

Abstract
Apolipoprotein A-V (apoA-V) and apoC-III are exchangeable constituents of VLDL and HDL. ApoA-V counteracts the effect of apoC-III on triglyceride (TG) metabolism with poorly defined mechanisms. To better understand the effects of apoA-V on TG and cholesterol metabolism, we delivered apoA-V cDNA into livers of hypertriglyceridemic APOC3 transgenic mice by adenovirus-mediated gene transfer. In response to hepatic apoA-V production, plasma TG levels were reduced significantly as a result of enhanced VLDL catabolism without alternations in VLDL production. This effect was associated with reduced apoC-III content in VLDL. Increased apoA-V production also resulted in decreased apoC-III and increased apoA-I content in HDL. Furthermore, apoA-V-enriched HDL was associated with enhanced LCAT activity and increased cholesterol efflux. This effect, along with apoE enrichment in HDL, contributed to HDL core expansion and α-HDL formation, accounting for significant increases in both the number and size of HDL particles. As a result, apoA-V-treated APOC3 transgenic mice exhibited decreased VLDL-cholesterol and increased HDL-cholesterol levels. ApoA-V-mediated reduction of apoC-III content in VLDL represents an important mechanism by which apoA-V acts to ameliorate hypertriglyceridemia in adult APOC3 transgenic mice. In addition, increased apoA-V levels accounted for cholesterol redistribution from VLDL to larger HDL particles. These data suggest that in addition to its TG-lowering effect, apoA-V plays a significant role in modulating HDL maturation and cholesterol metabolism
Supplementary key words: apolipoprotein A-V, apolipoprotein C-III, hypertriglyceridemia, lipoprotein metabolism, very low density lipoprotein, high density lipoprotein
Hypertriglyceridemia is a hallmark of the metabolic syndrome that is closely associated with obesity and type 2 diabetes, and it is characterized by a triad plasma lipid profile [i.e., increased triglyceride (TG) and LDL levels and decreased HDL levels] (1, 2). Because of its atherogenic potential, hypertriglyceridemia is considered an independent risk factor for coronary artery disease (3, 4). Nevertheless, the pathophysiology of hypertriglyceridemia remains poorly understood.
Recent studies have identified apolipoprotein A-V (apoA-V) and apoC-III as key regulators of plasma TG metabolism. ApoA-V and apoC-III are exchangeable constituents of VLDL and HDL with opposing effects on plasma TG metabolism (5). In humans, genetic variants of apoC-III or apoA-V genes are linked with altered VLDL-TG metabolism (5–14). Furthermore, APOA5 haplotypes are associated with abnormal TG metabolism in hyperlipidemic subjects (15). In animals, transgenic apoC-III production results in hypertriglyceridemia as a result of delayed catabolism of VLDL and chylomicron (16–18). In contrast, transgenic apoA-V expression is associated with hypotriglyceridemia as a result of enhanced clearance of TG-rich particles in mice (6). On the other hand, apoC-III deficiency is associated with significantly lower plasma TG levels (19), whereas apoA-V knockout mice manifest hypertriglyceridemia attributable to decreased lipolysis of TG-rich lipoproteins (6, 20). These data are indicative of an antagonistic interplay between apoC-III and apoA-V in plasma TG metabolism. In support of this notion, genetically engineered mice with cotransgenic expression or double knockout of apoA-V and apoC-III genes exhibit normal plasma TG metabolism (21, 22).
However, the mechanism underlying the apoA-V-mediated TG-lowering effect remains incompletely understood. Although transgenic apoA-V expression prevents the development of hypertriglyceridemia in APOC3 transgenic mice (21), it remains unknown whether augmented apoA-V production can reverse hypertriglyceridemia in adult mice with palpable dyslipidemia. Unlike apoC-III, which is a major constituent of VLDL and an inhibitor of LPL, apoA-V is present mainly in HDL (23, 24). The effect of apoA-V on HDL-cholesterol metabolism remains undetermined. To address these issues, we delivered mouse apoA-V cDNA by adenovirus-mediated gene transfer into livers of hypertriglyceridemic APOC3 transgenic mice. Hepatic apoA-V production ameliorated hypertriglyceridemia in APOC3 transgenic mice, as shown by significantly reduced plasma VLDL-TG levels and improved postprandial TG response. This effect correlated with reduced apoC-III content in VLDL and improved the catabolism of TG-rich particles in apoA-V vector-treated hypertriglyceridemic mice. Furthermore, increased apoA-V levels were shown to enhance LCAT activity and promote cholesterol efflux from cultured macrophage cells. This effect, together with increased apoE and apoA-I content in HDL, contributed to HDL core expansion and α-HDL formation, resulting in a marked increase in both the number and size of HDL. As a result, apoA-V vector-treated APOC3 transgenic mice displayed decreased VLDL-cholesterol and increased HDL-cholesterol levels without changes in total plasma cholesterol levels. These data validated the concept that increased apoA-V production ameliorated hypertriglyceridemia in adult APOC3 transgenic mice with overt dyslipidemia. In addition, our studies elucidated an important role of apoA-V in HDL-cholesterol homeostasis by modulating the composition of apolipoproteins in HDL.
MATERIALS AND METHODS
Adenoviral vectors
Mouse apoA-V cDNA (1.2 kb) was obtained by RT-PCR from C57BL/6J mouse liver RNA using specific primers (forward reaction, 5′-GCAGCCTGAGTTCCAGGTTC-3′; reverse reaction, 5′-CCTAATAGACCATGCTAGCG-3′). ApoA-V cDNA was cloned into plasmid pENTR-1A (Invitrogen, Carlsbad, CA), which was used as an entry vector for the generation of adenovirus Ad-CMV-apoA-V using the ViraPower Adenoviral Expression System (Invitrogen). The control adenovirus Ad-RSV-LacZ expressing the bacterial β-gal gene has been described (25, 26). Adenoviruses were produced in HEK293 cells and purified by CsCl gradient centrifugation, as described (26).
Animal studies
Transgenic mice expressing human apoC-III in the C57BL/6J background have been described (16). Mice were fed standard rodent chow and water ad libitum in sterile cages with a 12 h light/dark cycle. Under this condition, apoC-III transgenic mice spontaneously developed hypertriglyceridemia with markedly increased plasma TG content (>300 mg/dl at 12 weeks old), compared with gender- and age-matched littermates (plasma TG levels, 60–120 mg/dl). For adenovirus administration, mice were injected via tail vein with 200 μl of either apoA-V or control LacZ adenovirus at 2.5 × 1012 viral particles/kg (equivalent to 1.5 × 1011 plaque-forming unit/kg), as described (27). This approach resulted in ~70% transduction of hepatocytes in liver, with little transduction in extrahepatic organs (26, 28). For blood chemistry measurement, treated mice were fasted overnight (16 h) and aliquots of tail vein blood (100 μl) were collected into capillary tubes precoated with potassium-EDTA (Sarstedt, Nümbrecht, Germany). Plasma levels of TG and cholesterol were determined using Thermo Infinity TG and cholesterol reagents (Thermo Electron, Melbourne, Australia). Plasma NEFA levels were determined using the Wako NEFA assay kit (Wako Chemical USA, Richmond, VA). All procedures were approved by the Institutional Animal Care and Usage Committee of Children’s Hospital of Pittsburgh (protocol 41-04).
Fast-protein liquid chromatography fractionation of lipoproteins
Aliquots (250 μl) of plasma pooled from a given group of mice were applied to two head-to-tail linked Tricorn high-performance Superose S-6 10/300GL columns using a fast-protein liquid chromatography system (Amersham Biosciences), followed by elution with PBS at a constant flow rate of 0.25 ml/min. Fractions (500 μl) were eluted and assayed for TG and cholesterol concentrations using the Thermo Infinity TG and cholesterol reagents (Thermo Electron), as described (29).
Fat tolerance test
Mice were fasted overnight, followed by oral gavage of olive oil at the dose of 10 μl/g body weight. Before and at different times after oral fat administration, aliquots of tail vein blood (25 μl) were sampled for the determination of plasma TG levels. Area under the curve of plasma TG profiles during the fat tolerance test was calculated using KaleidaGraph software (Synergy Software, Reading, PA).
VLDL-TG production assay
Mice were fasted for 5 h, followed by intraperitoneal injection of 500 mg/kg Triton WR1339 (Sigma-Aldrich, St. Louis, MO) to inhibit plasma VLDL-TG hydrolysis and clearance. Aliquots of blood (25 μl) were collected at 20, 40, 60, and 80 min after Triton WR1339 injection. Plasma TG levels were determined and plotted as a function of time. The rates of hepatic VLDL-TG production, defined as milligrams of TG produced per minute per kilogram of body weight, were calculated.
Lipoprotein lipase assay
Mice were injected intravenously with 300 IU heparin/kg body weight, and tail vein blood (20 μl) was sampled at 10 min after heparin infusion. Heparinized sera were prepared for the determination of LPL activity using the LPL activity kit (Roar Biochemical, Inc.), as described previously (29).
Western blot analysis
Aliquots of plasma at a fixed concentration of 15 μg of protein per lane were applied to 4–20% polyacrylamide gels. The gels were subjected to Western blot assay using goat anti-apoC-III antibody (1:1,000 dilution; Abcam, Cambridge, MA). This antibody cross-reacts with both human and mouse apoC-III protein. The secondary antibody was rabbit anti-goat IgG conjugated with HRP (1:6,000 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA). ApoC-III proteins were visualized by ECL detection reagents, and the relative intensities of protein bands were quantified by densitometry, as described (29). Likewise, plasma apoA-V levels were determined by Western blot assay using rabbit anti-apoA-V antibody. This antibody was developed in our laboratory by immunizing rabbits with the mouse apoA-V-specific peptide (amino acids 113–128, VGWNLEGLRQQLK-PYT) (Genemed Synthesis, Inc., San Francisco, CA). For the determination of apoB and apoE levels, rabbit anti-apoB (1:1,000 dilution) and anti-apoE (1:1,000 dilution) antibodies were obtained from Abcam.
To determine hepatic protein levels, aliquots (20 mg) of liver tissue were homogenized in 400 μl of ice-cold M-PER mammalian protein extraction buffer (Pierce, Rockford, IL) supplemented with 4 μl of protease inhibitor cocktail (Pierce). Aliquots (20 μg) of liver protein extracts were resolved on 4–20% SDS-polyacrylamide gels, followed by immunoblot analysis using rabbit anti-HMG-CoA reductase (catalog No. 07-457; Upstate Biotechnology, Lake Placid, NY) and goat anti-actin (sc-1615; Santa Cruz Biotechnology).
For the detection of low density lipoprotein receptor (LDLR) and scavenger receptor class B type I (SR-BI), plasma membrane proteins of liver were extracted using the Mem-PER membrane protein extraction kit (Pierce). Aliquots of liver tissue (20 mg) was homogenized in 500 μl of TBS (10 mM Tris-HCl, pH 7.0, 10 mM NaCl, and 25 mM EDTA) supplemented with 5 μl of protease inhibitor cocktail (Pierce). The samples were centrifuged at 5,000 g at 4°C for 5 min. The supernatants were centrifuged at 14,000 rpm at 4°C for 45 min. The pellets containing enriched plasma membrane proteins were subjected to Western blot analysis using rabbit anti-SR-BI (NB400-104; Novus Biologicals, Littleton, CO) and chicken anti-LDLR antibody (NB300–338; Novus Biologicals). As a control, monoclonal mouse antibody against the plasma membrane-specific marker sodium potassium ATPase (ab7671; Abcam) was used. The hepatic abundance of individual proteins was quantified by semiquantitative immunoblot assay using β-actin or sodium potassium ATPase as an internal control, as described (29).
Pre-β-HDL and α-HDL determination
Native agarose gel electrophoresis was used to determine plasma preβ-HDL (also known as lipid-poor HDL) and α-HDL levels, as described (30). Aliquots of plasma (40 μg of protein) were applied to 0.7% agarose gels. After electrophoresis for 2.5 h in 60 mM sodium barbital buffer, pH 8.6 (Sigma-Aldrich), in a 4°C room, proteins were transferred to a nitrocellulose membrane in deionized water by capillary blotting for 16 h. The membrane was probed with rabbit anti-apoA-I antibody (Biodesign, Saco, ME), followed by incubation with anti-rabbit IgG conjugated with HRP (Amersham Biosciences, Piscataway, NJ). ApoA-I proteins were visualized by ECL detection reagents and quantified by densitometry, as described (29).
LCAT activity assay
Plasma LCAT activity was assayed using the Calbiochem fluorometric LCAT assay kit (EMD Bioscience, San Diego, CA). This assay is based on the hydrolysis of an artificial LCAT substrate that fluoresces at 470 nm, resulting in a product that fluoresces at 390 nm. Aliquots (2 μl) of serum in both control and apoA-V groups were mixed with 1 μl of fluorescent LCAT substrate and 200 μl of LCAT assay buffer, followed by incubation for 2 h at 37°C. The reaction was stopped by adding 300 μl of READ reagent (provided in the kit) to 100 μl of the reaction mixture, followed by fluorometry at 390 and 470 nm. LCAT activity is defined as the change in the ratio of 390/470 nm fluorescence emission intensities.
Cholesterol efflux assay
Cholesterol efflux was measured as described (31). Mouse macrophage cells (RAW-264.7; American Type Culture Collection, Manassas, VA) were cultured in DMEM supplemented with 10% (v/v) FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin on six-well plates. Cells grown at ~75% confluence were incubated with [1,2-3H]cholesterol (1 μCi/well; specific activity, 40 Ci/mmol; Perkin-Elmer, Boston, MA) in 2 ml of culture medium for 24 h at 37°C. Cells were washed three times with prewarmed PBS and incubated in serum-free medium for 8 h to equilibrate the intracellular cholesterol pool. Afterward, cells were washed three times with PBS and incubated in serum-free DMEM supplemented with 5 μl of serum from individual mice in both control and apoA-V groups. Four hours later, the conditioned medium was collected and centrifuged at 13,000 rpm in a microfuge for 5 min to remove potential cell contamination. Cells were washed four times with PBS and collected in 1 ml of 1 N NaOH. After incubation at 60°C for 1 h, the cell lysates were centrifuged at 13,000 rpm for 5 min. The radioactivity of conditioned medium and cell lysates was counted in a scintillation counter (Wallac 1410 Liquid Scintillation Counter; Perkin-Elmer). Cholesterol efflux was calculated as the percentage of radioactivity in conditioned medium out of total radioactivity in both conditioned medium and cells.
Statistics
Statistical analyses of data were performed by ANOVA using the JMP statistics software (Cary, NC). Dunnett’s test was performed to study the significance of differences between control and apoA-V groups. Data are expressed as means ± SEM. P < 0.05 was considered statistically significant.
RESULTS
Effect of apoA-V on plasma TG metabolism in APOC3 transgenic mice
To investigate whether apoA-V counteracts the effect of apoC-III on TG metabolism, we transferred apoA-V cDNA by adenovirus-mediated gene delivery to APOC3 transgenic mice. This APOC3 transgenic line expresses human apoC-III under the control of its native promoter in the C57BL/6J background (16). When fed regular chow, APOC3 transgenic mice developed hypertriglyceridemia at 4 weeks of age, as reflected in markedly increased plasma TG levels with normal blood glucose (80–140 mg/dl). In this study, male APOC3 transgenic mice (12 weeks old; mean body weight, 27 ± 3.1 g) were stratified by plasma TG levels and randomly assigned to two groups to ensure similar mean nonfasting plasma TG levels (670 ± 43). Mice were treated with either control LacZ or apoA-V vector for 2 weeks.
As shown in Fig. 1A, hepatic apoA-V production ameliorated hypertriglyceridemia, as indicated by an ~3-fold reduction in plasma TG levels in APOC3 transgenic mice. In contrast, hypertriglyceridemia persisted in control vector-treated APOC3 transgenic mice. To study the effect of apoA-V on cholesterol and NEFA metabolism, we determined total plasma cholesterol and NEFA levels. No significant differences in plasma cholesterol (Fig. 1B) and NEFA (Fig. 1C) levels were detected between apoA-V and control vector-treated APOC3 transgenic mice.
Fig. 1.

Lipid profiles. APOC3 transgenic mice treated with apolipoprotein A-V (apoA-V) and control (Ctrl) vectors were euthanized on day 14 after 16 h of fasting. Plasma levels of triglyceride (TG) (A), cholesterol (B), and NEFA (C) were determined. Aliquots of plasma (20 μl) of individual mice in capillaries were photographed (D). Data are expressed as means ± SEM. * P < 0.05.
The effect of hepatic apoA-V production on plasma TG metabolism was underpinned by the examination of sera extracted from control and apoA-V vector-treated mice. As shown in Fig. 1D, sera of control vector-treated APOC3 transgenic mice appeared milky, which is characteristic of severe hypertriglyceridemia. In contrast, sera of apoA-V vector-treated APOC3 transgenic mice were relatively clear, correlating with their significantly reduced plasma TG levels after 2 weeks of hepatic apoA-V production.
In addition, we determined the body weights of individual mice in both control and apoA-V vector-treated APOC3 transgenic groups. No significant differences in body weight between apoA-V (28.8 ± 2.4 g) and control (29.3 ± 1.5 g) groups of mice were detected during the course of this study. These results rule out the possibility that the reduction in plasma TG levels in apoA-V vector-treated APOC3 transgenic mice was attributable to body weight changes.
To correlate the reduction in plasma TG levels with hepatic apoA-V production, we determined plasma apoA-V, apoC-III, and apoB protein levels in both control and apoA-V vector-treated APOC3 transgenic mice. As shown in Fig. 2A, APOC3 transgenic mice exhibited relatively low basal plasma apoA-V levels, in agreement with previous observations that apoA-V is one of the least abundant apolipoproteins in plasma in mice and humans (5, 22). However, after 2 weeks of hepatic apoA-V production, ~4-fold higher levels of plasma apoA-V protein were detected in apoA-V vector-treated APOC3 transgenic mice (Fig. 2A). In addition, we detected a significant reduction in plasma apoC-III levels in apoA-V vector-treated APOC3 transgenic mice (Fig. 2B). In contrast, plasma apoB levels remained unaltered in response to hepatic apoA-V production (Fig. 2C).
Fig. 2.
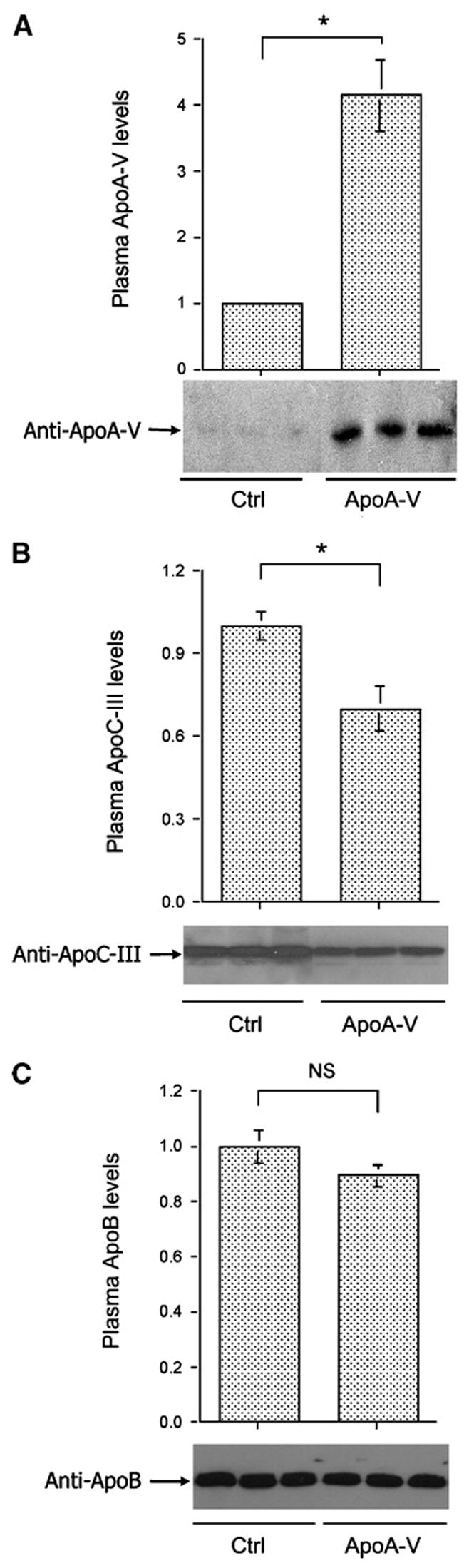
Plasma apolipoprotein levels. Aliquots of plasma (20 μg of protein) from euthanized APOC3 transgenic mice in control (Ctrl) and apoA-V groups were resolved on 4–20% SDS-polyacrylamide gels, which were subjected to immunoblot assay. Plasma levels of apoA-V (A), apoC-III (B), and apoB (C) were determined. Data are expressed as means ± SEM. * P < 0.05.
Effect of apoA-V on VLDL-TG production versus catabolism
To characterize the mechanism of apoA-V-mediated amelioration of hypertriglyceridemia in APOC3 transgenic mice, we determined hepatic VLDL-TG production versus catabolism. Mice in both groups at day 8 were fasted for 5 h, followed by intraperitoneal injection of 500 mg/kg Triton WR1339 to inhibit VLDL-TG hydrolysis and clearance. Aliquots of blood (25 μl) were collected from tail veins for the determination of plasma TG levels at 20 min intervals. As shown in Fig. 3A, plasma TG levels in both control and apoA-V vector-treated APOC3 transgenic mice increased after Triton WR1339 administration. The rates of VLDL-TG production, defined as milligrams of TG per minute per kilogram of body weight, were statistically indistinguishable between apoA-V and control groups (Fig. 3B). These results were consistent with the lack of alterations in plasma apoB levels in response to hepatic apoA-V production (Fig. 2C).
Fig. 3.
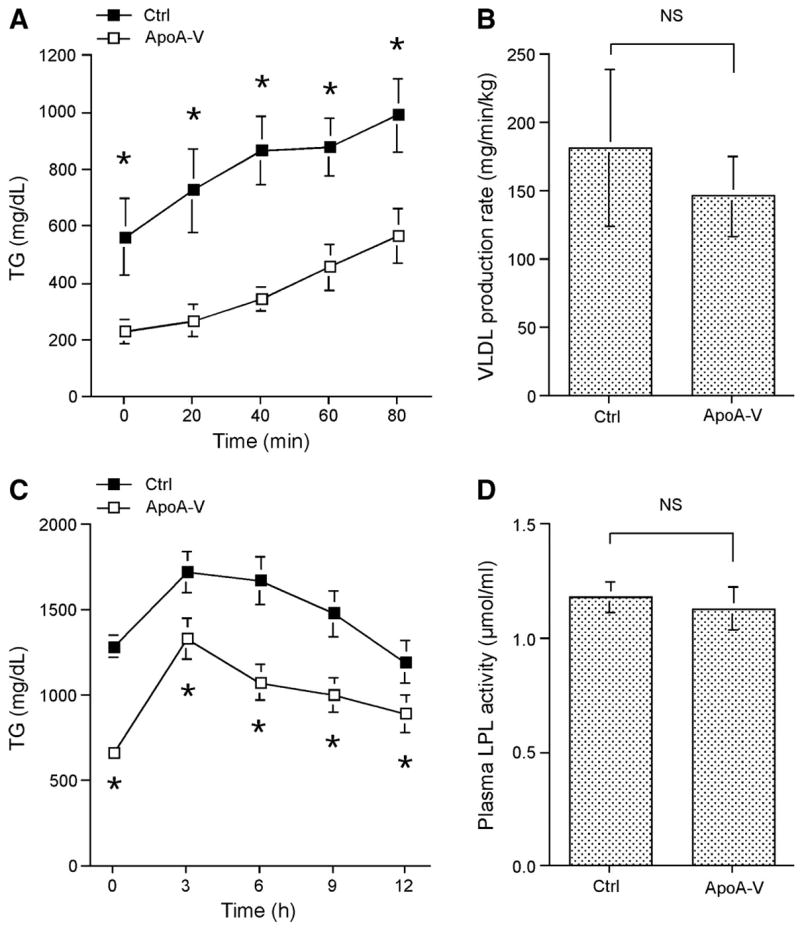
Hepatic VLDL-TG production versus catabolism. A: Hepatic VLDL-TG production. APOC3 transgenic mice treated with apoA-V and control (Ctrl) vectors were fasted at day 9 after vector administration for 5 h, followed by intraperitoneal injection of 500 mg/kg Triton WR1339. Aliquots of blood (25 μl) were collected at 20, 40, 60, and 80 min for the determination of plasma TG levels. B: The rates of hepatic VLDL-TG production, defined as milligrams of TG produced per minute per kilogram of body weight, were calculated based on the data in panel A. C: Postprandial TG response. Treated mice were fasted at day 5 after vector administration for 16 h, followed by an oral bolus of olive oil (10 μl/g body weight). Aliquots of tail vein blood (25 μl) were sampled at different times for the determination of plasma TG levels. D: Postheparin LPL activity. Treated APOC3 transgenic mice at day 7 were injected intravenously with 300 IU heparin/kg body weight, and tail vein blood (20 μl) was sampled at 10 min after heparin infusion. Heparinized sera were prepared for the determination of LPL activity. Data are expressed as means ± SEM.*P < 0.01.
To determine the effect of apoA-V on VLDL-TG catabolism, we performed oral fat tolerance tests. Mice were fasted overnight, followed by an oral bolus of olive oil. Aliquots of tail vein blood (25 μl) were sampled for the determination of plasma TG levels before and at different times after fat administration. As shown in Fig. 3C, control vector-treated APOC3 transgenic mice were associated with severely impaired fat tolerance. Compared with apoA-V vector-treated APOC3 transgenic mice, significantly higher plasma TG levels were detected at all time points in control vector-treated APOC3 transgenic mice (Fig. 3C). At 6 h after fat administration, plasma TG levels were still maintained at markedly higher levels in control vector-treated APOC3 transgenic mice (1,674 ± 142 mg/dl vs. 1,028 ± 108 mg/dl in the apoA-V group; P < 0.01).
To study the impact of hepatic apoA-V production on plasma LPL activity, we heparinized mice in both groups by intravenous injection of heparin. Aliquots of tail vein blood were collected to measure plasma LPL activity. As shown in Fig. 3D, no significant differences in postheparin plasma LPL activities were detected between control and apoA-V vector-treated APOC3 transgenic mice, suggesting that the observed reduction in plasma TG levels in apoA-V vector-treated mice was not attributable to increased LPL levels in plasma.
Impact of apoA-V on lipoprotein and cholesterol metabolism
To study the effect of apoA-V on lipoprotein metabolism, aliquots of plasma (250 μl) pooled from individual mice in the control or apoA-V group were subjected to gel filtration column chromatography for the fractionation of lipoprotein particles. ApoA-V vector-treated APOC3 transgenic mice were associated with reduced VLDL-TG levels (Fig. 4A), which were consistent with their significantly decreased plasma TG levels (Fig. 1A). Increased apoA-V production also resulted in decreased VLDL-cholesterol levels, but the fractional concentrations of cholesterol in HDL1, HDL2, and HDL3 were markedly increased in apoA-V vector-treated APOC3 transgenic mice (Fig. 4B). Thus, despite the lack of changes in total plasma cholesterol levels (Fig. 1B), increased apoA-V levels were associated with skewed cholesterol distribution from VLDL to large HDL particles (Fig. 4B).
Fig. 4.
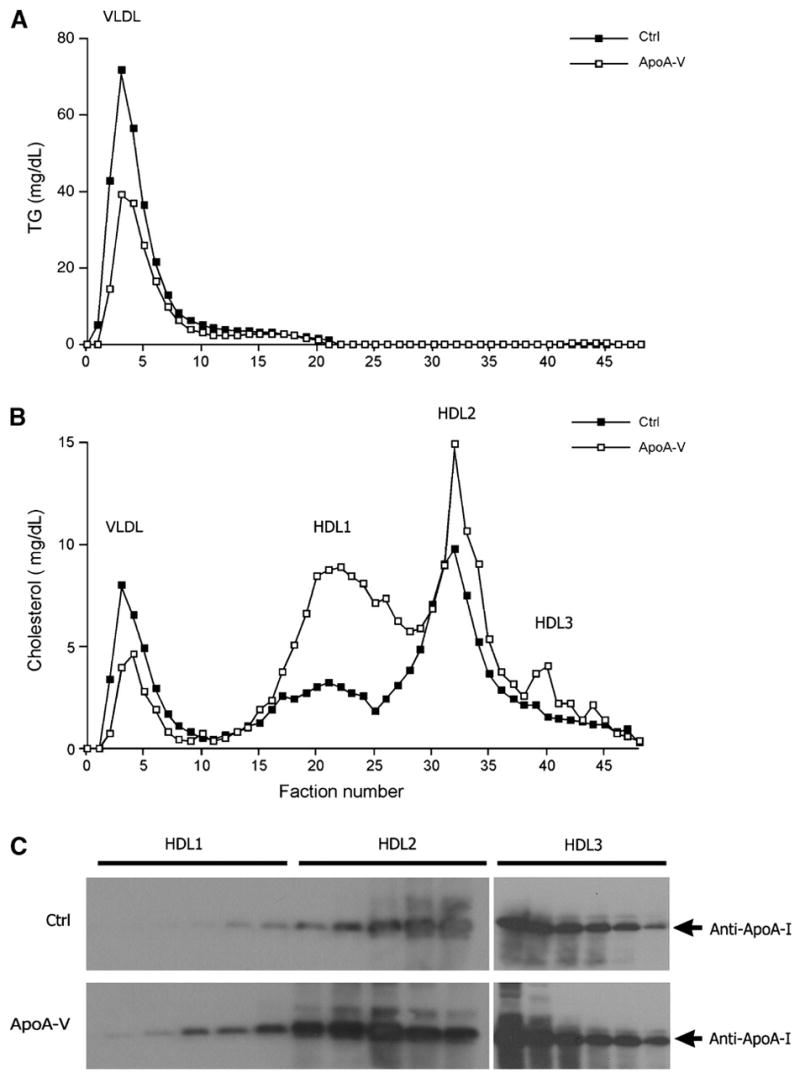
Plasma lipoprotein profiles. Mice were euthanized after a 16 h fast at 2 weeks after vector administration. Aliquots (250 μl) of plasma pooled from each group of mice were fractionated by column gel filtration chromatography. Fractions (500 μl) were eluted and assayed for TG (A) and cholesterol (B) levels. In addition, aliquots of HDL1 and HDL2 fractions (15 μl) were resolved on 4–20% SDS-polyacrylamide gels, which were subjected to immunoblot assay using anti-apoA-I antibody (C). Ctrl, control.
To account for these observations, we performed immunoblot assays to determine the fractional concentrations of apoA-I, a major constituent of HDL particles. As shown in Fig. 4C, significantly higher levels of apoA-I were detected in HDL1, HDL2, and HDL3 fractions of apoA-V vector-treated APOC3 transgenic mice. Thus, hepatic apoA-V production resulted in increased apoA-I levels, contributing to increased HDL-cholesterol levels in apoA-V vector-treated APOC3 transgenic mice. These results raise the possibility that apoA-V modulates nascent HDL formation and maturation.
To address this possibility, we used native agarose gel electrophoresis to separate nascent preβ-HDL from α-HDL particles, followed by immune detection and quantification. Preβ-HDL levels remained unchanged in response to hepatic apoA-V production (Fig. 5A, B). Instead, we detected a significant increase in α-HDL levels (Fig. 5A, C), correlating with the relatively higher HDL-cholesterol levels in apoA-V vector-treated APOC3 transgenic mice (Fig. 4B). Together, these results indicate that increased apoA-V production promoted α-HDL formation, resulting in a significant increase in both the size and number of HDL particles.
Fig. 5.
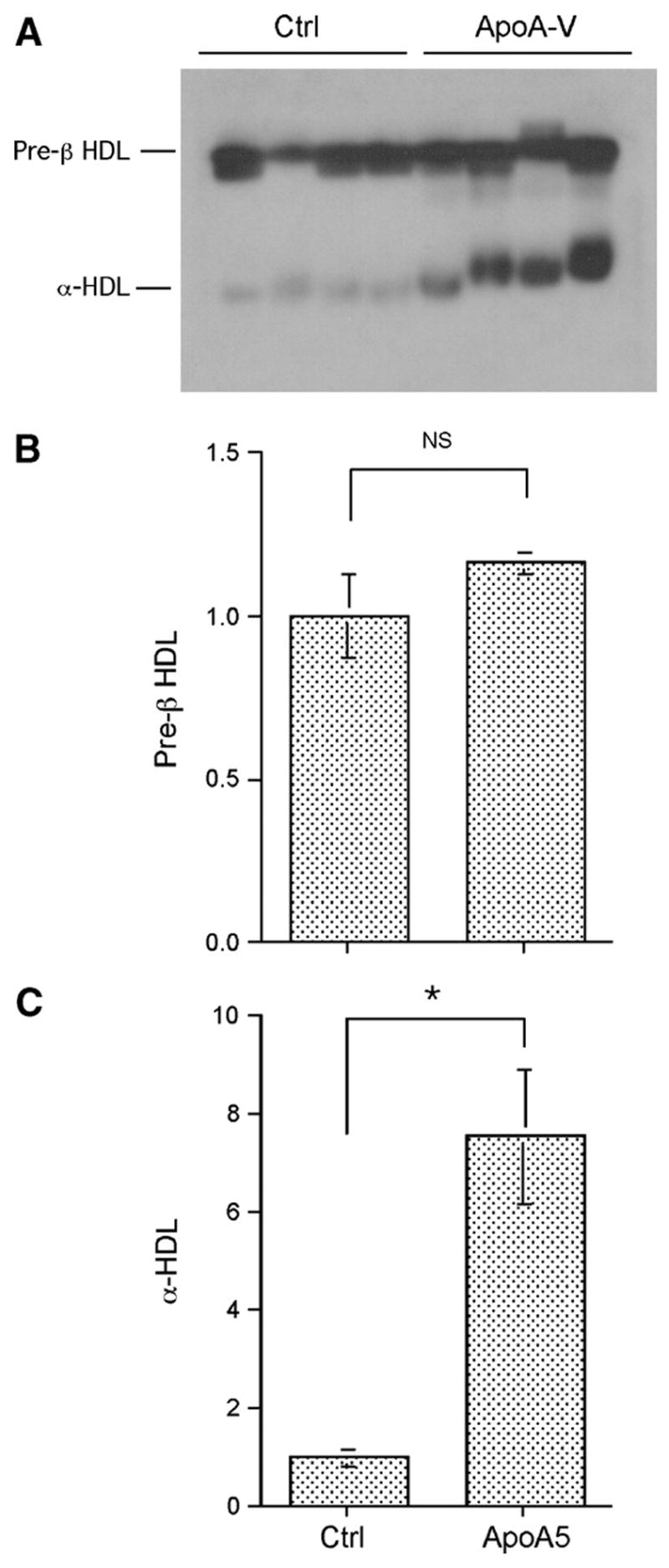
Effect of apoA-V on preβ-HDL and α-HDL metabolism. Aliquots of plasma (40 μg of protein) from euthanized mice were resolved on 0.7% agarose gels (A). Lipoproteins were transferred to nitrocellulose membrane by capillary blotting, followed by immune detection using rabbit anti-apoA-I antibody. Protein bands corresponding to preβ-HDL (B) and α-HDL (C) were quantified by densitometry. Data are expressed as means ± SEM. * P < 0.001. Ctrl, control.
Mechanism of apoA-V-mediated improvement in VLDL-TG catabolism
To probe the molecular basis underlying apoA-V-mediated improvement in VLDL-TG catabolism, we determined apoC-III and apoA-V content in lipoprotein particles by semiquantitative immunoblot assay. As shown in Fig. 6A, hepatic apoA-V production resulted in a significant reduction in apoC-III content in VLDL particles, correlating with the reduction in plasma VLDL-TG levels in apoA-V vector-treated APOC3 transgenic mice. Similarly, we detected a significant reduction in apoC-III content in HDL2 particles in response to hepatic apoA-V production (Fig. 6A). Interestingly, this observed reduction in apoC-III content in HDL was accompanied by a corresponding increase in apoA-V levels in both HDL1 and HDL2 fractions in apoA-V vector-treated APOC3 transgenic mice (Fig. 6B). ApoA-V levels in VLDL fractions were undetectable in both control and apoA-V vector-treated APOC3 transgenic mice.
Fig. 6.
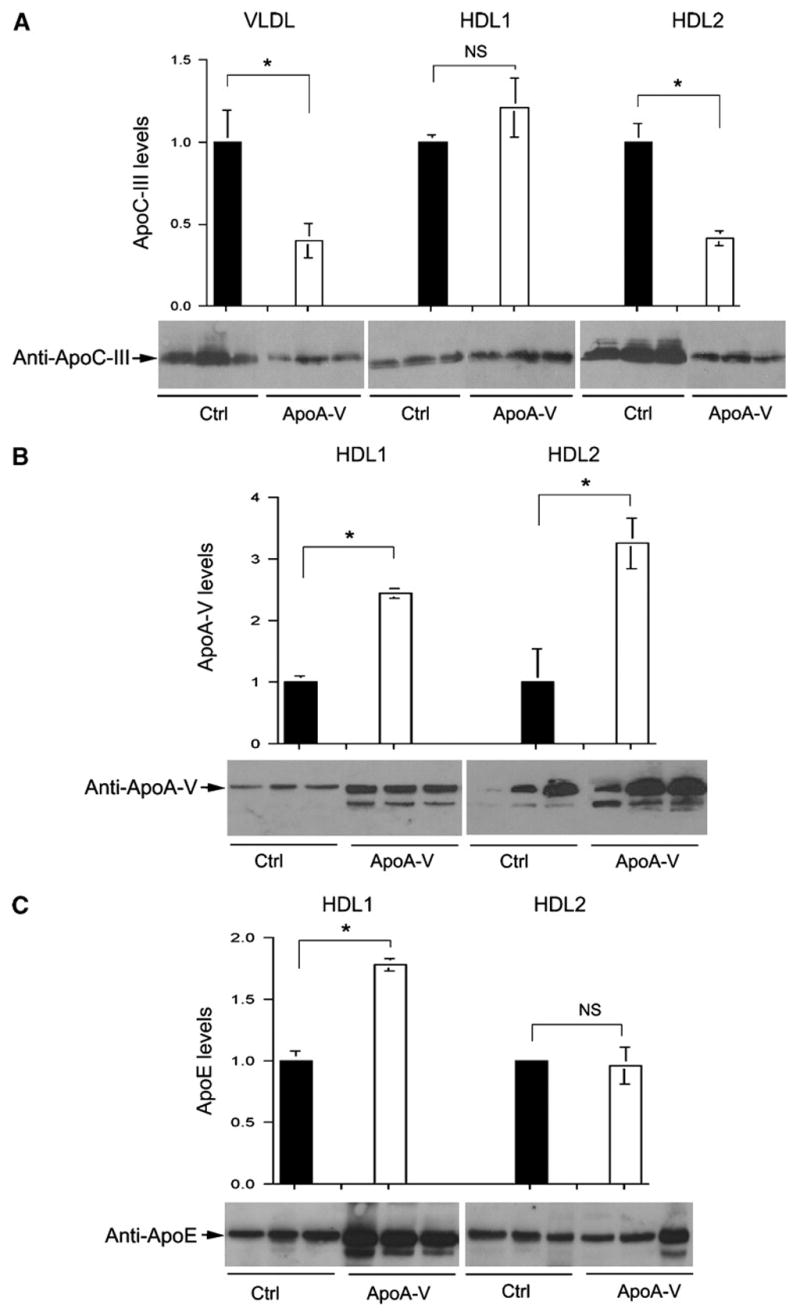
ApoC-III and apoA-V levels in lipoproteins. Aliquots of peak fractions (15 μl) in VLDL, HDL1, and HDL2 fractions were resolved on 4–20% SDS-polyacrylamide gels, followed by semiquantitative immunoblot assay using antibodies against apoC-III (A), apoA-V (B), and apoE (C). ApoA-V and apoE were undetectable in VLDL fractions using the same immunoblot assay. Protein bands were quantified by densitometry. Data are expressed as means ± SEM. * P < 0.05. Ctrl, control.
In addition, we detected a significant increase in apoE levels in HDL1 fractions (Fig. 6C), correlating with increased HDL-cholesterol levels in apoA-V vector-treated APOC3 transgenic mice. Together, these results suggest that in addition to its beneficial effect on VLDL-TG catabolism, apoA-V appears to regulate HDL-cholesterol metabolism by modulating the composition of apolipoproteins in HDL particles.
Mechanism of apoA-V-mediated increase in HDL particles
To account for the increase in the size and number of HDLs in apoA-V vector-treated APOC3 transgenic mice, we determined the activity of LCAT, a plasma protein that plays a key role in HDL maturation. LCAT catalyzes the conversion of free cholesterol to cholesteryl esters, resulting in the accumulation of cholesteryl esters in the core of HDL. As shown in Fig. 7A, plasma LCAT activity was increased significantly in response to increased apoA-V production. This effect mirrored the enrichment of apoE in HDL and the increases in the size and number of HDL particles in apoA-V vector-treated APOC3 transgenic mice (Fig. 6).
Fig. 7.

A: Plasma LCAT activity. Plasma was prepared from 16 h fasted mice after 2 weeks of hepatic apoA-V production and subjected to LCAT activity assay. The relative LCAT activity was compared between apoA-V and control (Ctrl) groups. B: Cholesterol efflux in macrophages. The ability of apoA-V-enriched HDL to conduct cholesterol efflux was assayed in cultured macrophages using 5 μl of plasma from apoA-V and control groups of fasted mice at the end of the 2 week study. C: Hepatic scavenger receptor class B type I (SR-BI) levels. D: Hepatic low density lipoprotein receptor (LDLR) levels. E: Hepatic HMG-CoA reductase levels. Aliquots of 20 mg of liver tissue were homogenized for either the preparation of total liver protein extracts or the enrichment of plasma membrane proteins, which were subjected to semiquantitative Western blot analysis. The relative levels of hepatic proteins SR-BI, HMG-CoA, and LDLR were compared between apoA-V and control groups using actin or the membrane-specific marker sodium potassium ATPase as an internal control. Data are expressed as means ± SEM. * P < 0.05.
In parallel, we determined the effect of apoA-V on cholesterol efflux in macrophages. As shown in Fig. 7B, increased apoA-V levels in plasma were associated with increased efflux of [3H]cholesterol from macrophages in culture. This effect, along with increased LCAT activity, indicates that apoA-V-enriched HDL particles are associated with an enhanced ability to conduct cholesterol efflux from peripheral tissue, contributing to increased plasma HDL-cholesterol levels in apoA-V vector-treated APOC3 transgenic mice.
To gain insight into hepatic HDL uptake and clearance, we determined hepatic expression levels of SR-BI, an acceptor for selective HDL uptake and clearance (32–34). As shown in Fig. 7C, we detected a small, but insignificant, increase in hepatic SR-BI protein level in response to increased apoA-V production. Likewise, similar levels of hepatic LDLR protein were detected in apoA-V and control groups (Fig. 7D). These results indicate that hepatic LDLR and SR-BI expression levels were not altered significantly in response to increased apoA-V production in liver.
To address whether increased plasma HDL-cholesterol levels were attributable to increased cholesterol synthesis in liver, we determined the expression levels of HMG-CoA reductase, an enzyme that controls the rate-limiting step in de novo cholesterol synthesis (35). As shown in Fig. 7E, no significant differences in hepatic HMG-CoA reductase levels were detected between apoA-V and control groups. These data suggest that increased HDL-cholesterol levels might not be the result of increased hepatic cholesterol synthesis in apoA-V vector-treated APOC3 transgenic mice.
DISCUSSION
In this study, we determined the effect of apoA-V on plasma TG and cholesterol metabolism in APOC3 transgenic mice. APOC3 transgenic mice fed regular chow develop hypertriglyceridemia as a result of retarded VLDL-TG catabolism (16, 17). Our goal in this study was 2-fold: 1) to address whether apoA-V can counteract apoC-III in improving VLDL-TG metabolism in adult mice with frank hypertriglyceridemia; and 2) to determine whether apoA-V exerts an effect on HDL-cholesterol metabolism.
Using an adenovirus-mediated gene expression approach, we achieved a 4-fold increase in plasma apoA-V levels in adult hypertriglyceridemic APOC3 transgenic mice. We show that hepatic apoA-V production resulted in significant reductions in plasma TG levels without alterations in total plasma cholesterol and NEFA levels. APOC3 transgenic mice with increased apoA-V levels exhibited significantly reduced VLDL-TG levels and improved postprandial TG response. This effect correlated with decreased apoC-III and increased apoA-V content in VLDL-TG particles in apoA-V vector-treated APOC3 transgenic mice. These results indicate that increased apoA-V levels modified VLDL apolipoprotein composition by decreasing apoC-III content, which favored VLDL-TG catabolism in apoA-V vector-treated hypertriglyceridemic mice. Using a transgenic approach, Fruchart-Najib et al. (21) showed that transgenic apoA-V expression prevented the development of hypertriglyceridemia in APOC3 transgenic mice. These results, together with our data, support the notion that apoA-V counterbalances the effect of apoC-III on VLDL-TG catabolism, contributing to the amelioration of hypertriglyceridemia in APOC3 transgenic mice.
Despite the improvement in VLDL-TG catabolism, plasma LPL activity remained unchanged in apoA-V vector-treated APOC3 transgenic mice. These results were consistent with previous data that neither apoA-V overexpression nor deficiency is associated with significant alterations in plasma LPL activity (36). How does apoA-V act to improve the clearance of TG-rich particles in the absence of altered LPL activity? To address this fundamental question, Merkel and Heeren (37) postulated that apoA-V augments plasma TG hydrolysis by fostering the interaction of TG-rich lipoproteins with endothelium-bound LPL. In support of this model, apoA-V has been shown to accelerate the hydrolysis of TG-rich lipoproteins in the presence of proteoglycan-bound LPL (36). In the absence of proteoglycans, apoA-V derived from TG-rich lipoproteins and apoA-V transgenic mouse HDL fractions did not affect the LPL-mediated hydrolysis of TG-rich particles (36, 38). This model helps explain the TG-lowing effect of apoA-V, but the underlying molecular basis remains to be elucidated (37, 38).
Although it is accepted that apoA-V-mediated reduction of plasma TG levels is through an enhanced clearance of TG-rich particles, there is a lack of consensus in the literature regarding the inhibitory effect of apoA-V on hepatic VLDL-TG production. We show that increased apoA-V production did not result in significant changes in hepatic VLDL-TG production, correlating with the lack of alterations in plasma apoB levels in apoA-V vector-treated APOC3 transgenic mice. Consistent with our observations, three independent studies have demonstrated in apoA-V transgenic or knockout mice that apoA-V does not affect hepatic VLDL-TG secretion in mice (20, 21, 36). In contrast, Schaap et al. (39) showed that adenovirus-mediated apoA-V production inhibits hepatic VLDL-TG production in addition to its stimulatory effect on VLDL-TG hydrolysis. Apart from the difference in mouse strains, a potential contributing factor to this discrepancy is the level of hepatic apoA-V production. In our study, we increased plasma apoA-V levels by ~4-fold in hypertriglyceridemic APOC3 transgenic mice, whereas a 10-fold increase of plasma apoA-V levels was achieved in normal C57BL/6J mice in the study by Schaap et al. (39).
In addition to its effect on plasma TG levels, our data revealed a profound effect of apoA-V on HDL metabolism. Increased apoA-V production did not affect preβ-HDL levels but promoted α-HDL formation, resulting in significant increases in both the number and size of HDL particles, as shown by increased HDL1- and HDL2-cholesterol levels in apoA-V vector-treated APOC3 transgenic mice. This effect correlated with increased apoA-V and decreased apoC-III contents in HDL particles. Furthermore, increased apoA-V levels resulted in increased apoA-I levels in HDL1 and HDL2 fractions. In contrast, VLDL-cholesterol levels were markedly reduced in response to hepatic apoA-V production. This reduction in VLDL-cholesterol levels was unlikely to be caused by decreased cholesterol synthesis, as hepatic protein levels of HMG-CoA reductase remained unchanged in apoA-V versus control groups. Thus, despite the lack of changes in total plasma cholesterol levels, increased apoA-V levels contributed to HDL remodeling and expansion with cholesterol, resulting in skewed cholesterol distribution toward large HDLs. It appeared that HDL particles with increased apoA-V content were associated with increased cholesterol-loading capacity.
Consistent with this idea is our observation that larger HDL1 particles in apoA-V vector-treated APOC3 transgenic mice were enriched with apoE, an apolipoprotein that plays an obligatory role in HDL particle expansion (40, 41). In addition, increased apoA-V levels were associated with enhanced LCAT activity and increased cholesterol efflux from cultured macrophages. A significant increase in LCAT activity, together with augmented efflux of cholesterol from peripheral tissues, contributed to HDL core expansion and enlargement of HDL particles in apoA-V vector-treated APOC3 transgenic mice. In accordance with these observations, increased plasma LCAT activity, resulting from transgenic LCAT expression in both mice and rabbits, is associated with enlarged HDL particles (42, 43). This property of apoA-V in modulating HDL remodeling, along with its ability to improve VLDL-TG catabolism, presages a potential beneficial effect of apoA-V on atherosclerosis. However, because of the infrastructural constraint in our animal facility, we are limited to conducting radioisotope tracer assays to determine the effect of apoA-V on hepatic HDL uptake and clearance in mice. Thus, whether apoA-V can improve reverse cholesterol transport and ameliorate atherosclerotic lesions remains to be determined in animal models with premature atherosclerosis.
In conclusion, our data show that apoA-V at higher levels is able to modulate the apolipoprotein composition of VLDL, resulting in enhanced VLDL-TG catabolism and contributing to the amelioration of hypertriglyceridemia in adult APOC3 transgenic mice. Furthermore, apoA-V was shown to promote α-HDL formation, accounting at least in part for skewed cholesterol redistribution to large HDL particles. These data underscore the functional importance of apoA-V in plasma VLDL-TG and HDL-cholesterol metabolism. Augmented apoA-V production should be considered a modality for the pharmacological management of hypertriglyceridemia associated with the metabolic syndrome.
Acknowledgments
The authors thank Drs. Steve Ringquist, Adama Kamagate, and Sandra Slusher for critical reading of the manuscript. This study was supported in part by the American Diabetes Association and by National Institutes of Health Grant DK-066301.
Abbreviations
- apoA-V
apolipoprotein A-V
- LDLR
low density lipoprotein receptor
- SR-BI
scavenger receptor class B type I
- TG
triglyceride
Footnotes
This study was presented at the 66th American Diabetes Association Scientific Session, Washington, DC, June 9–13, 2006.
References
- 1.Rapp RJ. Hypertriglyceridemia: a review beyond low-density lipoprotein. Cardiol Rev. 2002;10:163–172. doi: 10.1097/00045415-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Moller DE, Kaufman KD. Metabolic syndrome: a clinical and molecular perspective. Annu Rev Med. 2005;56:45–62. doi: 10.1146/annurev.med.56.082103.104751. [DOI] [PubMed] [Google Scholar]
- 3.Hopkins PN, Heiss G, Ellison RC, Province MA, Pankow JS, Eckfeldt JH, Hunt SC. Coronary artery disease risk in familial combined hyperlipidemia and familial hypertriglyceridemia: a case-control comparison from the National Heart, Lung, and Blood Institute Family Heart Study. Circulation. 2003;108:519–523. doi: 10.1161/01.CIR.0000081777.17879.85. [DOI] [PubMed] [Google Scholar]
- 4.Hopkins PN, Wu LL, Hunt SC, Brinton EA. Plasma triglycerides and type III hyperlipidemia are independently associated with premature familial coronary artery disease. J Am Coll Cardiol. 2005;45:1003–1012. doi: 10.1016/j.jacc.2004.11.062. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien PJ, Alborn WE, Sloan JH, Ulmer M, Boodhoo A, Knierman MD, Schultze AE, Konrad RJ. The novel apolipoprotein A5 is present in human serum, is associated with VLDL, HDL, and chylomicrons, and circulates at very low concentrations compared with other apolipoproteins. Clin Chem. 2005;51:351–359. doi: 10.1373/clinchem.2004.040824. [DOI] [PubMed] [Google Scholar]
- 6.Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 7.Talmud PJ, Hawe E, Martin S, Olivier M, Miller GJ, Rubin EM, Pennacchio LA, Humphries SE. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet. 2002;11:3039–3046. doi: 10.1093/hmg/11.24.3039. [DOI] [PubMed] [Google Scholar]
- 8.Aouizerat BE, Kulkarni M, Heilbron D, Drown D, Raskin S, Pullinger CR, Malloy MJ, Kane JP. Genetic analysis of a polymorphism in the human apoA-V gene: effect on plasma lipids. J Lipid Res. 2003;44:1167–1173. doi: 10.1194/jlr.M200480-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Lai CQ, Tai ES, Tan CE, Cutter J, Chew SK, Zhu YP, Adiconis X, Ordovas JM. The APOA5 locus is a strong determinant of plasma triglyceride concentrations across ethnic groups in Singapore. J Lipid Res. 2003;44:2365–2373. doi: 10.1194/jlr.M300251-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Li GP, Wang JY, Yan SK, Chen BS, Xue H, Wu G. Genetic effect of two polymorphisms in the apolipoprotein A5 gene and apolipoprotein C3 gene on serum lipids and lipoprotein levels in a Chinese population. Clin Genet. 2004;65:470–476. doi: 10.1111/j.1399-0004.2004.00251.x. [DOI] [PubMed] [Google Scholar]
- 11.Klos KL, Hamon S, Clark AG, Boerwinkle E, Liu K, Sing CF. APOA5 polymorphisms influence plasma triglycerides in young, healthy African Americans and whites of the CARDIA Study. J Lipid Res. 2005;46:564–571. doi: 10.1194/jlr.M400437-JLR200. [DOI] [PubMed] [Google Scholar]
- 12.Wright WT, I, Young S, Nicholls DP, Patterson C, Lyttle K, Graham CA. SNPs at the APOA5 gene account for the strong association with hypertriglyceridaemia at the APOA5/A4/C3/A1 locus on chromosome 11q23 in the Northern Irish population. Atherosclerosis. 2006;185:353–360. doi: 10.1016/j.atherosclerosis.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 13.Eichenbaum-Voline S, Olivier M, Jones EL, Naoumova RP, Jones B, Gau B, Patel HN, Seed M, Betteridge DJ, Galton DJ, et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 2004;24:167–174. doi: 10.1161/01.ATV.0000099881.83261.D4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mar R, Pajukanta P, Allayee H, Groenendijk M, Dallinga-Thie G, Krauss RM, Sinsheimer JS, Cantor RM, de Bruin TW, Lusis AJ. Association of the APOLIPOPROTEIN A1/C3/A4/A5 gene cluster with triglyceride levels and LDL particle size in familial combined hyperlipidemia. Circ Res. 2004;94:993–999. doi: 10.1161/01.RES.0000124922.61830.F0. [DOI] [PubMed] [Google Scholar]
- 15.Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. 2002;11:3031–3038. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- 16.Aalto-Setala K, Fisher EA, Chen X, Chajek-Shaul T, Hayek T, Zechner R, Walsh A, Ramakrishnan R, Ginsberg HN, Breslow JL. Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J Clin Invest. 1992;90:1889–1900. doi: 10.1172/JCI116066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aalto-Setala K, Weinstock PH, Bisgaier CL, Wu L, Smith JD, Breslow JL. Further characterization of the metabolic properties of triglyceride-rich lipoproteins from human and mouse apoC-III transgenic mice. J Lipid Res. 1996;37:1802–1811. [PubMed] [Google Scholar]
- 18.Ito YN, Azrolan A, O’Connell A, Walsh A, Breslow JL. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990;249:790–793. doi: 10.1126/science.2167514. [DOI] [PubMed] [Google Scholar]
- 19.Maeda N, Li H, Lee D, Oliver P, Quarfordt SH, Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J Biol Chem. 1994;269:23610–23616. [PubMed] [Google Scholar]
- 20.Grosskopf I, Baroukh N, Lee SJ, Kamari Y, Harats D, Rubin EM, Pennacchio LA, Cooper AD. Apolipoprotein A-V deficiency results in marked hypertriglyceridemia attributable to decreased lipolysis of triglyceride-rich lipoproteins and removal of their remnants. Arterioscler Thromb Vasc Biol. 2005;25:2573–2579. doi: 10.1161/01.ATV.0000186189.26141.12. [DOI] [PubMed] [Google Scholar]
- 21.Fruchart-Najib J, Bauge E, Niculescu LS, Pham T, Thomas B, Rommens C, Majd Z, Brewer B, Pennacchio LA, Fruchart JC. Mechanism of triglyceride lowering in mice expressing human apolipoprotein A5. Biochem Biophys Res Commun. 2004;319:397–404. doi: 10.1016/j.bbrc.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Baroukh N, Bauge E, Akiyama J, Chang J, Afzal V, Fruchart JC, Rubin EM, Fruchart-Najib J, Pennacchio LA. Analysis of apolipoprotein A5, C3, and plasma triglyceride concentrations in genetically engineered mice. Arterioscler Thromb Vasc Biol. 2004;24:1297–1302. doi: 10.1161/01.ATV.0000130463.68272.1d. [DOI] [PubMed] [Google Scholar]
- 23.Rensen PC, van Dijk KW, Havekes LM. Apolipoprotein AV: low concentration, high impact. Arterioscler Thromb Vasc Biol. 2005;25:2445–2447. doi: 10.1161/01.ATV.0000193889.65915.f9. [DOI] [PubMed] [Google Scholar]
- 24.Jakel H, Nowak M, Helleboid-Chapman A, Fruchart-Najib J, Fruchart JC. Is apolipoprotein A5 a novel regulator of triglyceride-rich lipoproteins? Ann Med. 2006;38:2–10. doi: 10.1080/07853890500407488. [DOI] [PubMed] [Google Scholar]
- 25.Nakae J, Kitamura T, Silver DL, Accili D. The fork-head transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altomonte J, Richter A, Harbaran S, Suriawinata J, Nakae J, Thung SN, Meseck M, Accili D, Dong H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol. 2003;285:E718–E728. doi: 10.1152/ajpendo.00156.2003. [DOI] [PubMed] [Google Scholar]
- 27.Dong H, Altomonte J, Morral N, Meseck M, Thung SN, Woo SLC. Basal insulin gene expression significantly improves conventional insulin therapy in type 1 diabetic rats. Diabetes. 2002;51:130–138. doi: 10.2337/diabetes.51.1.130. [DOI] [PubMed] [Google Scholar]
- 28.Huard J, Lochmüller H, Acsadi G, Jani A, Massie B, Karpati G. The route of administration is a major determinant of the transduction efficiency of rat tissues by adenoviral recombinants. Gene Ther. 1995;2:107–115. [PubMed] [Google Scholar]
- 29.Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. Foxo1 mediates insulin action on apoC-III and triglyceride metabolism. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Kane MJ, Wisdom GB, McEneny J, McFerran NV, Trimble ER. Pre-beta high-density lipoprotein determined by immunoblotting with chemiluminescent detection. Clin Chem. 1992;38:2273–2277. [PubMed] [Google Scholar]
- 31.de la Llera Moya M, Atger V, Paul JL, Fournier N, Moatti N, Giral P, Friday KE, Rothblat G. A cell culture system for screening human serum for ability to promote cellular cholesterol efflux. Relations between serum components and efflux, esterification, and transfer. Arterioscler Thromb. 1994;14:1056–1065. doi: 10.1161/01.atv.14.7.1056. [DOI] [PubMed] [Google Scholar]
- 32.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 33.Huby T, Doucet C, Dachet C, Ouzilleau B, Ueda Y, Afzal V, Rubin E, Chapman MJ, Lesnik P. Knockdown expression and hepatic deficiency reveal an atheroprotective role for SR-BI in liver and peripheral tissues. J Clin Invest. 2006;116:2767–2776. doi: 10.1172/JCI26893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chroni A, Nieland TJ, Kypreos KE, Krieger M, Zannis VI. SR-BI mediates cholesterol efflux via its interactions with lipid-bound apoE. Structural mutations in SR-BI diminish cholesterol efflux. Biochemistry. 2005;44:13132–13143. doi: 10.1021/bi051029o. [DOI] [PubMed] [Google Scholar]
- 35.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 36.Merkel M, Loeffler B, Kluger M, Fabig N, Geppert G, Pennacchio LA, Laatsch A, Heeren J. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280:21553–21560. doi: 10.1074/jbc.M411412200. [DOI] [PubMed] [Google Scholar]
- 37.Merkel M, Heeren J. Give me A5 for lipoprotein hydrolysis! J Clin Invest. 2005;115:2694–2696. doi: 10.1172/JCI26712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan RO. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem. 2005;280:25383–25387. doi: 10.1074/jbc.M501589200. [DOI] [PubMed] [Google Scholar]
- 39.Schaap FG, Rensen PC, Voshol PJ, Vrins C, van der Vliet HN, Chamuleau RA, Havekes LM, Groen AK, van Dijk KW. ApoAV reduces plasma triglycerides by inhibiting very low density lipoproteintriglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem. 2004;279:27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 40.Koo C, Innerarity TL, Mahley RW. Obligatory role of cholesterol and apolipoprotein E in the formation of large cholesterol-enriched and receptor-active high density lipoproteins. J Biol Chem. 1985;260:11934–11943. [PubMed] [Google Scholar]
- 41.Mahley RW, Huang Y, Weisgraber KH. Putting cholesterol in its place: apoE and reverse cholesterol transport. J Clin Invest. 2006;116:1226–1229. doi: 10.1172/JCI28632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brousseau ME, Santamarina-Fojo S, Vaisman BL, Applebaum-Bowden D, Berard AM, Talley GD, Brewer HB, Jr, Hoeg JM. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J Lipid Res. 1997;38:2537–2547. [PubMed] [Google Scholar]
- 43.Berard AM, Foger B, Remaley A, Shamburek R, Vaisman BL, Talley G, Paigen B, Hoyt RF, Jr, Marcovina S, Brewer HB, Jr, et al. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744–749. doi: 10.1038/nm0797-744. [DOI] [PubMed] [Google Scholar]