtle changes in synaptic structure. Many synapses in aged SMG-CG have an apparent increase in the number of synaptic vesicles and an increased amount of osmiophilic unstructured cytoplasm (Fig. 3A) compared to those of young mouse ganglia. These synapses (arrow, Fig. 3B) are more likely to have irregular shapes compared with the typical spherical appearance of synapses in young mice. In contrast to the separation of active zone synaptic vesicles from those in the remainder of the presynaptic axon terminal in normal synapses, collections of vesicles often formed only a single population merging with large numbers of vesicles occupying the rest of presynaptic axon terminals (Fig. 3A). Most nerve terminals contained two major types of vesicles: small agranular forms that were most frequent and larger vesicles with dense cores. Aged presynaptic axonal boutons also contained larger agranular vesicles that had more heterogeneous shapes (arrows, Fig. 3C, D) that were 5- to 6-fold more frequent in aged mice than in young controls (Table 2). As in normal young mouse ganglia, postsynaptic dendritic elements (Fig. 3A, C) typically were more electron lucent than presynaptic axon terminals in aged mice.
Figure 3.

Presynaptic axon terminals of aged mouse SMG-CG. (A) Presynaptic nerve terminals in aged SMG-CG are typically larger than young controls, with numerous synaptic vesicles frequently without separation of a reserve from active zone, seen adjacent to a postsynaptic dendrite (*). Magnification bar: 500 nm. (B) Abnormal presynaptic terminals may be convoluted in shape (arrow) with several separate active zones. Magnification bar: 500 nm. (C, D) In addition to small, agranular synaptic vesicles and dense core vesicles, this axon terminal contains heterogeneously enlarged vesicles, seen at higher magnification in D (arrows, C, D), adjacent to a small dendrite (*, C). Magnification bars: C, 500 nm; D, 100 nm.
A distinctive alteration in the presynaptic axonal termini of aged mice was the presence of anastomosing tubulovesicular elements (Fig. 4); this alteration was more than 30-fold more frequent in aged than in young SMG-CG (Table 2). The most subtle, likely initial, alteration was the development of interconnected tubules (arrows, Fig. 4A–C) that appear to be intimately related to active zones and are frequently admixed with amorphous fuzzy material and agranular and granular synaptic vesicles. Occasionally, tubulovesicular elements were intimately admixed with vesicles, suggesting the possibility of fusion of vesicular elements with anastomosing tubulovesicular networks (Fig. 4D, E), or they showed a complex connection with the plasma membrane (arrow, Fig. 4F). Eventually, accumulations of subcellular elements appear to result in swollen presynaptic axon terminals in which tubulovesicular elements dominated the other accumulated organelles (Fig. 4G). Finally, there were markedly swollen, frankly dystrophic axons containing a complex admixture of subcellular organelles in which the demonstration of the presynaptic specialization was usually no longer possible (arrow, Fig. 4H). It was not uncommon for collections of similarly altered nerve terminals to be distributed around individual neurons as though they had originated from a single parent axon as a terminal spray.
Figure 4.
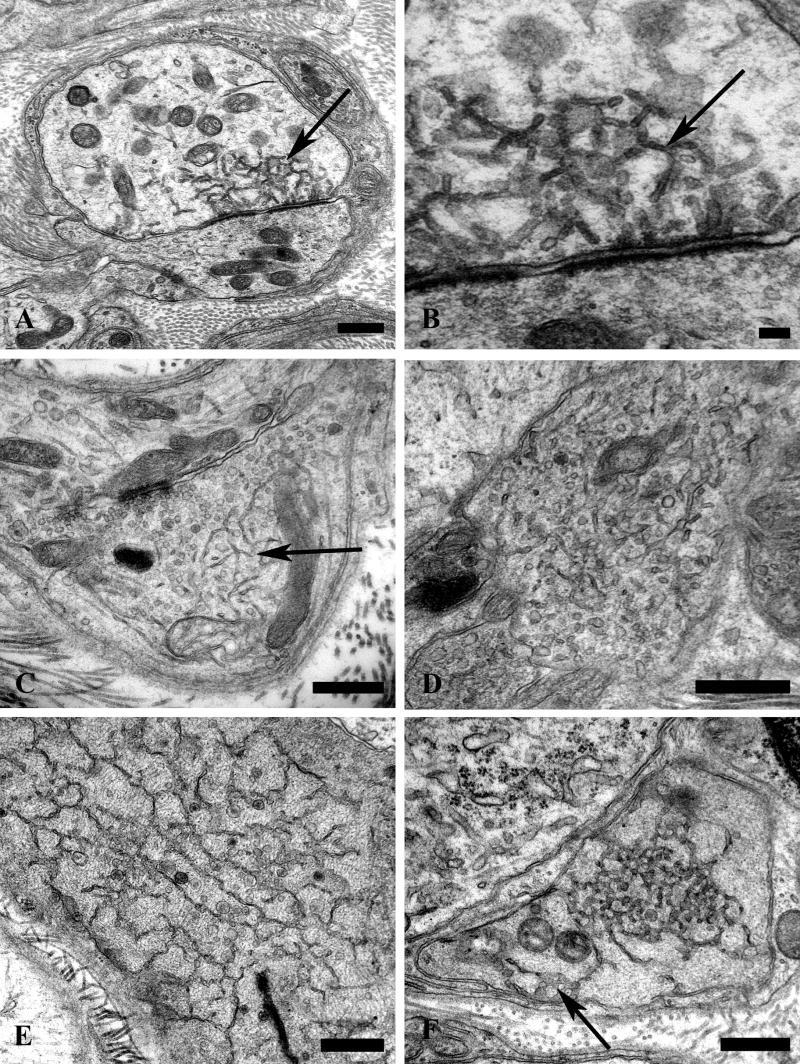

Accumulation of delicate tubulovesicular elements in presynaptic axon terminals in aged SMG-CG. (A–C) Presynaptic nerve terminals show anastomosing tubulovesicular elements (arrows) in close apposition to the active zone and admixed with agranular synaptic vesicles. Magnification bars: A, 500 nm; B, 100 nm; C, 500 nm. (D, E) Intimate association of agranular and heterogeneous agranular elements with tubulovesicular elements. Magnification bars: D, E, 500 nm. (F) Tubulovesicles may form connections with the axolemma (arrow). Magnification bar: 500 nm. (G) Progressive enlargement of presynaptic nerve terminals is dominated by delicate tubulovesicular elements. Magnification bar: 500 nm. (H) Typical dystrophic axon (arrow) is large with compact tubulovesicular elements. Magnification bar: 2 μm.
Enlarged presynaptic axon terminals containing numerous synaptic vesicles also often contained rare (arrow, Fig. 5A) to numerous autophagosomes or multivesicular bodies in which ghosts of synaptic vesicles could be identified (arrows, Fig. 5B). Autophagosomes were also more than 30-fold more numerous in presynaptic axon terminals of aged mice compared to young controls (Table 2). A continuum of involvement ranged from individual terminals with a single MVB to those with numerous autophagic bodies culminating in typical dystrophic axon terminals (Fig. 5C) in which marked swelling of the presynaptic element made the demonstration of the original synaptic specialization difficult.
Figure 5.

Accumulation of autophagosomes in presynaptic axonal terminals in aged SMG-CG. (A, B) Presynaptic nerve terminal with scattered autophagosomes with collections of synaptic vesicles (arrow, B), common in early dystrophic lesions. Magnification bars: A, 500 nm; B, 100 nm. (C) Dystrophic swellings surround occasional perikarya and typically contain the same aggregates of organelles, suggesting a possible spray of terminals originating from the same parent axon. Magnification bar: 2 μm.
The rarest lesion pattern, seen only in aged ganglia (Table 2), was the deposition of aggregates of amorphous osmiophilic material (Fig. 6) within the axon terminal (arrow, Fig. 6A) or admixed with tubulovesicular elements in markedly enlarged dystrophic forms (arrows, Fig. 6B, C). Accumulated material was the approximate consistency of the content of dense core granules (Fig. 6C) and eventually aggregated into large osmiophilic extravesicular bodies (Fig. 6D).
Figure 6.

Amorphous granular debris in presynaptic nerve terminals of aged mouse SMG-CG. (A) Presynaptic axonal terminal containing assorted subcellular organelles and small amounts of granular osmiophilic debris (arrow). (B, C) Marked swelling, no longer with synaptic specialization visible in this cross section contains multivesicular bodies and large aggregates of granular osmiophilic debris (arrows, B, C), seen better at higher magnification in C. (D) Dystrophic axon with large collections of osmiophilic material admixed with islands of tubulovesicular elements. Magnification bars: A–D, 500 nm.
Postsynaptic Dendritic Alterations
Several patterns of dendritic pathology were seen in aged ganglia. Scattered swollen neurites contained lucent cytoplasm with large numbers of tubulovesicular elements devoid of ribosomes (Fig. 7A, B) or large numbers of small mitochondria (Fig. 7C), although they were significantly less frequent in aged compared to diabetic ganglia in which we characterized them most fully (see below). Rare megamitochondria were found in dendrites of aged mice (arrows, Fig. 7D).
Figure 7.

Dendritic alterations in aged mouse SMG-CG. (A, B) The neuropil contains an enlarged dendrite with numerous lucent small tubulovesicular elements seen at higher magnification in B. Magnification bars: A, 2 μm; B, 500 nm. (C, D) Mitochondriopathy is represented by collections of large numbers of mitochondria smaller than those of adjacent cell body (arrow, C) and megamitochondria (arrow, D). Magnification bars: C, D, 2 μm.
Pathologic Alterations in Neuronal Cell Bodies
Our previous studies showed little reproducible pathologic alteration in neuronal cell bodies comprising prevertebral sympathetic ganglia other than the development of abnormal contours as the result of apparent distortion by adjacent dystrophic elements located within their satellite cell sheaths. In the present study, our focus on mitochondrial elements resulted in the demonstration of previously unnoticed collections of very large spherical mitochondria (“megamitochondria” ≥ 2 microns in diameter; arrows, Fig. 8A) that were often interspersed with a range of smaller mitochondria in the perikarya of aged SMG-CG neurons (Fig. 8). The internal structure of megamitochondria also varied from those with nearly normal density of cristae (arrow, Fig. 8B) to those with few short cristae (arrows, Fig. 8C) or internal membranous vacuoles embedded within granular intramitochondrial matrix. Megamitochondria were found in as many as 10% of aged neurons (aged: 5.2 ± 1.1%, mean + SEM, 54 ± 15 neurons examined per ganglion, n = 7 mice) and were not observed in ganglia of young animals (young: 0 ± 0%, 58 ± 10 neurons examined per ganglion, n = 4 mice).
Figure 8.

Mitochondriopathy in the perikarya of sympathetic neurons of aged mouse SMG-CG. (A) Enlarged mitochondria involving the perikaryon and adjacent proximal dendrites. Magnification bar: A, 10 μm. (B, C) Variation in the appearance of megamitochondria includes enlarged forms with relatively well maintained cristae (B), shortened cristae with lucent or granular matrix (C) or intramitochondrial vacuoles. Magnification bars: B, C, 2 μm.
DIABETIC MICE
Presynaptic Nerve Terminals
Synaptic ultrastructural pathology was nearly identical in aged and diabetic mice, but changes in diabetic animals developed much more quickly than in aged mice, reaching dramatic levels in mice after as little as 2 to 4 weeks of diabetes. Morphometric studies demonstrate overall increases in presynaptic axon terminal size of the SMG-CG of all type 1 diabetic models (Table 3); a size-frequency histogram of (NOD) diabetic presynaptic axonal terminal surface area shows a shift from small to larger boutons (Fig. 9A), similar to that of aged mice (Fig. 2).
Table 3.
Effect of Diabetes on Mean Presynaptic Axon Terminal Cross Sectional Area of Sympathetic Ganglia
| Experimental Group | Age | Diabetes (duration) | Synapses (#) | Presynaptic Axon Terminal Cross Sectional Area (square microns) |
|---|---|---|---|---|
| NOD Mice | ||||
| SMG-CGs | ||||
| Control (n = 6) | 6 months | 74 ± 12 | 0.75 ± 0.06 | |
| Diabetics (n = 5) | 6 months | 3–5 weeks | 92 ± 4 | 1.33 ± 0.14* |
| SCG | ||||
| Controls (n = 3) | 6 months | 60 ± 6 | 0.97 ± 0.02 | |
| Diabetics (n = 3) | 6 months | 3–5 weeks | 48 ± 10 | 0.82 ± 0.03# |
| B6D2F1 + STZ Exp | ||||
| Controls (n = 3) | 6.5 months | 111 ± 18 | 0.75 ± 0.05 | |
| Diabetics (n = 4) | 6.5 months | 5 months | 97 ± 12 | 1.28 ± 0.08* |
| C57BL6 + STZ | ||||
| Controls (n = 3) | 4–6 months | 83 ± 16 | 0.76 ± 0.04 | |
| Diabetics (n = 5) | 4–6 months | 3–4 months | 59 ± 3 | 1.15 ± 0.06* |
| NOD-SCID + STZ | ||||
| Controls (n=4) | 3 mo | 75 ± 10 | 0.91 ± .04 | |
| Diabetics (n=5) | 3 mo | 2–4 weeks | 88 ± 7 | 1.28 ± 0.04* |
Values represent the means ± SEM of the mean cross-sectional area of 50 to 125 synapses sampled from each of n mice. Statistical comparison:
p ≤ 0.01;
p ≤ 0.05.
Figure 9.


Diabetic and age-matched control mouse SMG-CG presynaptic axon terminal cross sectional area size-frequency histograms. (A) NOD mouse SMG-CG presynaptic axon terminal cross sectional area. (B) NOD mouse SCG presynaptic axon terminal cross sectional area.
The presynaptic axonal termini of diabetic mice showed the presence of anastomosing tubulovesicular elements (Fig. 10A, B), a finding which was essentially limited to diabetic mice compared to age-matched controls (Table 4). Enlarged presynaptic axon terminals also contained single to numerous autophagosomes or multivesicular bodies (Fig. 11A, B) in which ghosts of synaptic vesicles (Fig. 11C) and tubulovesicular elements (Fig. 11D) were visible.
Figure 10.

Accumulation of delicate tubulovesicular elements in presynaptic axon terminals of diabetic mouse SMG-CG. (A, B) Delicate tubulovesicular elements are intimately associated with active zones and adjacent axon terminal boutons. Magnification bar: A, B, 500 nm.
Table 4.
Distribution of Synaptic Pathology in Diabetic Mouse SMG-CG
| Experiment | Synapses (#/mouse) | Heterogeneous Vesicles (% total) | Tubulovesicles (% total) | Autophagosomes (% total) | Amorphous Debris (% total) |
|---|---|---|---|---|---|
| NOD-SCID + STZ | |||||
| Control (n = 4) | 75 ± 10 | 1.7 ± 0.3 | 0 ± 0 | 0 ± 0 | 0 + 0 |
| Diabetic (n = 5) | 88 ± 7 | 9.3 ± 1.0* | 2.5 ± 0.4* | 4.3 ± 1.1# | 0 + 0 |
Synapses were classified into subgroups on the basis of ultrastructural criteria described in the text and expressed as the percentage of the total number of synapses examined. The number of synapses represents the mean number ± SEM of synapses examined in each of n mice.
Statistical comparison:
=p ≤ 0.01;
p ≤ 0.05 vs. respective non-diabetic controls.
Figure 11.

Accumulation of autophagosomes in diabetic presynaptic dystrophic axon terminals. (A, B) Presynaptic nerve terminal contents range from single multivesicular bodies (MVBs) (arrow, A) to large forms with numerous MVBs admixed with mitochondria (B). Magnification bars: A, 500 nm; B, 500 nm. (C, D) MVBs consist of collections of synaptic vesicles (arrow, C), common in early dystrophic lesions, to tubulovesicular elements in later dystrophic terminals (arrows, D). Magnification bars: C, 500 nm; D, 500 nm.
Our previous studies failed to show dystrophic neurites in the diabetic mouse paravertebral SCG, even in the same mice in which the SMG-CG was dramatically involved (17). As anticipated, neither measurement of cross sectional area of (NOD) mouse SCG presynaptic axon terminals (Table 3) nor size-frequency histograms of (NOD) mouse SCG (Fig. 9B) showed an effect of diabetes on the SCG comparable to the increased size of (NOD) mouse SMG-CG synapses; indeed, the mean presynaptic axon terminal area actually decreased 15% in diabetic SCG compared to age-matched non-diabetic siblings (Table 3).
Postsynaptic Dendritic Alterations
We focused on structures clearly identified as dendrites by the presence of synapses on their surface (i.e. they were post-synaptic to axon terminals with well formed active zones) and their content of ribosomes or lipopigment. The most frequent alteration in SMG-CG dendrites of type 1 diabetic mouse prevertebral ganglia, but not the SCG, consisted of a mitochondriopathy characterized by the accumulation of increased numbers of small diameter mitochondria (Fig. 12). Small processes containing mitochondrial aggregates (Fig. 12A, B) were frequently postsynaptic to axonal termini, thereby identifying them as dendrites. Although the axonal termini adjacent to mitochondria-rich dendrites may be abnormal (e.g. the enlarged form containing heterogeneous vesicles; arrow, Fig. 12B), most presynaptic axon terminals ending on mitochondria filled dendrites were not compellingly dystrophic themselves. Other mitochondria-laden dystrophic dendrites (arrow, Fig. 12C) were of such size and located within the satellite cell sheath that they altered the contours of adjacent neurons (Fig. 12C, D). The mitochondria that accumulated in dystrophic dendrites were typically smaller in diameter and more osmiophilic (arrow, Fig. 12D) than those in adjacent perikarya (arrowheads, Fig. 12D). Although these changes were seen in both aged and diabetic ganglia, dendritic terminals with collections of minute mitochondria were particularly numerous in diabetic mice (Fig. 12E).
Figure 12.

Mitochondriopathy in diabetic mouse SMG-CG. (A, B) Accumulation of mitochondria in a small process (arrow, A) within the perineuronal satellite cell sheath is postsynaptic to an enlarged axonal terminus (arrow, B). Magnification bars: A, 2 μm; B, 500 nm. (C, D) A markedly enlarged dendrite (arrow, C) whose synaptic specializations are not identified at this level, distorts the contours of the adjacent principal sympathetic neuron, and contains large numbers of mitochondria (arrow, D) that are considerably smaller and hyperchromatic than mitochondria in adjacent perikarya (arrowheads, D). Magnification bars: C, 2 μm; D, 500 nm. (E) Large numbers of mitochondria-laden dendritic processes may be particularly numerous in diabetic mouse SMG-CG. Magnification bar: 2 μm.
In diabetic mouse ganglia there were populations of electron-lucent dendrites that ranged in size from small forms within the ganglionic neuropil (arrows, Fig. 13A) to enlarged elements that rivaled adjacent perikarya in diameter (arrow, Fig. 13B). These dendritic swellings contained a mixed population of tubulovesicular elements as well as variable numbers of small, hyperchromatic mitochondria. Enormously dilated forms (Fig. 13C, D) and minimally enlarged dendrites (Fig. 13E) occasionally had identified associated synapses (arrows, Fig. 13C, E). The majority of these dendrites had a combination of vesicular (Fig. 13F) and tubulovesicular (Fig. 13G, H) forms composed of thin walled, anastomosing, tubulovesicles that had irregular shapes. These structures were occasionally clathrin-coated with complex interconnections (arrows, Fig. 13G) between coated vesicles and coated pits or evidence of uptake into autophagosomes (arrowhead, Fig. 13G). In some cases tubulovesicles contained electron dense material (Fig. 13H) and were intimately related to the dendritic plasmalemma. The origins of these elements included dendritic plasmalemmal invaginations of tubulovesicular elements with a coated surface (arrow, Fig. 13I). Others appeared to be derived from rough endoplasmic reticulum with partial loss of ribosomes (arrows, Fig. 13J). In these processes the overall amount of rough endoplasmic reticulum was markedly decreased compared to controls.
Figure 13. Dendritic tubulovesicular processes in diabetic mouse SMG-CG.
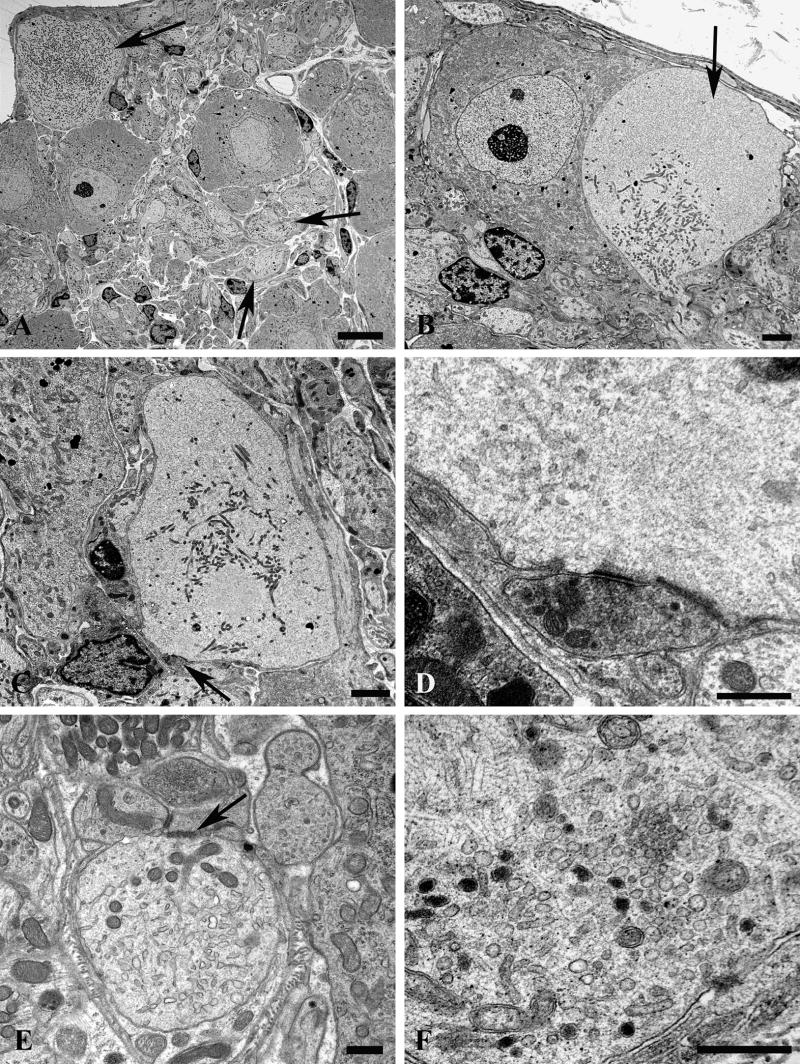

(A) The neuropil of this diabetic mouse ganglion contains both markedly enlarged neurites as well as numerous lucent small dendritic processes (arrows). Magnification bar: 10 μm. (B) An enlarged neurite within a satellite cell sheath results in the distortion of the contours of the adjacent neuronal cell body. Magnification bar: 2 μm. (C, D) A markedly dilated tubulovesicular-laden process shows a maintained presynaptic axon at its lower margin (arrow, C) seen at higher magnification in D. Magnification bars: C, 2 μm; D, 500 nm. (E) A small dendrite with a maintained synapse shows accumulated lucent tubulovesicular processes. Magnification bar: 500 nm. (F–H) The accumulated tubulovesicular elements are heterogeneous with lucent tubules and vesicles interspersed with cytoplasm (F), complex collections of coated vesicles forming unusual shapes (arrow, G), others part of multivesicular bodies (arrowhead, G), or delicate anastomosing tubulovesicular elements with proteinaceous intraluminal content (arrow, H). Magnification bars: F, 500 nm; G, 100 nm; H, 500 nm. (I, J) Occasional tubulovesicular elements appear to arise from the plasmalemma with partial clathrin coats (arrow, I) or contain residual ribosomes that may derive from rough endoplasmic reticulum (arrows, J). Magnification bars: I, 100 nm; J, 500 nm.
DISCUSSION
Mammalian prevertebral sympathetic ganglia are complex relay and integrative centers for projections from parasympathetic, sensory, spinal cord-derived motor and retrograde intestinofugal sources (19, 20). In addition, intrinsic principal sympathetic neurons are known to give rise to intraganglionic synapses, dendrodendritic and dendrosomatic interactions with neighboring neurons (22–25), some forming complex dendritic perisomal nests (26). Intracellular labeling studies have shown that dendrodendritic and dendrosomatic appositions of sympathetic neurons terminate on neighboring neurons rather than on the originating neuron (27). In experiments with surgically denervated sympathetic ganglia, intraganglionic projections of primary sympathetic neurons have been proposed to give rise to noradrenaline-mediated lateral inhibitory control of adjacent neighboring neurons (28, 29). Earlier immunohistochemical studies have also shown that there are numerous dystrophic swellings in prevertebral ganglia of aged and diabetic human subjects that are immunoreactive for dopamine-beta-hydroxylase, consistent with an origin from sympathetic, likely intraganglionic, neurons (8). Non-synaptic exocytic neurotransmitter release from neuronal cell bodies and dendrites in response to nonspecific depolarization or cholinergic agonists does occur in rat sympathetic ganglia (30).
Axonal Pathology
We have proposed that dystrophic axonal swellings in aged and diabetic prevertebral sympathetic ganglia represent the abnormal outcome of cycles of axonal terminal degeneration and regeneration (13) that occur in normal synapses (31) or as the result of insult-related terminal axonal injury (32, 33). Early studies demonstrated that dystrophic elements retract from the postsynaptic element in response to distal axotomy resulting in loss of the amplitude of excitatory postsynaptic potentials recorded in principal neurons in response to maximal preganglionic stimulation (32). We expected that structural alterations would initially develop in delicate (≤1 μm diameter) presynaptic axonal terminals that would then progress through intermediate forms to culminate in the development of large dystrophic swellings. Our current investigation demonstrates that alterations in the ultrastructure of presynaptic axonal termini are a consistent finding in aged and type 1 diabetic mouse prevertebral sympathetic ganglia. The initial alteration appears to represent an increase in synaptic vesicle number in presynaptic axonal termini followed by the accumulation of heterogeneous vacuoles, autophagosomes and abnormal anastomosing tubulovesicular elements. There is an apparent continuum of ultrastructural changes originating in presynaptic axon terminals that culminates in the formation of markedly enlarged dystrophic axons, a characteristic hallmark of aging and diabetes in humans and in a variety of rodent models (14). The frequency of axonal terminal pathology in mouse ganglia far exceeds the frequency of giant swellings of neuroaxonal dystrophy, however. Based on the results of our current study, extrapolation of the results of prior studies that focused on dystrophic axons as a pathologic endpoint may actually also indirectly provide more general information on the presence and extent of an underlying more insidious synapse-directed process. Morphometric analysis of synapse size suggests that there are also alterations in small presynaptic axonal termini, which would not be considered dystrophic by any previously established structural criteria, but that shift their size-frequency histogram to somewhat larger forms. Ultrastructural and immunohistochemical studies of Alzheimer disease and the Ts65Dn mouse model of Down syndrome have shown a similar increase in synaptic size and appositional length (34–36), possibly reflecting compensatory changes in residual synapses remaining after the loss of others. Focusing on alteration of early changes in axon terminals provides evidence that initial pathology occurs on axonal terminals that are still connected to the postsynaptic dendrite/dendritic spine and does not require retraction of axons that typically occurs following postganglionic crush injury (37) and spontaneously in aged mouse parasympathetic ganglia (38).
Microarray Studies of Synaptic Pathology in Diabetic Ganglia
An important role for diabetes-induced synaptic changes in the development of axonal dystrophy is also supported by our microarray studies (39). Within a few weeks of onset of diabetes in the STZ-diabetic rat (i.e. prior to the development of dystrophic structural pathology) gene expression patterns of diabetic rat SMG-CG differ significantly from age-matched non-diabetic controls and from changes in the paravertebral SCG of diabetic rats, which does not develop similar dystrophic changes. Genes that are preferentially altered in expression in diabetic SMG-CG compared to control SMG-CG encode proteins involved in nerve terminal structure and function, including synaptic vesicle binding to actin as part of stimulus-dependent tethering of vesicles (synapsin 2), catecholamine uptake and storage (chromaffin granule amine transporter, secretogranin 2, chromogranin B), docking, vesicle clustering (annexin V), targeting to the active zone, fusion (synaptotagmin), membrane flux (rab 3A, rab 11B), synaptic vesicle recovery (NSF), dense core granule storage (VGF) and function (GIT1, which regulates β-2 adrenergic receptor endocytosis). In diabetic rats neuroaxonal dystrophy develops within prevertebral sympathetic ganglia as well as the distal portions of their postganglionic projections to the alimentary tract and, therefore, changes in gene expression in prevertebral ganglia likely reflect alterations at both sites.
The enigmatic sparing of the SCG from pathology comparable to the SMG-CG in aging and diabetic man and rodent models remains unexplained. However, a variety of studies have shown additional substantial differences in pre- and paravertebral “chain” ganglia in electrophysiology, morphology, immunohistochemistry, sensitivity to antisera to NGF and response to guanethidine (40–42). Recent studies of mutant mice lacking the calcium independent phospholipase A2β (iPLA2β) gene also show marked neuroaxonal dystrophy (43), which is similarly confined to the SMG-CG, sparing the SCG. In that study we proposed that altered membrane composition due to changes in classes of lipid molecules could result in membrane instability and interference with membrane recycling. It is possible that an abnormality in membrane recycling, involving synaptic vesicles, endosomes, and endoplasmic reticulum, is the critical defect in aged and diabetic mice as well; in the absence of additional proof, however, this remains speculation at this time.
Autophagy
Autophagic vacuoles containing agranular synaptic vessels or tubulovesicular elements are prominent parts of early age and diabetes related synaptic pathology in our study as well as in a variety of human and experimental neurodegenerative diseases (see 44 for review). An active role for autophagy in the degeneration of terminal portions of neurites has been described in mouse sympathetic neurons in vitro in response to NGF withdrawal, a result prevented by inhibition of autophagy with 3-methyladenine or knocking down the key autophagy-related genes Atg7 and Beclin1 (45). The accumulation of distal axonal autophagosomes in Zn+2 chelation-induced degeneration is associated with a “dying-back” or distal axonopathy, a pattern of injury which resembles that of aging and diabetes (45), and is also similar to the damage produced by inhibition of the ubiquitin-proteosome pathway (46). The development of autophagosomes in the dystrophic axons of Purkinje cells of the Lurcher mouse mutant is thought to represent an early stress response that may participate in the further remodeling of subcellular organelles (47), possibly in anticipation of regeneration.
Dendritic and Perikaryal Pathology
Pale swollen dendrites are a conspicuous part of age- and diabetes-related alterations in prevertebral sympathetic ganglia. These changes are found immediately adjacent to cell bodies within their satellite cell sheaths as well as within the neuropil. They were never found connected to the immediately adjacent neuronal cell body and presumably represent the dendritic projections of adjacent intraganglionic neurons, which are known to exist in sympathetic ganglia (20, 22). Dendritic changes consist of the development of swellings composed of delicate tubulovesicular elements often connected in complex association, occasionally with numerous admixed coated vesicles. Dendritic membranous aggregates are not as compressed as those of dystrophic axons; they are typically larger than the tubulovesicular elements in dystrophic axon terminals and are often connected with the dendritic plasma membrane. Since similar structures have been described following axotomy in guinea pig sympathetic ganglia (37), dendritic swellings or retraction of processes may also develop in normal sympathetic ganglia in response to axonal injury. After initial retraction the dendritic arbor reexpands as reinnervation proceeds (48). Dystrophic dendrites typically lack membrane-attached ribosomes, which are numerous in the cytoplasm of normal dendrites.
Afferents to sympathetic ganglia primarily form synapses on dendritic spines and dendrites compared to cell bodies (49), and changes in dendritic size or structure may influence neuronal function since the extent of the dendritic arbor permits the integration of many weak inputs from diffuse sources (50). It is known that the size of the dendritic arbor plays an important role in determining the number of axonal terminals forming synapses on a principal sympathetic neuron (51), and that significant differences in the size of dendritic fields may exist even within individual prevertebral ganglia (52) or with changes in target size (53). Significant dendritic plasticity and rearrangements occur throughout life in sympathetic ganglia examined in vivo (54); some but not all subpopulations of sympathetic neurons may undergo NGF-sensitive dendritic atrophy (55–57). Large dendritic swellings are likely to interfere with synapses forming on the swollen dendritic segment as well as the central propagation of signals arising distal to the dilatation. The lack of membrane-bound ribosomes in dendritic processes is particularly poised to contribute to the loss of plasticity in dendrites and dendritic spines.
Mitochondriopathy
Although mitochondria accumulate in small numbers in aging and diabetic mouse presynaptic axonal termini, we found that mitochondrial alterations are most prominent in dendrites, a site where mitochondria are more highly charged, metabolically active (58) and more threadlike than in axons and presynaptic terminals (59). We noted two patterns of dendritic mitochondriopathy in aged and diabetic sympathetic ganglia: the presence of collections of small mitochondria and the formation of markedly enlarged mitochondria (“megamitochondria”); the latter seen only in dendrites and perikarya of aged ganglia. The significance of these changes in the number and size of mitochondria is unknown; however, it has been recently appreciated that mitochondrial fission and fusion have significant roles in mitochondrial number, size and function and may underlie the development of neurodegenerative processes (60–62). It is possible that the accumulation of small mitochondria in dendritic sites reflects foci of increased energy demand. Mitochondria are known to exist in neural tissue as elongated forms that fuse and divide as part of a dynamic network (63), particularly at sites of energy demand in response to ATP/ADP gradients, redistributing into dendritic protrusions in response to synaptic excitation and correlating with synaptogenesis and spine formation. Significant functional differences, such as susceptibility to calcium overload, have also been described between synaptic and nonsynaptic mitochondria (64). Large aggregates of axonal mitochondria have also been identified in DRG of diabetic rats in vivo and in DRG cultured in high glucose media, a result proposed to result from the upregulation of the fission-related protein Drp1 (65). Expression of neuropathy-associated forms of the fusion-inducing protein MFN2 in cultured DRG neurons induced defects in axonal transport of mitochondria, resulting in abnormal clustering of small fragmented mitochondria in both neuronal cell bodies and proximal axons (62–66).
Mitochondriopathy in Diabetes
Aggregates of mitochondria may also contribute to local pathology as the result of increased production of reactive oxygen species, loss of normal energy production or abnormality of calcium handling ability. Studies of cultured rat liver cells or rat myoblasts demonstrated that mitochondrial fragmentation is necessary for high glucose-induced increase in respiration with concomitant overproduction of reactive oxygen species (67). Mitochondrial dysfunction resulting in hyper- and depolarization of the transmembrane gradient has been proposed as a significant pathogenetic mechanism in diabetic neuropathy (65, 68, 69).
Age-Related Mitochondriopathy
Occasional dendrites and perikarya of aged mouse SMG-CG contained markedly enlarged megamitochondria. Although numerous small mitochondria could represent the substrate for the formation of megamitochondria as the result of fusion, megamitochondria were not seen in diabetic ganglia (in which small mitochondria were most numerous), but rather in aged ganglia. Megamitochondria have also been described in the livers of experimental animals treated with the copper-chelating agent cuprizone (70), as well as ethanol, hydrazine, hydrogen peroxide and chloramphenicol in response to apparent oxidative stress that may be prevented by free radical scavengers (71). Although oxidative stress has been proposed as a mechanism resulting in diabetic neuropathy, its role in the pathogenesis of diabetic autonomic neuropathy is currently unproven.
Summary
Our current studies demonstrate that aging and diabetes selectively target pre- and post-synaptic elements in the sympathetic autonomic nervous system and that early synaptic changes represent the precursor of axonal and dendritic dystrophy, a classical neuropathologic finding in a large number and variety of human, veterinary and experimental disorders in the central and peripheral nervous systems (14, 15). Our ultrastructural findings have also provided possible mechanistic insights into the pathogenesis of these alterations by identifying changes in synaptic vesicle and membrane structure in the nerve terminal, a role for autophagy and substantial alterations in mitochondrial size, number and ultrastructure in postsynaptic dendrites and cell bodies. Although it is unlikely that the same pathogenetic mechanisms underlie all conditions in which dystrophic elements are found, elucidating the final pathway to this common pathologic finding represents the first step in understanding a critically placed pattern of injury.
Acknowledgments
We would like to thank Eugene M. Johnson, Jr. for his helpful criticism.
Support: NIH awards R37 DK19645 and AG10299; Juvenile Diabetes Research Foundation Grant 1-2005-1085.
References
- 1.Dyck PJ, Jedrzejowska H, Karnes J, et al. Reconstruction of motor, sensory, and autonomic neurons based on morphometric study of sampled levels. Muscle Nerve. 1979;2:399–405. doi: 10.1002/mus.880020513. [DOI] [PubMed] [Google Scholar]
- 2.Jarvi R, Helen P, Pelto-Huikko M, et al. Age-related changes of enkephalinergic innervation of human sympathetic neurons. Mech Ageing Dev. 1988;44:143–51. doi: 10.1016/0047-6374(88)90086-3. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt RE, Plurad SB, Parvin CA, et al. Effect of diabetes and aging on human sympathetic autonomic ganglia. Am J Pathol. 1993;143:143–53. [PMC free article] [PubMed] [Google Scholar]
- 4.Kuntz A. Histological variations in autonomic ganglia and ganglion cells associated with age and disease. Am J Pathol. 1938;14:783–95. [PMC free article] [PubMed] [Google Scholar]
- 5.Duchen LW, Anjorin A, Watkins PJ, et al. Pathology of autonomic neuropathy in diabetes mellitus. Ann Intern Med. 1980;92:301–3. doi: 10.7326/0003-4819-92-2-301. [DOI] [PubMed] [Google Scholar]
- 6.Helen P. Fine-structural and degenerative features in adult and aged human sympathetic ganglion cells. Mech Ageing Dev. 1983;23:161–75. doi: 10.1016/0047-6374(83)90065-9. [DOI] [PubMed] [Google Scholar]
- 7.Hervonen A. Age related neuropathological changes in human sympathetic ganglia (Abstract) Soc Neurosci. 1984;10:451. [Google Scholar]
- 8.Schmidt RE. Age-related sympathetic ganglionic neuropathology: Human pathology and animal models. Auton Neurosci. 2002;96:63–72. doi: 10.1016/s1566-0702(01)00372-1. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt RE. Neuropathology and pathogenesis of diabetic autonomic neuropathy. In: Tomlinson DR, editor. Neurobiology of Diabetic Neuropathy. Amsterdam: Academic Press; 2002. pp. 267–92. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt RE. Neuronal preservation in the sympathetic ganglia of rats with chronic streptozotocin-induced diabetes. Brain Research. 2001;921:256–59. doi: 10.1016/s0006-8993(01)03155-9. [DOI] [PubMed] [Google Scholar]
- 11.Herman MM, Huttenlocher PR, Bensch KG. Electron microscopic observations in infantile neuroaxonal dystrophy. Report of a cortical biopsy and review of the recent literature. Arch Neurol. 1969;20:19–34. doi: 10.1001/archneur.1969.00480070029004. [DOI] [PubMed] [Google Scholar]
- 12.Sotelo C, Palay SL. Altered axons and axon terminals in the lateral vestibular nucleus of the rat. Possible example of axonal remodeling. Lab Invest. 1971;25:653–71. [PubMed] [Google Scholar]
- 13.Schmidt RE. Synaptic dysplasia in sympathetic autonomic ganglia. J Neurocytol. 1996;25:777–91. doi: 10.1007/BF02284841. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt R. Neuroaxonal dystrophy in aging rodent and human sympathetic autonomic ganglia-Synaptic pathology as a common theme in neuropathology. Adv Pathol Lab Med. 1993;6:505–22. [Google Scholar]
- 15.Jellinger K. Neuroaxonal dystrophy: Its natural history and related disorders. Prog Neuropathol. 1973;2:129–80. [Google Scholar]
- 16.Schmidt RE, Beaudet L, Plurad SB, et al. Pathologic alterations in pre- and postsynaptic elements in aged mouse sympathetic ganglia. J Neurocytol. 1995;24:189–206. doi: 10.1007/BF01181534. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt RE, Dorsey DA, Beaudet LN, et al. Non-obese diabetic mice rapidly develop dramatic sympathetic neuritic dystrophy: A new experimental model of diabetic autonomic neuropathy. Am J Pathol. 2003;163:2077–91. doi: 10.1016/S0002-9440(10)63565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blunt T, Gell D, Fox M, et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the SCID mouse. Proc Natl Acad Sci U S A. 1996;93:10285–90. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibbins IL, Morris JL. Structure of peripheral synapses: Autonomic ganglia. Cell Tissue Res. 2006;326:205–20. doi: 10.1007/s00441-006-0233-1. [DOI] [PubMed] [Google Scholar]
- 20.Miller SM, Hanani M, Kuntz SM, et al. Light, electron, and confocal microscopic study of the mouse superior mesenteric ganglion. J Comp Neurol. 1996;365:427–44. doi: 10.1002/(SICI)1096-9861(19960212)365:3<427::AID-CNE7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt RE, Dorsey DA, Beaudet LN, et al. Experimental rat models of types 1 and 2 diabetes differ in sympathetic neuroaxonal dystrophy. J Neuropathol Exp Neurol. 2004;63:450–60. doi: 10.1093/jnen/63.5.450. [DOI] [PubMed] [Google Scholar]
- 22.Matthews M. The ultrastructure of junctions in sympathetic ganglia of mammals. In: Elfvin L-G, editor. Autonomic Ganglia. New York: Wiley and Sons; 1983. pp. 27–66. [Google Scholar]
- 23.Elfvin LG. Ultrastructural studies on the synaptology of the inferior mesenteric ganglion of the cat. I J Ultrastruct Res. 1971;37:432–48. doi: 10.1016/s0022-5320(71)80137-5. [DOI] [PubMed] [Google Scholar]
- 24.Elfvin LG. Ultrastructural studies on the synaptology of the inferior mesenteric ganglion of the cat. I. Observations on the cell surface of the postganglionic perikarya. J Ultrastruct Res. 1971;37:411–25. doi: 10.1016/s0022-5320(71)80135-1. [DOI] [PubMed] [Google Scholar]
- 25.Kondo H, Dun NJ, Pappas GD. A light and electron microscopic study of the rat superior cervical ganglion cells by intracellular HRP-labeling. Brain Res. 1980;197:193–99. doi: 10.1016/0006-8993(80)90444-8. [DOI] [PubMed] [Google Scholar]
- 26.Kawai Y. Ultrastructure of neuronal circuitry in sympathetic ganglia. Microsc Res Tech. 1996;35:146–56. doi: 10.1002/(SICI)1097-0029(19961001)35:2<146::AID-JEMT5>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 27.Kiraly M, Favrod P, Matthews MR. Neuroneuronal interconnections in the rat superior cervical ganglion: Possible anatomical bases for modulatory interactions revealed by intracellular horseradish peroxidase labeling. Neuroscience. 1989;33:617–42. doi: 10.1016/0306-4522(89)90413-2. [DOI] [PubMed] [Google Scholar]
- 28.Kawai Y, Senba E. Correlation between dendrodendritic synapses of adrenergic type and synaptically evoked hyperpolarization in the sympathetic ganglion of adult rats. Neuroscience. 1995;68:925–35. doi: 10.1016/0306-4522(95)00202-t. [DOI] [PubMed] [Google Scholar]
- 29.Kawai Y, Senba E. Noradrenaline-mediated lateral inhibition in the sympathetic ganglion. Neurosci Lett. 1997;238:87–89. doi: 10.1016/s0304-3940(97)00854-9. [DOI] [PubMed] [Google Scholar]
- 30.Zaidi ZF, Matthews MR. Exocytotic release from neuronal cell bodies, dendrites and nerve terminals in sympathetic ganglia of the rat, and its differential regulation. Neuroscience. 1997;80:861–91. doi: 10.1016/s0306-4522(96)00664-1. [DOI] [PubMed] [Google Scholar]
- 31.Cotman CW, Nieto-Sampedro M, Harris EW. Synapse replacement in the nervous system of adult vertebrates. Physiol Rev. 1981;61:684–784. doi: 10.1152/physrev.1981.61.3.684. [DOI] [PubMed] [Google Scholar]
- 32.Nja A, Purves D. The effects of nerve growth factor and its antiserum on synapses in the superior cervical ganglion of the guinea-pig. J Physiol. 1978;277:53–75. [PMC free article] [PubMed] [Google Scholar]
- 33.Ohara S, Beaudet LN, Schmidt RE. Transganglionic response of GAP-43 in the gracile nucleus to sciatic nerve injury in young and aged rats. Brain Res. 1995;705:325–31. doi: 10.1016/0006-8993(95)01164-1. [DOI] [PubMed] [Google Scholar]
- 34.Scheff SW, Price DA. Synaptic pathology in Alzheimer’s disease: A review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–46. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Belichenko PV, Masliah E, Kleschevnikov AM, et al. Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol. 2004;480:281–98. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- 36.Geula C, Nagykery N, Nicholas A, et al. Cholinergic neuronal and axonal abnormalities are present early in aging and in Alzheimer’s disease. J Neuropathol Exp Neurol. 2008;67:309–18. doi: 10.1097/NEN.0b013e31816a1df3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purves D. Functional and structural changes in mammalian sympathetic neurones following interruption of their axons. J Physiol. 1975;252:429–63. doi: 10.1113/jphysiol.1975.sp011151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coggan JS, Grutzendler J, Bishop DL, et al. Age-associated synapse elimination in mouse parasympathetic ganglia. J Neurobiol. 2004;60:214–26. doi: 10.1002/neu.20022. [DOI] [PubMed] [Google Scholar]
- 39.Carroll SL, Byer SJ, Dorsey DA, et al. Ganglion-specific patterns of diabetes-modulated gene expression are established in prevertebral and paravertebral sympathetic ganglia prior to the development of neuroaxonal dystrophy. J Neuropathol Exp Neurol. 2004;63:1144–54. doi: 10.1093/jnen/63.11.1144. [DOI] [PubMed] [Google Scholar]
- 40.Hill CE, Hendry IA, Ngu MC, van Helden DF. Subpopulations of sympathetic neurones differ in their sensitivity to nerve growth factor antiserum. Brain Res. 1985;355:121–30. doi: 10.1016/0165-3806(85)90011-2. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt RE, McAtee SJ, Plurad DA, et al. Differential susceptibility of prevertebral and paravertebral sympathetic ganglia to experimental injury. Brain Res. 1988;460:214–26. doi: 10.1016/0006-8993(88)90366-6. [DOI] [PubMed] [Google Scholar]
- 42.Jobling P, Gibbins IL. Electrophysiological and morphological diversity of mouse sympathetic neurons. J Neurophysiol. 1999;82:2747–64. doi: 10.1152/jn.1999.82.5.2747. [DOI] [PubMed] [Google Scholar]
- 43.Malik I, Turk J, Mancuso DJ, et al. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy due to PLA2G6 Mutations. Am J Pathol. 2008;172:406–16. doi: 10.2353/ajpath.2008.070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- 45.Yang Y, Kawataki T, Fukui K, et al. Cellular Zn2+ chelators cause “dying-back” neurite degeneration associated with energy impairment. J Neurosci Res. 2007;85:2844–55. doi: 10.1002/jnr.21411. [DOI] [PubMed] [Google Scholar]
- 46.Laser H, Mack TG, Wagner D, et al. Proteasome inhibition arrests neurite outgrowth and causes “dying-back” degeneration in primary culture. J Neurosci Res. 2003;74:906–16. doi: 10.1002/jnr.10806. [DOI] [PubMed] [Google Scholar]
- 47.Wang QJ, Ding Y, Kohtz DS, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057–68. doi: 10.1523/JNEUROSCI.2261-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Purves D, Snider WD, Voyvodic JT. Trophic regulation of nerve cell morphology and innervation in the autonomic nervous system. Nature. 1988;336:123–28. doi: 10.1038/336123a0. [DOI] [PubMed] [Google Scholar]
- 49.Forehand CJ. Density of somatic innervation on mammalian autonomic ganglion cells is inversely related to dendritic complexity and preganglionic convergence. J Neurosci. 1985;5:3403–8. doi: 10.1523/JNEUROSCI.05-12-03403.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkman HP, Stapelfeldt WH, Miller SM, et al. Enteric connection with peripheral sympathetic neurons. In: Tache Y, Wingate DL, Burks FT, editors. Innervation of the Gut-Pathophysiological Implications. Boca Raton: CRC Press; 1994. pp. 149–56. [Google Scholar]
- 51.Purves D, Lichtman JW. Geometrical differences among homologous neurons in mammals. Science. 1985;228:298–302. doi: 10.1126/science.3983631. [DOI] [PubMed] [Google Scholar]
- 52.Gibbins IL, Teo EH, Jobling P, Morris JL. Synaptic density, convergence, and dendritic complexity of prevertebral sympathetic neurons. J Comp Neurol. 2003;455:285–98. doi: 10.1002/cne.10404. [DOI] [PubMed] [Google Scholar]
- 53.Voyvodic JT. Peripheral target regulation of dendritic geometry in the rat superior cervical ganglion. J Neurosci. 1989;9:1997–2010. doi: 10.1523/JNEUROSCI.09-06-01997.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purves D, Hadley RD, Voyvodic JT. Dynamic changes in the dendritic geometry of individual neurons visualized over periods of up to three months in the superior cervical ganglion of living mice. J Neurosci. 1986;6:1051–60. doi: 10.1523/JNEUROSCI.06-04-01051.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrews TJ, Cowen T. Nerve growth factor enhances the dendritic arborization of sympathetic ganglion cells undergoing atrophy in aged rats. J Neurocytol. 1994;23:234–41. doi: 10.1007/BF01275527. [DOI] [PubMed] [Google Scholar]
- 56.Andrews TJ, Li D, Halliwell J, et al. The effect of age on dendrites in the rat superior cervical ganglion. J Anat. 1994;184:111–17. [PMC free article] [PubMed] [Google Scholar]
- 57.Andrews TJ, Cowen T. Nerve growth factor enhances the dendritic arborization of sympathetic ganglion cells undergoing atrophy in aged rats. J Neurocytol. 1994;23:234–41. doi: 10.1007/BF01275527. [DOI] [PubMed] [Google Scholar]
- 58.Overly CC, Rieff HI, Hollenbeck PJ. Organelle motility and metabolism in axons vs. dendrites of cultured hippocampal neurons. J Cell Sci. 1996;109:971–80. doi: 10.1242/jcs.109.5.971. [DOI] [PubMed] [Google Scholar]
- 59.Muller M, Mironov SL, Ivannikow MV, et al. Mitochondrial organization and motility probed by two-photon microscopy in cultured mouse brainstem neurons. Exp Cell Res. 2005;303:114–27. doi: 10.1016/j.yexcr.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 60.Frank S. Dysregulation of mitochondrial fusion and fission: an emerging concept in neurodegeneration. Acta Neuropathol (Berl) 2006;111:93–100. doi: 10.1007/s00401-005-0002-3. [DOI] [PubMed] [Google Scholar]
- 61.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 62.Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- 63.Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–80. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- 64.Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–68. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 65.Leinninger GM, Backus C, Sastry AM, et al. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 66.Baloh RH, Schmidt RE, Pestronk A, et al. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci. 2007;27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA. 2006;103:2653–58. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 69.Huang TJ, Sayers NM, Verkhratsky A, et al. Neurotrophin-3 prevents mitochondrial dysfunction in sensory neurons of streptozotocin-diabetic rats. Exp Neurol. 2005;194:279–83. doi: 10.1016/j.expneurol.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 70.Guerineau M, Guerineau S, Gosse C. Abnormal mitochondrial DNA molecules in megamitochondria from cuprizone-treated rats. Eur J Biochem. 1974;47:313–19. doi: 10.1111/j.1432-1033.1974.tb03695.x. [DOI] [PubMed] [Google Scholar]
- 71.Wakabayashi T, Adachi K, Matsuhashi T, et al. Suppression of the formation of megamitochondria by scavengers for free radicals. Mol Aspects Med. 1997;18 (Suppl):S51–61. doi: 10.1016/s0098-2997(97)00033-2. [DOI] [PubMed] [Google Scholar]