

Abstract
A central aim of cancer research has been to identify the mutated genes that are causally implicated in oncogenesis (‘cancer genes’). After two decades of searching, how many have been identified and how do they compare to the complete gene set that has been revealed by the human genome sequence? We have conducted a ‘census’ of cancer genes that indicates that mutations in more than 1% of genes contribute to human cancer. The census illustrates striking features in the types of sequence alteration, cancer classes in which oncogenic mutations have been identified and protein domains that are encoded by cancer genes.
Numerous alterations in DNA sequence underlie the development of every neoplasm. These sequence variants can be transmitted through the germline and result in susceptibility to cancer, or can arise by somatic mutation. A central aim of cancer research has been to identify the mutated genes that are causally implicated in oncogenesis (referred to here as ‘cancer genes’). Since the advent of recombinant-DNA technology more than two decades ago, there has been considerable success in this enterprise. Following the first report of a somatic mutation in a human cancer gene — the conversion of amino-acid 12 of HRAS from glycine to valine in the human bladder carcinoma cell line T24/EJ1 — a substantial number of human cancer genes have been identified, and their biological properties have been assiduously investigated. The proteins that are encoded by cancer genes normally regulate cell proliferation, cell differentiation and cell death. Mutations underlying oncogenesis also occur in genes that mediate DNA-repair processes3-5. These result in an increased rate of somatic mutation, which, in turn, might increase the likelihood of a growth-control gene being mutated. We have compiled a list of cancer genes from the published literature. The census illustrates several striking features with respect to the types of sequence alteration, cancer classes in which oncogenic mutations have been identified and protein domains that are encoded by cancer genes.
Criteria for inclusion in the cancer-gene census
We have exclusively listed genes in which mutations that are causally implicated in oncogenesis have been reported (see supplementary information S1 (table)) . Most cancer genes have been identified and initially reported on the basis of genetic evidence (that is, the presence of somatic or germline mutations) and without biological information supporting the oncogenic effects of the mutations. The underlying rationale for interpreting a mutated gene as causal in cancer development is that the number and pattern of mutations in the gene are highly unlikely to be attributable to chance. So, in the absence of alternative plausible explanations, the mutations are likely to have been selected because they confer a growth advantage on the cell population from which the cancer has developed. There often exists additional biological information supporting the role of a mutated gene in oncogenesis. However, if the genetic data is convincing (see below), we have regarded it as sufficient on its own for inclusion of the gene in the census (BOX 1).
Box 1. Genes that have been included in the cancer-gene census.
Genes with the following types of mutations have been included:
Base substitutions that lead to missense amino-acid changes, nonsense changes and alterations in the well-conserved positions of splice sites.
Insertions or deletions in coding sequences or splice sites that might cause in-frame or frameshifting alterations in the protein.
Rearrangements because of chromosomal translocations that lead to chimeric transcripts or to deregulation of genes through apposition to novel promoter or enhancer regions.
Copy-number increases and decreases.
Genes have been included in the census if there exist at least two independent reports showing mutations in primary patient material. In considering somatic mutations for inclusion in the census, we included only genes for which there was evidence of the somatic origin of at least a subset of mutations, based on analysis of normal tissue from the same individuals. In practice, this is most important for the interpretation of putative somatic base substitutions and small insertions/deletions, because these are also common as germline sequence variants. By contrast, translocations and copy-number changes are relatively uncommon as germline variants. Therefore, we have included some cancer genes that are altered by translocation or copy-number change for which definitive evidence of the somatic origin of their mutations is unavailable.
Summary.
We have conducted a census from the literature of genes that are mutated and causally implicated in cancer development (‘cancer genes’).
So far, 291 cancer genes have been reported, more than 1% of all the genes in the human genome.
90% of cancer genes show somatic mutations in cancer, 20% show germline mutations and 10% show both.
The most common mutation class among the known cancer genes is a chromosomal translocation that creates a chimeric gene or apposes a gene to the regulatory elements of another gene.
Many more cancer genes have been found in leukaemias, lymphomas and sarcomas than in other types of cancer, despite the fact that they represent only 10% of human cancer. These genes are usually altered by chromosomal translocation.
The most common domain that is encoded by cancer genes is the protein kinase. Several domains that are involved in DNA binding and transcriptional regulation are common in proteins that are encoded by cancer genes.
The census does not include genes that are only expressed at altered levels in cancer cells, without any mutation in DNA. Although alterations in expression might be part of the reconfiguration of cell biology following the acquisition of a mutation in a cancer gene, they are usually not the primary change. Indeed, in some cases they might be consequences of the neoplastic phenotype rather than determinants of it.
The list also does not include genes in which changes in methylation of CpG dinucleotides within promoter regions have been the only reported abnormalities. Many such alterations are clearly associated with altered transcriptional regulation and, in some cases (for example, in the promoters of CDKN2A or MLH1), are likely to cause neoplastic transformation6,7. Methylation of promoter regions is, however, relatively plastic. It can change in association with age or in vitro culture, is widespread in some cancer genomes and can occur in CPG ISLANDS without any known consequences for transcriptional regulation8. It is therefore problematic to identify genes in which alterations in promoter methyation, without mutation, cause neoplastic change, so this form of genetic modification has not been included in our database.
It is likely that some cancer genes have inadvertently been missed, and opinions might differ about some genes that have been excluded and included. We are keen to be informed of these omissions and controversial cases in order to evaluate their status and will regularly update this list on the Wellcome Trust Sanger Institute web site (see online links box).
Problems encountered in compiling the census
We have attempted to be comprehensive in compilation of the cancer-gene list, but have also been conservative in our criteria for inclusion. Several genes that were considered were ultimately excluded. Below, we have reviewed some common problems that we encountered in determining whether a gene should be included as a cancer gene.
Genes with few reported mutations
All classes of mutation occur as more or less random processes in both normal and neoplastic cells. Indeed, it is likely that most somatic mutations do not confer a clonal growth advantage. So, cancer genomes carry several ‘PASSENGER’ OR ‘BYSTANDER’ MUTATIONS, in addition to the mutations that cause the neoplastic phenotype. Unfortunately, the prevalence of passenger mutations in the genomes of most cancer types is not known. Therefore, if somatic mutations in a putative cancer gene are found in a very small proportion of tumours, it is difficult to exclude the possibility that they represent chance clustering of passenger alterations. Consequently, we have excluded genes in which fewer than five unambiguous somatic mutations have been reported in primary neoplasms.
Cancers with large numbers of mutations
A particular problem arises in cancer cells that have genomes with a high prevalence of somatic mutations. For example, cancers with mismatch-repair (MMR) gene defects carry tens of thousands of small insertions and deletions in short tandem repeats (known as microsatellites)9. Most of these occur in intronic or intergenic DNA and are, therefore, almost certainly passenger mutations. However, several hundred are in coding sequences and will often result in translational frameshifts, with the consequence of premature protein termination or nonsense-mediated RNA decay. It is highly likely that a small percentage of these are causally involved in oncogenesis. It is equally probable, however, that most are not. In these circumstances, distinguishing mutated cancer genes from genes with clusters of passenger mutations is particularly problematic.
Several genes with small insertions and deletions of coding microsatellites/mononucleotide repeats that lead to protein truncation in MMR-deficient tumours have been proposed as cancer genes9. Attempts have been made to distinguish mutations that confer a clonal growth advantage from bystander mutations in MMR-deficient cells10. Transforming growth factor-β type II receptor (TGF-βRII), which mediates the effects of the growth-suppressing ligand TGF-β, is one example of a potential cancer gene that was discovered in MMR-deficient cells. Mutations in TGF-βRII are believed to be more than simple bystander mutations because of the biological function of its gene product, the high frequency of biallelic mutations in MMR-deficient cells and the presence of some biologically relevant somatic mutations in MMR-proficient cancers11. However, the distinction is still unclear for most genes that have microsatellite instability in their coding sequence. Therefore, genes in which somatic mutations occur exclusively or predominantly in MMR-deficient cells have been excluded from the cancer-gene census.
Putative cancer genes with particularly high mutation rates in cancer cells
A similar problem arises when there is an increased mutation rate restricted to a small region of the genome. For example, the fragile histidine triad gene (FHIT) and WW-domain-containing oxidoreductase (WWOX) genes each straddle a fragile site (FRA3B and FRA16D, respectively12,13). Fragile sites are genomic regions that are associated with a high frequency of chromosome breakage, and many were originally identified through chemical stress of normal lymphocytes14. The intrinsic fragility of these regions is frequently expressed in cancer cells as rearrangements and deletions, mutational patterns similar to those found in certain recessive cancer genes (for example, CDKN2A and PTEN15,16). Therefore, based simply on mutation clustering and pattern, it is not clear whether FHIT and WWOX are recessive cancer genes (and therefore causally implicated in oncogenesis) or fragile sites that are frequently rearranged in cancer cells because of defects in the DNA-repair process, or both. Most well-established recessive cancer genes also contain somatic base substitutions and small insertions/deletions that cause protein truncation. These are uncommon in FHIT and WWOX. So, although there are additional biological data that might support a role for genes such as FHIT and WWOX in oncogenesis, genes overlying fragile sites are not included in this census of cancer genes.
Mutations that encompass many genes
Most classes of mutations (such as base substitutions) only affect a single gene, or, at most, two genes (such as reciprocal chromosomal translocations). However, copy-number changes, such as gene amplification, can affect several megabases of DNA and encompass many genes. Therefore, solely on the basis of genetic evidence, it is not always clear which gene is the crucial target of the amplification, and it is conceivable that, in some cases, there is more than one target. Additional evidence that might be invoked in assessing which gene(s) on an amplicon mediates oncogenesis includes increased expression levels in cancer cells and functional effects. Chromosome defects, such as trisomy, that involve even larger regions of the genome pose even greater problems, as they might alter the copy number of thousands of genes. We have therefore only included in the census amplified genes for which there is reasonable consensus that the cancer-causing gene has been identified, although even these might be controversial. Many amplified genes that have not been included in the cancer-gene census clearly exist and might be included in the future.
Low-penetrance cancer-susceptibility genes
There is usually little ambiguity in the identification of mutated genes that are responsible for high-penetrance (high-risk) cancer-susceptibility syndromes or of mutated genes that are associated with characteristic non-neoplastic manifestations in addition to cancer predisposition. However, germline variants of many genes have recently been proposed as low-penetrance (low-risk) cancer-susceptibility alleles without additional non-neoplastic features. In some cases — for example, for the variants APC*I1307K17 and CHK2*1100delC18 — the evidence for this effect is strong. For most genes, however, the evidence is statistically weak and often conflicting. Therefore, for purposes of clarity, we have not included this type of cancer-susceptibility gene in the current census.
Accessory information in the cancer-gene census
To facilitate further analyses and to illustrate some striking patterns, we have appended additional information to the list of cancer genes — see supplementary information S1 (table). Fields in the census include the gene name, symbol, LocusLink number, protein accession number and chromosomal location. Further fields indicate whether the gene is somatically mutated in cancer or mutated in the germline predisposing to cancer (or both). Cancer types and syndrome names that are associated with somatic and germline mutations in each gene have been included. These lists are not exhaustive and are intended to present a general picture of the cancer types that are associated with individual mutated cancer genes. So, they include the main classes of neoplasm that have been reported, but omit tumour types that infrequently show mutations.
We have also included a field in which neoplasms are classified into four groups: leukaemias/lymphomas, tumours of mesenchymal origin (sarcomas and benign mesenchymal tumours), epithelial tumours and others. This classification is arbitrary, but it facilitates certain types of analysis and detection of some notable patterns. Cancer genes have also been classified according to whether, at the cellular level, mutations are dominant (that is, a single mutated allele is sufficient to contribute to oncogenesis) or recessive (that is, both alleles need to be mutated — sometimes termed ‘tumour-suppressor genes’). We recognize that this classification is sometimes presumptive and occasionally an oversimplification. Finally, there is a field indicating the types of mutation that are observed in each gene. For recessive cancer genes, two mutations will usually be present in a single tumour sample and these often involve two of the categories of mutation listed. The proportions of cancer genes that are mutated in somatic or germline cells (FIG. 1), that are mutated by translocation or by other types of mutation (FIG. 2), that act in a dominant or recessive manner (FIG. 3), and that are mutated in the various classes of neoplasm (FIG. 4) are shown.
Figure 1. Mutation types in human cancer.
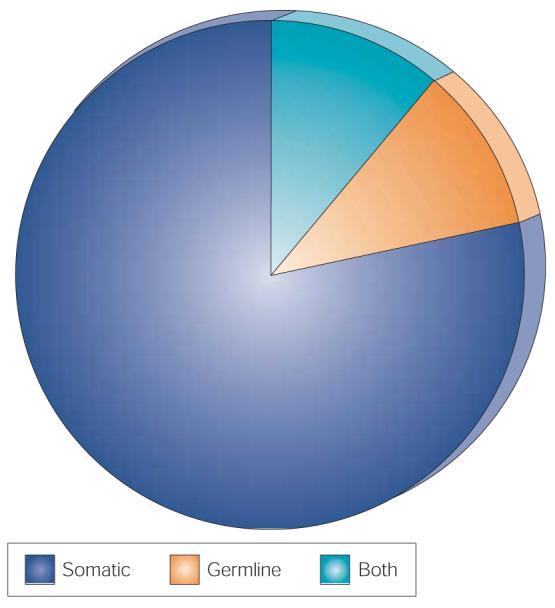
Cancer genes showing somatic mutations, germline mutations or both somatic and germline mutations in human cancers.
Figure 2. Proportion of translocated genes in human cancer.

Somatically mutated cancer genes with translocations, with mutations other than translocations or with both.
Figure 3. Dominant and recessive mutations in human cancer.
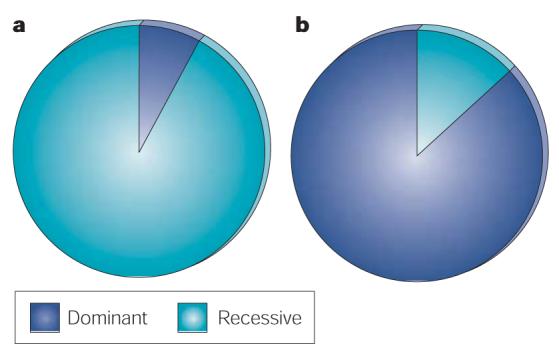
a | Cancer genes with germline mutations acting in a dominant or recessive manner. b | Cancer genes with somatic mutations acting in a dominant or recessive manner.
Figure 4. Proportion of genes associated with different tumour types.

a | Cancer genes with germline mutations by tumour class (for definitions see text and supplementary tables, the category ‘numerous’ refers to cancer genes with combinations of two or more classes). b | Cancer genes with somatic mutations by tumour class.
General features of cancer genes
Mutations in at least 291 human genes are causally implicated in oncogenesis (see supplementary information S1 (table)). All encode proteins. No mutated RNA genes have yet been shown to be involved in oncogenesis. Assuming that there are approximately 25,000 coding genes in the human genome — as indicated on the Ensembl Human Genome Browser web page (see online links box) — it seems that mutations in more than 1% genes are so far known to be involved in cancer pathogenesis.
Most were identified by positional cloning without any previous hypothesis of biological function. By this strategy, the cancer gene is first localized to a small part of the genome and, subsequently, genes within the delimited interval are screened for mutations. The primary positional clues have been diverse and include chromosomal rearrangements that are visible in metaphase spreads of cancer cells, DNA copy-number changes in cancer cells, (which are detected by various molecular approaches) and, for cancer susceptibility genes, genetic-linkage analyses of families with several cases of cancer.
A small number of cancer genes have been detected through biological assays. Most notable among these has been the NIH-3T3 transformation assay, in which total human DNA is introduced into a line of mouse fibroblasts, and cells that incorporate certain classes of mutated human cancer genes acquire the transformed phenotype19,20. Mutations in the remainder of human cancer genes have been identified through analysis of plausible candidates based on known biological features of cancer cells. However, these constitute a small minority of known cancer genes.
Approximately 90% of cancer genes show somatic mutations and 20% show germline mutations. Both somatic and germline mutations have been reported in 10% of all cancer genes (FIG. 1). In general, the spectrum of neoplasms that are associated with germline mutations in a particular gene is similar to that reported with somatic mutations. There are, however, several notable exceptions to this rule. For example, somatic mutations in TP53 are found in more than half of colorectal cancers, yet germline mutations do not apparently cause a predisposition to colorectal cancer21,22. Similarly, germline mutations in STK11 are associated with a predisposition to hamartomas of the gastrointestinal tract and to colorectal, pancreatic and ovarian neoplasms (the Peutz–Jegher syndrome)23. However, somatic mutations of STK11 have only been found in lung adenocarcinomas, which are not usually considered to be components of the Peutz–Jegher syndrome24. Several genes with germline mutations that cause cancer predisposition show very few, if any, somatic mutations in sporadic cancers of the same type, such as BRCA1 and BRCA2 in breast cancer25,26. The reasons for differences between the tumour spectrum that is associated with somatic mutations and the spectrum that is associated with germline mutations are generally unknown.
The most common class of somatic mutation that is registered in the cancer-gene census involves chromosomal translocations that result in a chimeric transcript or apposition of one gene to the regulatory regions of another gene — usually immunoglobulin or T-cell-receptor genes. This mutation type is common in leukaemias, lymphomas and mesenchymal tumours. However, several examples have now been reported among epithelial neoplasms, including thyroid papillary carcinoma (RET and NTRK1, both with several partners), thyroid follicular carcinoma (PAX8 and PPARγ), renal papillary carcinoma (PRCC and TFE3) and breast secretory carcinomas (ETV6 and NTR3)27-30. Because two genes are structurally rearranged in each chromosomal translocation, the number of translocated cancer genes, compared with other types of mutated cancer gene, is exaggerated in the census. Moreover, certain genes, such as MLL (mixed-lineage leukaemia), are highly promiscuous and form chimeric transcripts with a large number of partners31. As a consequence of this ability to form chimeric genes with more than one partner, ‘networks’ of translocation partners can be discerned. For example, MLL can form a chimeric cancer gene by chromosomal translocation with CREB-binding protein (CREBBP; also known as CBP). In addition to MLL, CBP can also form a chimeric cancer gene with RUNXBP2 (also known as ZNF220). RUNXBP2, in turn, can form a chimeric cancer gene with EP300, and this gene can from a chimeric cancer gene with MLL.
For some genes, several different types of mutations have been associated with cancer. Notably, many of the different classes of mutation are found in most of the recessive cancer genes, because the result is usually inactivation of the encoded protein, and there are many mutation types that can achieve this end. However, for dominantly acting cancer genes, in which the encoded protein is usually activated, the patterns of mutation are much more restricted, so only one class of mutation is usually found.
Indeed, if more than one class of mutation occurs in a dominantly acting cancer gene, each type of mutation might be associated with a particular type of cancer. For example, two classes of mutation result in constitutive activation of the RET kinase — chromosomal translocations and base substitutions that lead to missense amino-acid changes. Rearrangements of RET are found in papillary thyroid carcinoma, whereas base substitutions are found in medullary thyroid cancer32. Occasionally, a single allele of a cancer gene might require more than one mutation for full biological effect. For example, in-frame deletions within the extracellular domain of EGFR are common in malignant gliomas33. Usually, the rearranged EGFR allele is also amplified. Similar increases in copy number occur on alleles mutated by base substitutions, for example MET in renal papillary carcinoma34.
More than 70% of cancer genes with somatic mutations in the census are associated with leukaemias, lymphomas and mesenchymal tumours, even though these account for less than 10% of human cancer incidence. Why have so many more genes been associated with these relatively rare tumour types than with the more common epithelial cancers? Some of this imbalance is attributable to the number of genes that are subject to chromosomal translocations in leukaemias, lymphomas and mesenchymal tumours, with the attendant double counting of genes (see above). Another explanation is that it might have been easier, in the past, to identify cancer genes by studying leukaemias, lymphomas and mesenchymal neoplasms than by studying epithelial cancers. If this interpretation is correct, it indicates that many more cancer genes remain to be identified in association with common epithelial cancers. Alternatively, there might be fundamental biological differences between the group of leukaemias, lymphomas and mesenchymal tumours and the group of common epithelial neoplasms, such that the repertoire of mutated genes that is necessary to generate common epithelial neoplasms is more restricted.
90% of somatic mutations in cancer genes are dominant at the cellular level. Again, this is predominantly determined by the frequency of chromosomal translocations in leukaemias, lymphomas and mesenchymal tumours, almost all of which act in this way. If cancer genes that contribute to oncogenesis by chromosomal translocation are excluded, equal numbers of somatically mutated cancer genes are dominant and recessive at the cellular level. By contrast, 90% of germline mutations that result in cancer predisposition are recessive at the cellular level, presumably because many cancer-causing mutations that might act in a dominant fashion would cause embryonic lethality.
There is at least one example of a cancer gene that is able to act through both dominant and recessive mechanisms. PRKAR1A — a regulatory subunit of protein kinase A — is a translocation partner with RET. This translocation activates the RET kinase in papillary carcinoma of the thyroid35. By contrast, germline mutations that inactivate PRKAR1A are associated with predisposition to Carney complex, a rare syndrome that is characterized by endocrine tumours and MYXOMA of the heart36.
Biological features of cancer proteins
There are now over 2,600 classes of protein domain reported on Pfam (see online links box) — a manually curated database of protein-domain families — that are encoded by genes in the human genome37. Of these, we found 221 in proteins that are encoded by cancer genes. We compared Pfam domains that are encoded by cancer genes to Pfam domains that are encoded by the complete human gene set (see supplementary information S2 (table)). The analysis has also been applied to subgroups of the cancer-gene list (see supplementary information S3,S4 (tables)). Compared with their representation in proteins that are encoded by the complete human-gene set, at least 11 Pfam domains are clearly over-represented among proteins that are encoded by cancer genes (see supplementary information S2 (table)). These include the protein-kinase, bromodomain, helix–loop–helix (HLH), homeobox, carboxy-terminal DNA-binding (ETS), PAX, prolyl hydroxylase, MMR, HATPase_c, MYC amino-terminal and AF-4 domains.
The most commonly represented Pfam domain that is encoded by cancer genes is the protein kinase, and this is also the domain for which there is strongest evidence of over-representation — there are 27 cancer genes in the census that encode protein-kinase domains, compared with the 6.3 that would be expected in a random selection of the same number of genes from the complete set of human genes (see supplementary information S2 (table)). Some over-representation of protein-kinase domains might be attributable to ascertainment bias. Protein kinases have long been implicated in oncogenesis and, consequently, mutations in some have been identified because they were examined as plausible candidates. However, mutations in most protein kinases in the cancer-gene census were identified through positional-cloning approaches, so ascertainment bias is unlikely to account completely for their over-representation.
Most of the protein kinases in the cancer-gene census show somatic mutations in cancer, but there are several in which germline mutations cause predisposition to neoplasia, including MET, KIT, STK11, and CDK4. Most cancer genes that encode protein kinases contain activating mutations and are dominant at the cellular level. However, a minority act in a recessive manner at the cellular level. These include ATM, STK11, and BMPR1A, which are all inactivated by mutations38,23,39. Mutated protein kinases are particularly strongly over-represented among epithelial neoplasms, but are also found in leukaemias, lymphomas and mesenchymal tumours. Dominantly-acting mutated protein kinases are activated by diverse classes of mutations, including gene amplification, base substitution, in-frame large insertions and deletions (for example, FLT3 and EGFR, respectively)33,40, in-frame small deletions (for example, KIT, PDGFRA)41,42 and chromosomal translocation. Tyrosine kinases and serine/threonine kinases are both represented in the cancer-gene census. However, tyrosine kinases are over-represented compared with serine/threonine kinases, accounting for approximately one quarter of all the known protein kinases and two-thirds of the protein kinases that are encoded by cancer genes. Interestingly, phosphatases are not prominent in the cancer-gene census — one tyrosine phosphatase is listed, approximately the expected number in a random selection of genes from the complete set of human genes.
After protein kinases, the most frequently over-represented Pfam domains are those that broadly constitute components of proteins that are implicated in transcriptional regulation. These include HLH, ETS, PAX, homeobox, MYC N-terminal, bromodomain, AF-4 and PHD domains. Many of these (for example, PAX, ETS, AF-4, HLH, bromodomain and MYC N-terminal) are over-represented tenfold or more in the cancer-gene census, compared with the numbers expected from a random selection of human proteins. In contrast to the protein-kinase domain, most domains that are involved in transcriptional regulation are encoded by cancer genes activated by chromosomal translocations in leukaemias, lymphomas and mesenchymal tumours.
The final group of domains that are clearly over-represented among cancer genes is associated with DNA maintenance and repair (MMR and HATPase_c domains). Mutated cancer genes encoding these domains generally act in a recessive manner at the cellular level, are inactivated during oncogenesis (resulting in increased somatic mutation rates) and often have germline mutations that result in cancer predisposition. Indeed, a substantial proportion of germline-mutated genes that cause cancer predisposition are involved in DNA maintenance and repair.
Other Pfam domains that are frequently encoded by cancer genes are not necessarily over-represented in the cancer-gene census. For example, ten cancer genes encode C2H2 zinc-finger domains (which are implicated in DNA binding and transcriptional regulation). However, the C2H2 zinc finger is a common motif and this is the number that would be expected based on a random selection of human genes. Certain Pfam domains are, however, under-represented among cancer genes. For example, only one cancer gene encodes a rhodopsin-like seven-transmembrane domain, compared with nine expected (see supplementary information S2 (table)). Rhodopsin-like seven-transmembrane domains form a large class of G-protein-coupled receptors (GPCRs) that respond to a wide variety of signals. Their under-representation among cancer genes is perhaps surprising, given the over-representation of protein kinases, as both groups of proteins are involved in signal transduction. However, the results indicate that the normal metabolic connections of many GPCRs do not substantially influence the processes of cell proliferation, differentiation and death that underlie neoplastic change.
This census provides a detailed view of cancer-associated genes, the mutations that contribute to tumorigenesis and the functional consequences of the resulting structural abnormalities. Some clear patterns and questions emerge. Even with our relatively conservative inclusion criteria, we find more than 1% of genes in the human genome are involved in oncogenesis. The total number of human cancer genes remains a matter for speculation. For most individual adult epithelial cancers, it is not possible at present to identify the four to seven somatically mutated cancer genes that are usually proposed to be necessary (as a minimum) for cancer development. There also seem to be more cancer genes on the way to being identified by conventional strategies. For example, there are several recurrent copy-number abnormalities found in human cancer for which the target gene has yet to be definitively identified, and there could be many genes with germline sequence variants that confer a small additional risk of cancer (low-penetrance cancer-susceptibility genes). Moreover, positional-cloning strategies (in the past, the most influential approaches to cancer-gene identification) might have completely missed many mutated cancer genes simply because they do not yield informative positional cues. Finally, the full role of promoter methylation in cancer and the number of genes that contribute to oncogenesis when modified in this way is yet to be clarified. So, it is plausible (although unproven), that many more cancer genes remain to be identified. The finished human genome sequence now offers new opportunities for identifying cancer genes. It will be interesting to observe if the current patterns persist, or whether they predominantly reflect the technical opportunities and constraints that have prevailed in the past twenty years of cancer-gene identification.
Glossary
- CPG ISLANDS
GC-rich areas of the genome, usually of the order of a kilobase in size, often in and around the 5′ regions of genes, which retain an unusually high number of CpG dinucleotides.
- PASSENGER OR BYSTANDER MUTATIONS
Somatic mutations that are found in cancer cells that are not involved in generating the neoplastic phenotype.
- MYXOMA
A rare type of tumour that is usually composed of sparse mesenchymal cells interspersed amid large amounts of intercellular material.
Online Links
DATABASES
The following terms in this article are linked online to:
Cancer.gov: http://cancer.gov/
breast cancer | colorectal cancer | lung cancer
LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/
APC | ATM | BMPR1A | BRCA1 | BRCA2 | CDK4 | CDKN2A | CHK2 | CREBBP | EGFR | EP300 | ETV6 | FHIT | FLT3 | HRAS | KIT | MET | MLH1 | MLL | NTR3 | NTRK1 | PAX8 | PDGFRA | PPARγ | PRCC | PRKAR1A | PTEN | RET | RUNXBP2 | STK11 | TFE3 | TGF-β | TGF-βRII | TP53 | WWOX
OMIM: http://www.ncbi.nlm.nih.gov/omim/
Carney complex | Peutz–Jegher syndrome
FURTHER INFORMATION
Cancer Genome Project: http://www.sanger.ac.uk/CGP/
Ensembl Human Genome Browser web site: http://www.ensembl.org/Homo_sapiens/
Pfam home page: http://www.sanger.ac.uk/Software/Pfam/
Wellcome Trust Sanger Institute web site: http://www.sanger.ac.uk/
Access to this interactive links box is free online.
Footnotes
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature. 1982;300:149–152. doi: 10.1038/300149a0. [DOI] [PubMed] [Google Scholar]
- 2.Tabin CJ, et al. Mechanism of activation of a human oncogene. Nature. 1982;300:143–149. doi: 10.1038/300143a0. [References 1 and 2 are seminal papers that defined the existence of oncogene sequences in cancer-cell genomes and that first identified a mutation involved in human cancer.] [DOI] [PubMed] [Google Scholar]
- 3.Parsons R, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75:1227–1236. doi: 10.1016/0092-8674(93)90331-j. [DOI] [PubMed] [Google Scholar]
- 4.Fishel R, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 5.Leach FS, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 6.Merlo A, et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 7.Kane MF, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 8.Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299–305. doi: 10.1016/s1535-6108(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 9.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–2454. [PubMed] [Google Scholar]
- 10.Duval A, et al. Evolution of instability at coding and non-coding repeat sequences in human MSI-H colorectal cancers. Hum. Mol. Genet. 2001;10:513–518. doi: 10.1093/hmg/10.5.513. [DOI] [PubMed] [Google Scholar]
- 11.Grady WM, et al. Mutational inactivation of transforming growth factor β receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–324. [PubMed] [Google Scholar]
- 12.Ohta M, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996;84:587–597. doi: 10.1016/s0092-8674(00)81034-x. [DOI] [PubMed] [Google Scholar]
- 13.Bednarek AK, et al. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000;60:2140–2145. [PubMed] [Google Scholar]
- 14.Sutherland GR, Baker E, Richards RI. Fragile sites still breaking. Trends Genet. 1998;14:501–506. doi: 10.1016/s0168-9525(98)01628-x. [DOI] [PubMed] [Google Scholar]
- 15.Kamb A, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 16.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1946. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 17.Laken SJ, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nature Genet. 1997;17:79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- 18.Meijers-Heijboer H, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nature Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 19.Shih C, Shilo BZ, Goldfarb MP, Dannenberg A, Weinberg RA. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc. Natl Acad. Sci. USA. 1979;76:5714–5718. doi: 10.1073/pnas.76.11.5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krontiris TG, Cooper GM. Transforming activity of human tumor DNAs. Proc. Natl Acad. Sci. USA. 1981;78:1181–1184. doi: 10.1073/pnas.78.2.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker SJ, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–7722. [PubMed] [Google Scholar]
- 22.Hwang S-J, Lozano G, Amos CI, Strong LC. Germline p53 mutations in a cohort with childhood sarcoma: sex differences in cancer risk. Am. J. Hum. Genet. 2003;72:975–983. doi: 10.1086/374567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hemminki A, et al. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Cespedes M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- 25.Futreal PA, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–122. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 26.Lancaster JM, et al. BRCA2 mutations in primary breast and ovarian cancers. Nature Genet. 1996;13:238–240. doi: 10.1038/ng0696-238. [DOI] [PubMed] [Google Scholar]
- 27.Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr. Pathol. 2002;13:3–16. doi: 10.1385/ep:13:1:03. [DOI] [PubMed] [Google Scholar]
- 28.Kroll TG, et al. PAX8–PPARγ1 fusion in oncogene human thyroid carcinoma. Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- 29.Sidhar SK, et al. The t(X;1)(p11. 2;q21. 2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum. Molec. Genet. 1996;5:1333–1338. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 30.Tognon C, et al. Expression of the ETV6–NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2002;2:367–376. doi: 10.1016/s1535-6108(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 31.Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 32.Eng C, Mulligan LM. Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumours, and Hirschsprung disease. Hum. Mutat. 1997;9:97–109. doi: 10.1002/(SICI)1098-1004(1997)9:2<97::AID-HUMU1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 33.Wong AJ, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl Acad. Sci. USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuang Z, et al. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nature Genet. 1998;20:66–69. doi: 10.1038/1727. [DOI] [PubMed] [Google Scholar]
- 35.Bongarzone I, et al. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RIα of cyclic AMP-dependent protein kinase A. Molec. Cell. Biol. 1993;13:358–366. doi: 10.1128/mcb.13.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirschner LS, et al. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nature Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 37.Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 39.Howe JR, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nature Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 40.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 41.Hirota S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 42.Heinrich MC, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]