

Abstract
Background
Chronic heart failure is associated with inflammation and neurohormonal imbalance. The 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors, or statins, exert anti-inflammatory and vascular protective effects. We hypothesized that short-term statin therapy may have beneficial effects in patients with nonischemic heart failure.
Methods and Results
Sixty-three patients with symptomatic, nonischemic, dilated cardiomyopathy were randomly divided into 2 groups. One group received simvastatin (n=24), and the other group received placebo (n=27). The initial dose of simvastatin was 5 mg/d, which was increased to 10 mg/d after 4 weeks. After 14 weeks, patients receiving simvastatin exhibited a modest reduction in serum cholesterol level compared with patients receiving placebo (130±13 versus 148±18, P<0.05). Patients treated with simvastatin had a lower New York Heart Association functional class compared with patients receiving placebo (2.04±0.06 versus 2.32±0.05, P<0.01). This corresponded to improved left ventricular ejection fraction in the simvastatin group (34±3 to 41±4%, P<0.05) but not in the placebo group. Furthermore, plasma concentrations of tumor necrosis factor-α, interleukin-6, and brain natriuretic peptide were significantly lower in the simvastatin group compared with the placebo group.
Conclusions
Short-term statin therapy improves cardiac function, neurohormonal imbalance, and symptoms associated with idiopathic dilated cardiomyopathy. These findings suggest that statins may have therapeutic benefits in patients with heart failure irrespective of serum cholesterol levels or atherosclerotic heart disease.
Keywords: heart failure, statins, cholesterol, endothelium, inflammation
Current therapy for patients with chronic heart failure (CHF) includes angiotensin-converting enzyme (ACE) inhibitors, β-adrenergic receptor blockers, and diuretics.1 However, despite aggressive medical therapy, CHF remains a major cause of morbidity and mortality worldwide. CHF is characterized by impaired cardiac performance, inflammation, and neurohormonal imbalance.2 Indeed, increased levels of catecholamines, cytokines, and renin angiotensin are thought to play important roles in the pathophysiology and development of CHF.3,4
The 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors or statins improve endothelial function5 and decrease plasma levels of tumor necrosis factor-α) (TNF-α) in patients with coronary artery disease and hyperlipidemia.6 Statins decrease the incidence of CHF in hyperlipidemic patients probably by lipid lowering and reducing subsequent ischemic cardiac events.7 However, it is not known whether statins have additional beneficial effects in patients with established, nonischemic cardiomyopathy. Because statins improve endothelial function8 and suppress systemic inflammatory responses,9 we hypothesized that statins may improve cardiac function and neurohormonal imbalance in patients with symptomatic, nonischemic left ventricular dysfunction.
Methods
Participants
We studied 51 patients with symptomatic CHF in Japan. The criteria for enrollment in this study were clinical evidence of heart failure despite the conventional therapy and a left ventricular ejection fraction below 40%, as assessed by 2D echocardiography. All of the patients have New York Heart Association (NYHA) functional classification of II to III. There were 36 men and 15 women with a mean age of 54 years. All patients were diagnosed with idiopathic dilated cardiomyopathy. Ischemic and primary valvular heart disease were excluded by angiography and echocardiography. Exclusion criteria included chronic obstructive pulmonary disease, primary valvular heart disease, history or evidence of ischemic heart disease, systolic blood pressure >160 mm Hg or diastolic blood pressure >95 mm Hg, clinical conditions other than cardiomyopathy that could lead to increased cytokine levels (ie, rheumatoid arthritis and sepsis), pregnancy, and severe liver disease as defined by hepatic enzymes >2 times the upper limit of normal. Informed consent was obtained from all patients before participation in this study in accordance with institutional approved protocols.
Study Protocol
All patients were on optimal, stable doses of β-blockers for at least 3 months before screening echocardiography and randomization. Before receiving their randomized treatment assignment, 2 noncompliant patients were dropped from the study. Patients were randomly divided into 2 treatment groups, simvastatin (n=24) and placebo (n=27). Simvastatin was administered orally after a placebo run-in period of 2 weeks. The initial dose of simvastatin was 5 mg/d, which was increased to a final dose of 10 mg/d after 4 weeks. The type of β-blockers used was not different between the placebo and treatment groups (Table 1), and with the exception of 3 patients who discontinued β-blocker therapy because of hypotension, the doses of β-blockers, ACE inhibitors, and diuretics were not adjusted during the 14-week course.
TABLE 1.
Baseline Characteristics of Study Population
| Placebo (n=25) | Statin (n=23) | |
|---|---|---|
| Age, y | 53±4 | 55±3 |
| M/F, % | 18/7 (72/28) | 15/8 (65/35) |
| NYHA functional class II/III, % | 16/9 (64/36) | 14/9 (61/39) |
| NYHA functional class, mean | 2.36±0.07 | 2.39±0.07 |
| Body mass index, kg/m2 | 21±2 | 20±2 |
| Former smoking history, % | 4 (16) | 3 (13) |
| Hypertension, % | 2 (8) | 1 (4) |
| Diabetes mellitus, % | 2 (8) | 2 (9) |
| Hypercholesterolemia (total cholesterol >220), % | 16 (64) | 14 (61) |
| Duration of CHF, y | 3.0±2.4 | 3.2±2.7 |
| Systolic blood pressure, mm Hg | 114±14 | 116±15 |
| Diastolic blood pressure, mm Hg | 64±5 | 66±8 |
| Heart rate, bpm | 75±7 | 77±6 |
| Total cholesterol, mg/dL | 217±16 | 221±18 |
| LDL cholesterol, mg/dL | 144±13 | 154±18 |
| HDL cholesterol, mg/dL | 46±5 | 44±8 |
| Triglycerides, mg/dL | 125±16 | 129±18 |
| Ejection fraction, % | 33±5 | 34±4 |
| Concomitant drugs, No. (%) | ||
| Digoxin | 21 (84) | 19 (83) |
| Diuretics | 23 (94) | 22 (96) |
| Nitrates | 4 (16) | 3 (13) |
| β-blockers | 16 (64) | 14 (61) |
| ACE inhibitors | 23 (94) | 22 (96) |
Values are either numbers of each group, range, or mean±SEM. Ejection fraction=(left ventricle end-diastolic diameter–left ventricle end-systolic diameter)/left ventricle end-diastolic diameter.
Blood samples were collected in test tubes containing EDTA at baseline and after 14 weeks of treatment with patients in the supine position for at least 30 minutes. The plasma was separated from blood cells by centrifugation and frozen at −80°C. Plasma norepinephrine was measured by high-performance liquid chromatography. Plasma concentrations of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and aldosterone were measured using a specific immunoradiometric assay.10 TNF-α and interleukin-6 (IL-6) levels were measured using high-sensitivity ELISA (Quantikine HS, R&D Systems). The personnel performing these assays were blinded to the patients’ treatment assignment.
M-mode echocardiography was performed with 2D monitoring using a Sono layer phased-array sector scanner (model SONOS 5500, Hewlett Packard) before and after 14 weeks of treatment with simvastatin or matching placebo. Screening echocardiograms were performed after a placebo run-in period of 2 weeks to eliminate the effects of other drugs. Left ventricular volumes were calculated using Teichholtz’s formula and used to determine LVEF.11 All echocardiograms were read by the same physician, who was blinded to patients’ treatment, assignment, and time point.
Flow-mediated dilatation (FMD) of the brachial artery was determined according to established and validated methods.12 Images were obtained with 10-MHz linear-array transducer (Hewlett Packard). Imaging was performed in the morning with the patients in a fasting state, resting quietly for at least 10 minutes in a light- and temperature-controlled room. After baseline measurements of the brachial artery diameter, a blood pressure cuff was inflated to 200 mm Hg over the proximal portion of the right forearm for 5 minutes. FMD was determined 1 minute after release of the cuff.13 The brachial ultrasound studies were interpreted by the same physician blinded to patient treatment assignment and time point.
All of the patient providers, including the echocardiographer and ultrasonographer, were blinded to plasma lipid levels and drug allocation, and the same physician made the NYHA assignments at baseline and after 14 weeks of treatment.
Statistical Analysis
Baseline characteristics were compared by Fisher’s exact or Cochran-Mantel-Haenszel test for categorical variables. ANOVA was used to test for treatment group baseline differences for continuous variables. Hemodynamic and hormonal responses to various statins were compared by parametric ANCOVA, with the baseline level used as the covariate. Within-treatment analyses of changes were performed at 0.05 significance level by Student’s t test.
Results
Medications were well tolerated over the 14-week period. Forty-eight (94%) of 51 patients completed the protocol. Of the 3 patients who failed to complete the protocol, 1 received simvastatin and withdrew from the study because of gastrointestinal disturbance. One patient with placebo and 1 patient with simvastatin were admitted to the hospital for worsening congestive heart failure and were excluded from the study because of changes in dose and type of medications. No patients died during the 14-week study. In addition, with the exception of 3 patients who discontinued β-blocker therapy because of hypotension (1 patient in placebo group [carvedilol] and 2 patients in statin group [1 carvedilol and 1 bisoprolol]), the doses of β-blockers (carvedilol, 20 mg/d; metoprolol, 120 mg/d; bisoprolol, 5 mg/d), ACE inhibitors, and diuretics were not altered during the course of the entire study.
There were no differences in age, gender, NYHA functional class, body mass index, lipid profile, hemodynamic parameters, LVEF, or concurrent medications between the stain and placebo groups (Table 1). Of the patients taking β-blockers in the placebo and statin groups, there were no differences in the number of patients taking either carvedilol (13 versus 11 patients; 52% versus 48%, P>0.05), metoprolol (2 versus 1 patients; 8% versus 4%, P>0.05), or bisoprolol (1 versus 2 patients; 4% versus 9%, P>0.05). After 14 weeks of treatment, there was a modest reduction in total and LDL cholesterol without significant changes in triglycerides and HDL cholesterol in the statin group (Table 2).
TABLE 2.
Effects of Statin on the Hemodynamic and Hormonal Parameter
| Placebo (n=25) | Statin (n=23) | |
|---|---|---|
| NYHA functional class I/II/III/IV | 1/16/7/1 | 4/14/5/0 |
| NYHA functional class, mean | 2.32±0.05 | 2.04±0.06* |
| Body mass index, kg/m2 | 21±3 | 20±2 |
| Systolic blood pressure, mm Hg | 113±5 | 124±6 |
| Diastolic blood pressure, mm Hg | 65±4 | 70±4 |
| Heart rate, bpm | 77±3 | 72±4 |
| Total cholesterol, mg/dL | 223±18 | 201±14† |
| LDL cholesterol | 148±16 | 130±13† |
| HDL cholesterol | 48±7 | 52±4 |
| Triglycerides | 128±16 | 122±18 |
| Ejection fraction, % | 34±3 | 41±4† |
Values are either numbers of each group, range, or mean±SEM.
P<0.01,
P<0.05 vs control.
Blood pressure and heart rate were not different between the statin and placebo groups either before or after treatment. However, patients who received simvastatin demonstrated improved functional capacity compared with patients receiving placebo (Figure 1A). In the statin group, 39.1% of patients had an improved functional class, 56.6% remained unchanged, and 4.3% deteriorated. In contrast, in the placebo group, 16% of patients improved, 84% remained unchanged, and 12% deteriorated (P<0.01 between the groups). The functional improvement in the statin group was associated with improved cardiac performance. Compared with the placebo group, patients treated with simvastatin have higher LVEF (Figure 1B). The increase in LVEF was attributable predominantly to a decrease in left ventricular end-systolic volume (Figures 2A and 2B).
Figure 1.
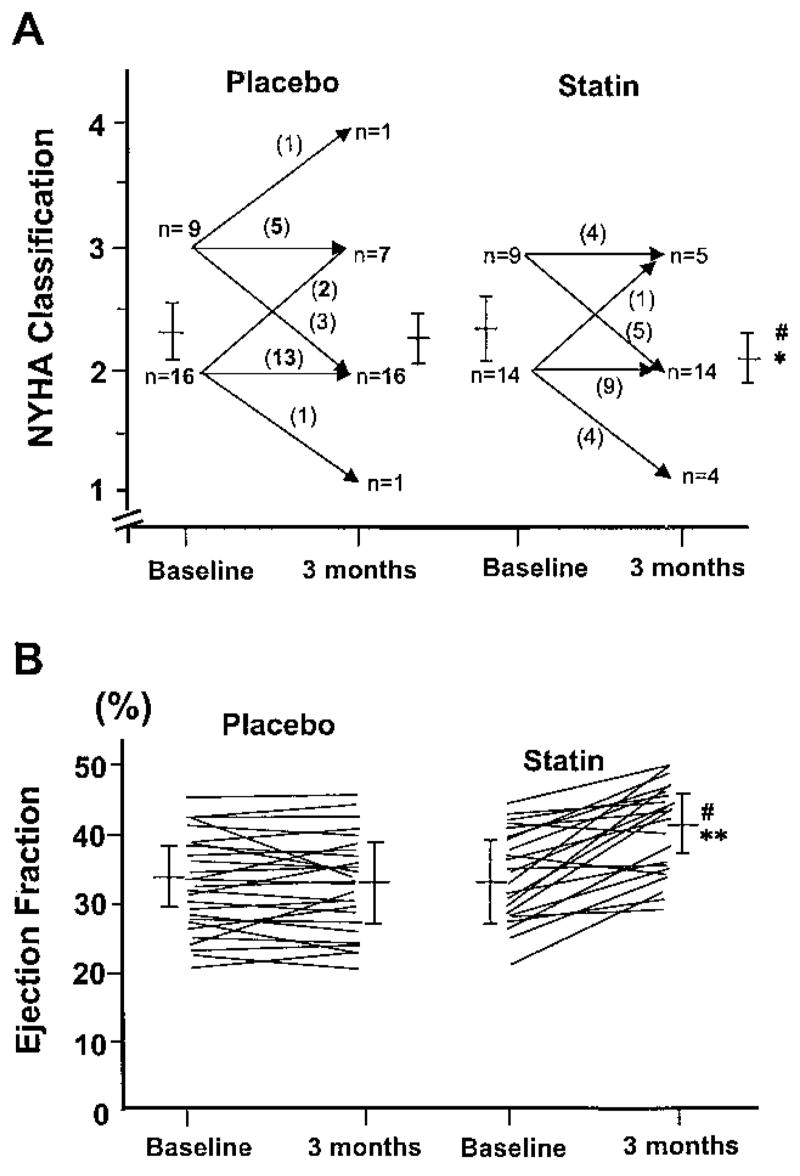
Changes in NYHA classification (A) and LVEF (B) before and after 14 weeks of treatment in placebo and statin groups. *P<0.01 and **P<0.05 vs placebo group.
Figure 2.

Changes in left ventricular end-diastolic volume (LVEDV) (A) or end-systolic volume (LVESV) (B) before and after 14 weeks of treatment in placebo and statin group. *P<0.05 vs placebo group.
Plasma levels of ANP, BNP, norepinephrine, aldosterone, or cytokines such as TNF-α and IL-6 were unchanged after 14 weeks in the placebo group. Plasma level of BNP, but not ANP (from 76.5±10.3 to 67.3±7.6 pg/mL), was decreased after statin therapy (Figure 3A). Plasma norepinephrine also tended to decline in patients treated with statins (from 420±49 to 398±67 pg/mL). This effect was not different from the changes observed in the placebo group, and there was no difference in aldosterone concentration between the 2 groups (placebo, from 62.4±9.2 to 67.1±8.2 pg/mL; statin group, from 74.3±21.7 to 63.3±19.2 pg/mL). Plasma levels of TNF-α and IL-6 were decreased in patients receiving simvastatin (Figures 3B and 3C).
Figure 3.
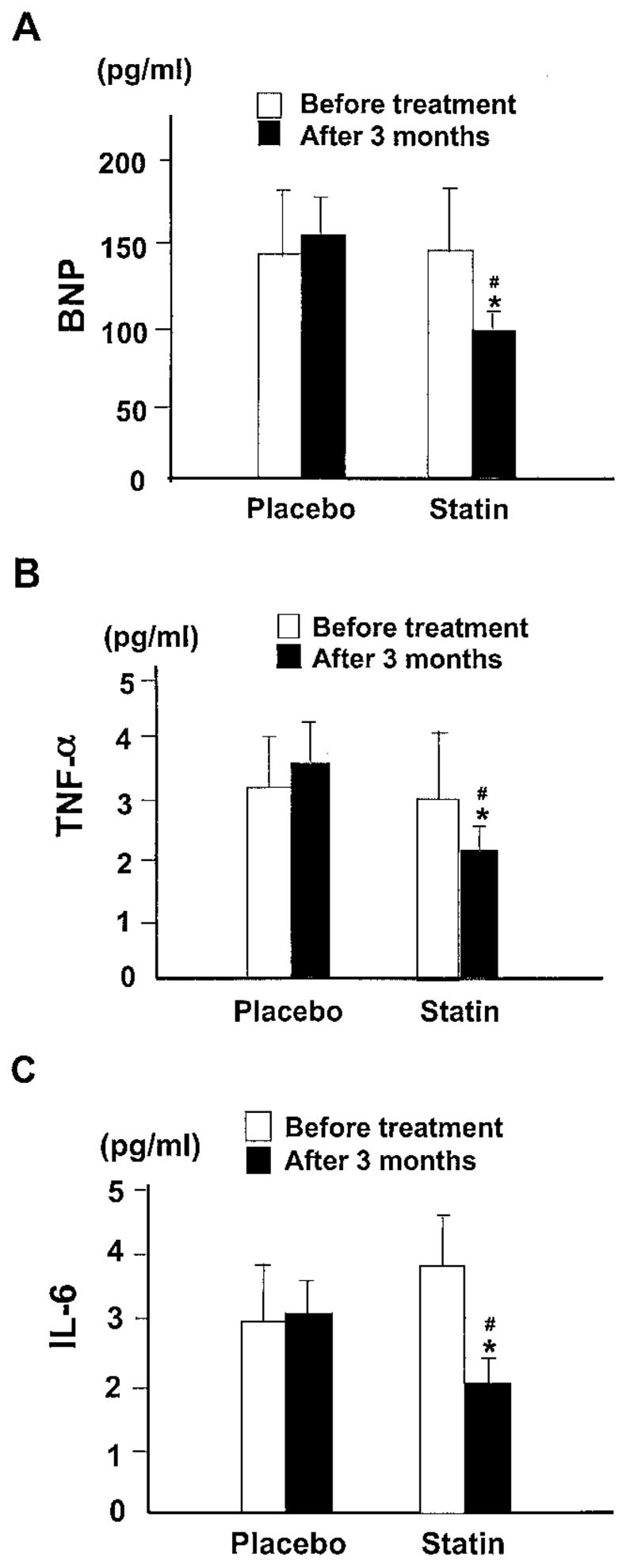
Comparison of the plasma levels of BNP (A), TNF-α (B), and IL-6 (C) before and after 14 weeks of treatment between the placebo and statin groups. *P<0.001 vs placebo group. #P<0.05 vs before treatment in the statin group.
Endothelial function was assessed using high-resolution B-mode ultrasonography of the brachial artery before and after cuff-induced transient ischemia of the forearm. After treatment with simvastatin, flow-mediated brachial artery vasodilation improved from 8±2% to 13±2% (P<0.01). No significant changes were observed in the placebo group (Figure 4). Multivariate analysis of changes in LDL cholesterol levels showed that serum cholesterol was not a significant predictor of statin-induced improvement in endothelium-dependent vasodilation in patients with CHF.
Figure 4.

Comparison of the flow-dependent dilatation of brachial artery before and after 14 weeks of treatment between the placebo and statin groups. *P<0.01 vs placebo group. #P<0.01 vs before treatment in the statin group.
There were positive correlations between changes in ejection fraction and percent improvement in IL-6 (γ=0.69, P<0.01), TNF-α (γ=0.61, P<0.05), and BNP (γ=0.72, P<0.01) concentrations before and after 14 weeks of statin therapy. These findings suggest that statin may improve cardiac function, in part, by modulating the inflammatory state.
Discussion
The present study demonstrates that short-term statin therapy has beneficial effects in patients with nonischemic, dilated cardiomyopathy. Statin decreased markers of inflammation and improved neurohormonal imbalance and cardiac function. These effects correlated with improved symptoms and suggest that statin may be therapeutically useful in patients with CHF with whom the medication may not otherwise be indicated.
Although we did not address the mechanism by which statins improve left ventricular function in this study, some of the beneficial effects could be explained, in part, by the reduction in systemic inflammation. For example, inflammatory cytokines, which are produced by activated macrophages, vascular wall cells, and cardiac myocytes, are elevated in CHF.14,15 These cytokines, especially TNF-α and IL-6, exert negative inotropic effects and induce apoptosis in cardiac myocytes.16 Although the anti-inflammatory effects of statins may contribute to some of their beneficial effects in CHF, we cannot exclude the possibility that plasma levels of TNF-α and IL-6 were reduced in response to improvement in cardiac function by statins.
Statins also inhibit the synthesis of isoprenoids, which are required for the posttranslational modification of important signaling molecules such as Rho, Rac, and Ras.17,18 In particular, inhibition of Rho geranylgeranylation leads to increased endothelial NO production19 and decreased endothelin-1 expression,20 factors that are favorable to improving endothelial function. Furthermore, inhibition of Rac by statins decreases vascular and myocardial oxidative stress by inhibiting Rac-induced NAD(P)H oxidase activity.21,22 Indeed, worsening heart failure is characterized by increased formation of oxygen-derived free radicals,23 which can scavenge and inactivate NO.
Finally, statins may modulate the remodeling process of heart failure through effects on matrix metalloproteinases (MMPs). For example, myocardial MMPs are increased during the development of dilated cardiomyopathy.24,25 Targeted deletion of the MMP-9 gene in mice reduces left ventricular dilatation after experimental myocardial infarction, suggesting that MMP-9 plays an important role in the progression to heart failure.26 Statins suppress growth of macrophages expressing MMP-9, MMP-3, and MMP-1.27,28 Thus, it is possible that statins improve cardiac function in patients with CHF by exerting inhibitory effects on MMPs.
Although we did not address the effects of statins on patient outcomes, our findings that short-term statin therapy improves cardiac function should pave the way for additional large-scale clinical trials to evaluate the long-term clinical benefits of statin therapy in normocholesterolemic patients with nonischemic, dilated cardiomyopathy.
Acknowledgments
This work was supported by the National Institutes of Health grant HL-52233, the Japan Research Foundation grant for Clinical Pharmacology and Vascular Function, and the Health Sciences Research grant from the Japan Ministry of Health and Welfare. Dr Liao is an Established Investigator of the American Heart Association.
References
- 1.Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation. 2000;102:14–23. doi: 10.1161/01.cir.102.suppl_4.iv-14. [DOI] [PubMed] [Google Scholar]
- 2.Torre-Amione G. The syndrome of heart failure: emerging concepts in the understanding of its pathogenesis and treatment. Curr Opin Cardiol. 1999;14:193–195. doi: 10.1097/00001573-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation. 1988;77:721–730. doi: 10.1161/01.cir.77.4.721. [DOI] [PubMed] [Google Scholar]
- 4.Drexler H, Hornig B. Importance of endothelial function in chronic heart failure. J Cardiovasc Pharmacol. 1996;27:S9–S12. doi: 10.1097/00005344-199600002-00003. [DOI] [PubMed] [Google Scholar]
- 5.Treasure CB, Klein JL, Weintraub WS, et al. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease. N Engl J Med. 1995;332:481–487. doi: 10.1056/NEJM199502233320801. [DOI] [PubMed] [Google Scholar]
- 6.Ferro D, Parrotto S, Basili S, et al. Simvastatin inhibits the monocyte expression of proinflammatory cytokines in patients with hypercholesterolemia. J Am Coll Cardiol. 2000;36:427–431. doi: 10.1016/s0735-1097(00)00771-3. [DOI] [PubMed] [Google Scholar]
- 7.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 8.Anderson TJ, Meredith IT, Yeung AC, et al. The effect of cholesterol-lowering and antioxidant therapy on endothelium-dependent coronary vasomotion. N Engl J Med. 1995;332:488–493. doi: 10.1056/NEJM199502233320802. [DOI] [PubMed] [Google Scholar]
- 9.Albert MA, Danielson E, Rifai N, et al. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE). A randomized trial and cohort study. JAMA. 2001;286:64–70. doi: 10.1001/jama.286.1.64. [DOI] [PubMed] [Google Scholar]
- 10.Braith RW, Wood CE, Limacher MC, et al. Abnormal neuroendocrine responses during exercise in heart transplant recipients. Circulation. 1992;86:1453–1463. doi: 10.1161/01.cir.86.5.1453. [DOI] [PubMed] [Google Scholar]
- 11.Goldstein M, Vincent JL, Kahn RJ. Evaluation of cardiac function by echo-Doppler studies in critically ill patients. Intensive Care Med. 1988;14:406–410. doi: 10.1007/BF00262897. [DOI] [PubMed] [Google Scholar]
- 12.Celermajer DS, Sorensen KE, Gooch VM, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340:1111–1115. doi: 10.1016/0140-6736(92)93147-f. [DOI] [PubMed] [Google Scholar]
- 13.Anderson TJ, Uehata A, Gerhard MD, et al. Close relation of endothelial function in the human coronary and peripheral circulations. J Am Coll Cardiol. 1995;26:1235–1241. doi: 10.1016/0735-1097(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 14.Packer M. Is tumor necrosis factor an important neurohormonal mechanism in chronic heart failure? Circulation. 1995;92:1379–1382. doi: 10.1161/01.cir.92.6.1379. [DOI] [PubMed] [Google Scholar]
- 15.Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 16.Pinsky DJ, Cai B, Yang X, et al. The lethal effects of cytokine-induced nitric oxide on cardiac myocytes are blocked by nitric oxide synthase antagonism or transforming growth factor beta. J Clin Invest. 1995;95:677–685. doi: 10.1172/JCI117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 18.Casey PJ. Protein lipidation in cell signaling. Science. 1995;268:221–225. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 19.Laufs U, La Fata V, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–2719. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wassmann S, Laufs U, Baumer AT, et al. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol. 2001;59:646–654. doi: 10.1124/mol.59.3.646. [DOI] [PubMed] [Google Scholar]
- 22.Takemoto M, Node K, Nakagami H, et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest. 2001;108:1429–1437. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiedermann CJ, Beimpold H, Herold M, et al. Increased levels of serum neopterin and decreased production of neutrophil superoxide anions in chronic heart failure with elevated levels of tumor necrosis factor-alpha. J Am Coll Cardiol. 1993;7:1897–1901. doi: 10.1016/0735-1097(93)90776-w. [DOI] [PubMed] [Google Scholar]
- 24.Thomas CV, Coker ML, Zellner JL, et al. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998;97:1708–1715. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 25.Spinale FG, Coker ML, Heung LJ, et al. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation. 2000;102:1944–1949. doi: 10.1161/01.cir.102.16.1944. [DOI] [PubMed] [Google Scholar]
- 26.Ducharme A, Frantz S, Aikawa M, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;11:1671–1678. doi: 10.1161/01.atv.18.11.1671. [DOI] [PubMed] [Google Scholar]
- 28.Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–283. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]