

Abstract
The distal to proximal degeneration of axons, or “dying back” is a common pattern of neuropathology in many diseases of the PNS and CNS. A long-standing debate has centered on whether this pattern of neurodegeneration is due to an insult to the cell body or to the axon itself, although it is likely that mechanisms are different for specific disease entities. We have addressed this question in a model system of vincristine-induced axonal degeneration. Here, we created a novel experimental apparatus combining a microfluidic divider with a multielectrode array substrate to allow for independent monitoring of injury-induced electrical activity from dorsal root ganglion (DRG) cell bodies and axons while isolating them into their own culture microenvironments. At specified doses, exposure of the cell body to vincristine caused neither morphological neurodegeneration nor persistent hyperexcitablility. In comparison, exposure of the distal axon to the same dose of vincristine first caused a decrease in the excitability of the axon and then axonal degeneration in a dying back pattern. Additionally, exposure of axons to vincristine caused an initial period of hyperexcitability in the cell bodies, suggesting that a signal is transmitted from the distal axon to the soma during the progression of vincristine-induced axonal degeneration. These data support the proposition that vincristine has a direct neurotoxic effect on the axon.
Keywords: injury signals, axonal degeneration, vincristine, compartmented cultures
I. Introduction
Axonal degeneration is a common feature of a variety of diseases of the CNS and PNS. Both clinically and pathologically, axonal degeneration frequently progresses from distal to proximal portions of the neuron, a pattern that is described as distal axonal degeneration or “dying back” [2, 16]. This pattern of disease in PNS and spinal cord disorders is well accepted, and there are decades of literature describing experimental studies aimed at determining whether the primary insult is to the axon or the cell body. There is recent evidence that this pattern of distal axonal degeneration also occurs in neurodegenerative disorders that typically are considered primary disorders of the cell body, including Alzheimer, Parkinson, and Huntington diseases, ALS and spinal muscular atrophies, and even in prion diseases [19]. In animal models of these disorders there is clear evidence of early pathology at axon terminals [3, 4, 9]. In addition, interventions that protect neuronal cell bodies do not necessarily protect the animals from clinical disease, raising the possibility that the axon may be a primary target in the pathogenesis of some neurodegenerative disorders [7, 13].
We have been using the model of vincristine neuropathy in order to study the process of axonal degeneration. Vincristine is a chemotherapeutic agent used to treat leukemias and other types of human cancer [5]. Vincristine is also a neurotoxin that causes distal axonal degeneration and peripheral neuropathy, making it a useful and relevant agent for investigations into the process of axonal degeneration [1, 6]. We have developed an in vitro model of vincristine neuropathy that recreates the dying back pattern of degeneration [18], and in a standard compartmented culture system we demonstrated that the distal axon is more susceptible than the cell body to the neurotoxic effect of vincristine [15]. These findings led us to examine the physiological pattern that accompanies distal axonal degeneration, asking whether there is evidence for dysfunction of the cell body that leads to dying back of the distal axon. Using a novel compartmented culture system that combines a microfluidic divider with an embedded multielectrode array, we found that exposure of axons but not cell bodies to vincristine caused electrophysiologic failure that preceded axonal degeneration. In addition, toxin exposure to axons caused hyperactivity in the cell bodies implying a retrograde signaling mechanism from axon to cell body.
II. Materials and Methods
System Assembly
The system was assembled as previously described [12]. Briefly, collagen was patterned on a glass electrode substrate using a polydimethylsiloxane mold (PDMS) with channels imprinted. This mold was removed, and the microfluidic divider was assembled on top of the multielectrode array under a dissection microscope (Fig. 1). The substrate was wetted with 0.4% methylcellulose between the two compartments and the divider was brought down on top of the multielectrode array (MEA) substrate.
Figure 1.
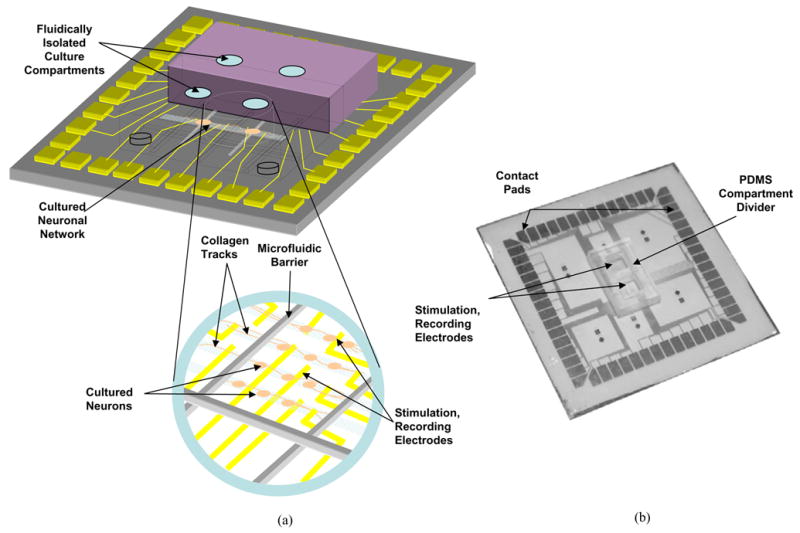
Schematic of integrated compartmented culture system, with microfluidic barriers and microelectrode array interfacing with cultured neurons. The multicompartment divider is aligned to and seated on the microelectrode array. Neurons are then plated in one or more of the compartments, after which they grow into adjacent compartments. (b) Fabricated version of the same system.
Culture Protocol
Explant cultures of E15 dorsal root ganglia (DRG) from Sprague-Dawley rats were prepared for culture in the microsystem as previously described in [11, 12]. Media was Eagle’s MEM supplemented with 1% N2 supplement and 7S NGF (Alomone Labs, Jerusalem, Israel) at 100ng/ml. For dissociated neuronal cultures, the DRGs were incubated in 100μl of 10X trypsin for 30 minutes, centrifuged, and washed twice with cell media. After the second rinse, the cells were suspended in 200μL of media and mechanically dissociated by trituration. The cells or explants were grown for 2 weeks in the system before the application of vincristine. To prepare the drug, vincristine sulfate salt (Sigma, St. Louis, Missouri) was mixed with cell media to achieve a 0.01μM concentration. Media was completely removed from one of the compartments and replaced by the solution with the drug.
Stimulation Protocol
DRG neurons were electrically stimulated by depolarization with KCl. A 500mM solution of KCl in standard culture media was applied to the neurons for one minute during which time electrical activity was recorded. All recordings were done with a Multichannel Systems (Reutlingen, Germany) 60-electrode preamplifier system. After the 1 minute recording window, the elevated K+ solution was replaced with normal media.
III. Results
A. Morphological Observations
After 10 days in culture in a two compartment system, a 0.01μM concentration of vincristine was applied either to the soma or axon compartment (Fig. 2). After the application of the drug, morphological assessments were made by measuring the distance of the axon from the cell body to its tip. These tests showed that only when the drug was applied to the axonal compartment was degeneration evident, leading us to believe that a mechanism local to the distal axon is responsible for vincristine neuropathy (see Table 1) [12, 15]. At this drug concentration, axonal degeneration started at day one and was complete (neurites retracted completely to the compartment divider) by the end of two days. On average, axons decreased in length 1–2mm per day. Degenerative changes were first noted at 18 hours after drug exposure, after which the rate of degeneration rapidly progressed. The sharp increase in the rate of degeneration at this time could result from the initiation of a critical signaling event local to the distal axon. When the same dose of drug was applied to the cell bodies, no axonal degeneration took place, and in fact, the axons continued to grow an average of 1 mm over the course of two days. At higher concentrations (≥ 0.5μM), axons and cell bodies degenerated regardless of where the drug was applied, and at lower concentrations (≤ 0.01μM), no degeneration was seen.
Figure 2.
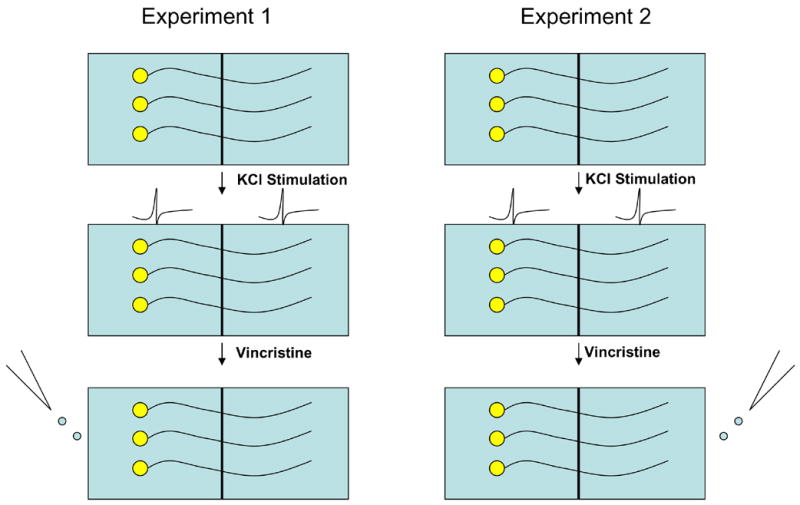
Two experimental protocols used to treated neurons with vincristine.
Table 1.
Morphological evidence of direct axonal effects in vincristine-induced axonal degeneration (n=6)
| Length of Axons after Drug Exposure (mm) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Start | 6 hrs | 12 hrs | 18 hrs | 24 hrs | 30 hrs | 36 hrs | 42 hrs | 48 hrs | |
| Control | 4.32±0.12 | 4.56±0.23 | 4.57±0.05 | 4.62±0.15 | 4.92±0.20 | 4.96±0.23 | 5.05±0.08 | 5.11±0.12 | 5.37±0.10 |
| Vincristine to Axons | 4.15±0.31 | 4.17±0.13 | 4.21±0.08 | 3.91±0.17 | 1.92±0.09 | 1.62±0.08 | 1.23±0.03 | 1.21±0.12 | 1.01±0.11 |
| Vincristine to Cell Bodies | 4.11±0.02 | 4.33±0.14 | 4.39±0.12 | 4.72±0.05 | 4.91±0.04 | 4.93±0.11 | 4.99±0.02 | 5.21±0.05 | 5.45±0.06 |
Values are mean ± SEM.
B. Electrophysiological Measurements
Electrophysiological recordings were performed at time points correlating with the onset and progression of axonal degeneration. After 10 days in culture, baseline electrical recordings were made from the somal and axonal compartments after depolarization with KCl. A 0.01μM solution of vincristine was then applied to either the somal or axonal compartment and KCl-stimulated electrical responses were recorded every 6 hours up to 48 hours (total 8 recordings). The data are summarized in Table 2. With addition of vincristine to the somal compartment, the electrophysiological response to KCl-mediated depolarization stayed constant throughout the two-day period. In contrast, addition of vincristine to the axonal compartment resulted in a progressive loss of the electrophysiological response beginning at 6 hours after addition of the drug, and culminating in complete electrical failure by about 30 hours. A comparison of the morphological and electrophysiological data demonstrated that the electrical abnormalities in the axons preceded the morphological changes, which were first noticeable at 18 hours after vincristine exposure. During the course of the two day experimental period, both the number and average amplitude of spikes decreased with stimulation of the axonal compartment, and only when vincristine was added to the axonal compartment. These findings suggest that the electrophysiological impairment of the axons precedes the retraction of the tip and is an earlier indictor of impaired neuronal health.
Table 2.
Electrophysiological activity during vincristine-induced axonal degeneration (n=6)
| No Vincristine (control) | Vincristine to Axons | Vincristine to Cell Bodies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Somal Compartment |
Axonal Compartment |
Somal Compartment |
Axonal Compartment |
Somal Compartment |
Axonal Compartment |
|||||||
| Time Point |
Number of Spikes |
Average Amplitude (μV) |
Number of Spikes |
Average Amplitude (μV) |
Number of Spikes |
Average Amplitude (μV) |
Number of Spikes |
Average Amplitude (μV) |
Number of Spikes |
Average Amplitude (μV) |
Number of Spikes |
Average Amplitude (μV) |
| Initial | 54±6 | 33.65±2.2 | 62±6 | 37.34±6.2 | 54±7 | 40.34±5.6 | 64±2 | 34.32±5.2 | 55±2 | 34.32±4.2 | 60±3 | 35.99±5.9 |
| 6hrs | 57±7 | 39.56±4.9 | 61±5 | 33.29±3.5 | 96±6 | 40.22±3.2 | 42±5 | 23.27±5.1 | 59±7 | 35.21±5.4 | 55±5 | 38.71±6.2 |
| 12hrs | 58±3 | 35.25±7.8 | 55±8 | 39.34±4.1 | 90±7 | 37.83±8.9 | 33±5 | 22.88±2.0 | 54±1 | 36.19±5.1 | 54±8 | 35.33±6.6 |
| 18hrs | 66±6 | 35.30±6.5 | 55±5 | 37.33±2.2 | 95±3 | 40.23±7.7 | 31±2 | 23.34±1.9 | 59±3 | 37.29±4.9 | 62±3 | 35.84±2.2 |
| 24hrs | 64±4 | 39.17±5.4 | 59±5 | 32.34±5.3 | 92±6 | 38.22±1.2 | 21±4 | 21.32±3.3 | 58±6 | 38.04±3.9 | 58±3 | 36.43±4.2 |
| 30hrs | 56±4 | 32.12±6.6 | 57±3 | 39.87±9.2 | 61±5 | 37.20±5.2 | 7±4 | 21.33±3.0 | 60±6 | 36.88±4.6 | 55±2 | 34.64±4.9 |
| 36hrs | 64±4 | 34.46±3.2 | 53±7 | 38.23±7.2 | 59±5 | 37.32±3.3 | 3±1 | 10.32±1.1 | 62±1 | 35.35±6.7 | 58±3 | 35.33±4.7 |
| 42hrs | 54±6 | 32.67±3.0 | 61±9 | 32.09±3.1 | 58±2 | 37.34±2.0 | 3±4 | 10.22±1.4 | 56±3 | 35.91±2.9 | 62±3 | 37.04±5.1 |
| 48hrs | 61±8 | 35.32±2.1 | 57±3 | 33.89±1.9 | 66±6 | 36.93±1.7 | 2±2 | 5.22±1.7 | 59±3 | 38.23±3.2 | 59±5 | 35.71±2.4 |
Values are mean ± SEM. P<0.001 when axonal compartments are compared at 48hrs. P<0.001 when somal compartments with vincristine at day 0 and day 1 are compared. P>0.05 when somal compartments are compared at 48hrs after drug exposure.
We found that 6 hours after vincristine exposure to the axonal compartment there was a relative increase in spike activity in the somal compartment as compared either to baseline or 48 hours after exposure. The increase in spike activity in the somal compartment persisted for about > 24 hours after axonal exposure to vincristine (p<0.001), and returned toward baseline by 36 hours. There was no increase in spike activity in the somal compartment with direct exposure of the cell bodies to vincristine. This perikaryal response to exposure of the axon to a neurotoxin suggests that an “injury signal” is transmitted from the axon to the cell body, but it remains unclear whether this response is important for the eventual death of the axon. Experiments that anesthetize the soma to observe changes in the distal axon could provide a definitive answer as to how important these signals are in axonal loss.
Finally, to monitor the spatial progression of the electrophysiological changes in response to vincristine exposure, each compartment was spatially divided into an inner, middle, and outer third containing ten electrodes each (see Fig. 3). This number of spikes and their amplitudes in each of the three compartments were quantified. Table 3a and 3b summarize these data. Four time points (initial, 6, 24, 36 hours) were chosen for spatial analysis. Initially, activity in each group was evenly distributed. As time progressed, the activity at the outer third decreased disproportionately compared to the other groups in each compartment (p<0.005). Moreover, the amplitudes of the degenerating axons declined, starting from the outer third of the axon compartment. Eventually, this decrease in both spike amplitude and number was seen in all of the axonal compartment and the somal compartment. These data support the conclusion that axonal degeneration due to vincristine begins at the distal axon and progresses centrally toward the cell body.
Figure 3.
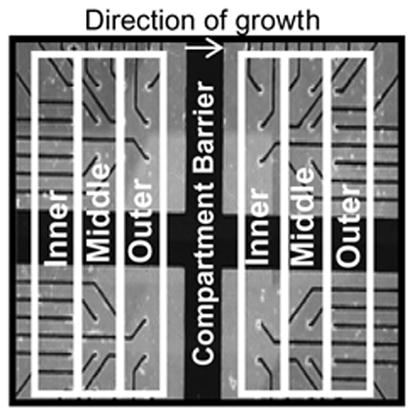
Grouping of inner, middle, and outer ten electrodes in each compartment.
Table 3a.
Distribution of activity among electrodes in the somal compartment (vincristine to axons), n=6
| Time Point After Drug Exposure | Inner 10 Electrodes | Middle 10 Electrodes | Outer 10 Electrodes | |||
|---|---|---|---|---|---|---|
| Number of Spikes | Average Amplitude (μV) | Number of Spikes | Average Amplitude (μV) | Number of Spikes | Average Amplitude (μV) | |
| Initial | 15±3 | 40.23±5.3 | 15±2 | 40.22±5.6 | 14±3 | 40.57±5.9 |
| 6 hrs | 33±4 | 40.38±3.2 | 29±3 | 40.36±3.2 | 34±2 | 39.92±3.2 |
| 24hrs | 35±2 | 42.26±1.2 | 39±2 | 38.18±1.3 | 20±2 | 34.22±1.2 |
| 36 hrs | 50±3 | 43.45±3.3 | 25±3 | 37.12±3.3 | 10±2 | 31.39±3.3 |
Values are mean ± SEM. P<0.001 when inner and outer 10 electrodes are compared at 36hrs after drug exposure.
Table 3b.
Distribution of activity among electrodes in the axonal compartment (vincristine to axons), n=6
| Time Point After Drug Exposure | Inner 10 Electrodes | Middle 10 Electrodes | Outer 10 Electrodes | |||
|---|---|---|---|---|---|---|
| Number of Spikes | Average Amplitude (μV) | Number of Spikes | Average Amplitude (μV) | Number of Spikes | Average Amplitude (μV) | |
| Initial | 22±3 | 34.3±5.2 | 22±3 | 34.34±5.3 | 20±3 | 34.32±5.1 |
| 6 hrs | 20±2 | 29.34±5.1 | 17±2 | 23.06±5.1 | 5±1 | 17.41±5.1 |
| 24hrs | 10±1 | 30.82±3.2 | 8±2 | 20.45±3.3 | 3±1 | 12.68±3.4 |
| 36 hrs | 3±1 | 13.34±0.9 | 1±0 | 10.38±1.1 | 1±0 | 7.24±1.3 |
Values are mean ± SEM. P<0.001 when inner and outer 10 electrodes are compared starting at 6hrs after drug exposure.
IV. Discussion
The experimental design provided us with morphological and electrical data taken at very specific and closely separated time intervals. As with our previous studies we found that there was differential susceptibility of axons and cell bodies to the neurotoxic effects of vincristine [15]. A specific concentration of vincristine added to the cell body compartment showed no effect on axonal survival or growth, whereas the same concentration in the axonal compartment caused progressive axonal degeneration. These data are consistent with our previous morphological data demonstrating direct vulnerability of the distal axon [15]. As detailed in Table 1, the course of axonal degeneration appeared to accelerate during the 18 and 24 hour time period, as compared to the first 12 hours. This observation, combined with the finding that no degeneration was seen with vincristine exposure to the cell body, suggests that a signaling event may occur in the axon, perhaps the triggering of microtubule depolymerization or some other event closely associated with it.
Our electrophysiological data adds an even richer picture of possible signaling mechanisms in vincristine neurotoxicity. With addition of vincristine to the axon compartment we observed an initial increase in spike activity in the somal compartment at 6 hours after drug exposure, long before any morphological changes were present. These data suggest that the proximal axon or cell body may be implicated in the signaling paradigm that leads to the retraction of the axon tip. Spatial analysis of electrode activity within the two compartments demonstrated conduction deficits in the distal portions of the axons prior to similar changes in proximal portions. Thus, the effect of vincristine seemed to impair the conduction of axons retrogradedly. Since the activity of the somal compartment begins to die down retrogradedly in the 6–24 hour time period, the hyperexcitability observed during this initial time period of vincristine toxicology is more likely to be from areas closer to the cell bodies than the proximal axons. Taken together, the morphological and electrophysiological data indicate that the signaling event for axonal degeneration could start in the cell body, but that only when the signal is transported from the retracting tip are the conditions ripe for producing the ensuing morphology.
Previous morphological observations on vincristine neuropathy in humans, animals, and in cell culture models have shown that the distal axon is particularly susceptible to degeneration [1, 14]. Electrophysiological correlates of this “dying back” phenotype are less straightforward. While some studies in animal models have shown a marked increase in the electrophysiological activity of neurons exposed to vincristine, others in humans have shown either no change or a decrease in the electrophysiogical health of the neuron [8, 10]. For instance, electrophysiological deficits have been recorded in clinical studies of human nerve fibers as evidenced by increases in distal latencies and decreases in the amplitudes of compound nerve action potentials [5]. However, other studies in rats have found that there is an increase in the excitability of DRG fibers during the early stages of vincristine neuropathy [17].
These results are not mutually exclusive, given that the time course could result in an initial hyperexcitability related to the feeling of chemotherapeutic pain followed by an eventual decrease in excitability due to axonal degeneration and loss of signal. However, they do lead to questions in how the neuron changes its electrical response during administration of the drug. The exact nature and localization of these electrophysiological changes could provide useful clues to the origin of this pathological condition. While the electrophysiological data here are consistent with the conclusion that the distal axon is the primary target, the data also suggest a role for other parts of the neuron in generating an initial phase of hyperexcitability. Reductions in action potential amplitudes as seen with this study and with numerous other studies could be attributed to the progressive retrograded death of the axonal compartment, but the initial periods of hyperexcitability could indicate a cell body-initiated nociceptive warning signal of axonal exposure to harmful stimuli. By manipulating the intensity of this signal through other pharmaceutical interventions, we can gain a better understanding of its importance in both the progression and prevention of the degenerative phenotype.
We found that the loss of inducible electrical activity in axons exposed to the vincristine precedes axonal degeneration as assessed morphologically. As electrophysiological signal transduction occurs more rapidly than signaling through metabolic transport mechanisms, it is not surprising to see that the changes in injury potentials precede those changes observed through morphology. However, the increase in the activity of the somal compartment prior to the loss of axonal integrity as measured electrically or morphologically suggests that the soma is involved in the process, perhaps in the transmission of an “injury signal” locally or back to the axon [17]. Given the complex changes in injury potentials occurring in both compartments, the data would also suggest that simple morphological observations may not provide an accurate representation of the progression of changes that occur in vincristine neuropathy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bradley WG, Lassman LP, Pearce GW, Walton JN. The neuromyopathy of vincristine in man: clinical, electrophysiological, and pathological studies. J Neurol Sci. 1970;10:107–131. doi: 10.1016/0022-510x(70)90013-4. [DOI] [PubMed] [Google Scholar]
- 2.Cavanagh JB. The dying back process: a common denominator in many naturally occurring and toxic neuropathies. Arch Pathol Lab Med. 1979;103:659–664. [PubMed] [Google Scholar]
- 3.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gidding CEM, Kellie SJ, Kamps WA, Graaf SSNd. Vincristine revisited. Crit Rev Oncol Hematol. 1999;29:267–287. doi: 10.1016/s1040-8428(98)00023-7. [DOI] [PubMed] [Google Scholar]
- 6.Goldstein BD, Lowndes HE, Cho E. Neurotoxicology of vincristine in the cat. Electrophysiological studies. Arch Toxicol. 1981;48:253–264. doi: 10.1007/BF00319653. [DOI] [PubMed] [Google Scholar]
- 7.Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guiheneuc P, Ginet J, Groleau JY, Rojouan J. Early phase of vincristine neuropathy in man. Electrophysiological evidence for a dying-back phenomenon, with transitory enhancement of spinal transmission of the monosynaptic reflex. J Neurol Sci. 1980;45:355–366. doi: 10.1016/0022-510x(80)90179-3. [DOI] [PubMed] [Google Scholar]
- 9.Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neuron loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- 10.McLeod JG, Penny R. Vincristine neuropathy: an electrophysiological and histological study. J Neurol Neurosurg Psychiatry. 1969;32:297–304. doi: 10.1136/jnnp.32.4.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravula SK, McClain MA, Wang MS, Glass JD, Frazier AB. A multielectrode microcompartment platform for studying signal transduction in the nervous system. Lab on a Chip. 2006;6:1530–1536. doi: 10.1039/b612684g. [DOI] [PubMed] [Google Scholar]
- 12.Ravula SK, Wang MS, Asress SA, Glass JD, Frazier AB. A compartmented neuronal culture system in microdevice format. J Neurosci Methods. 2007;159:78–85. doi: 10.1016/j.jneumeth.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Sagot Y, Dubois-Dauphin M, Tan SA, Bilbao Fd, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schlaepfer WW. Vincristine-induced axonal alterations in rat peripheral nerve. J Neuropathol Exp Neurol. 1971;30:488–505. doi: 10.1097/00005072-197107000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Silva A, Wang Q, Wang M, Ravula SK, Glass JD. Evidence for direct axonal toxicity in vincristine neuropathy. J Peripher Nerv Syst. 2006;11:211–216. doi: 10.1111/j.1529-8027.2006.0090.x. [DOI] [PubMed] [Google Scholar]
- 16.Spencer PS, Schaumburg HH. Central-peripheral distal axonopathy--the pathogenesis of dying-back polyneuropathies. In: Zimmerman H, editor. Progress in Neuropathology. Vol. 3. Grune and Stratton; New York: 1976. pp. 253–295. [Google Scholar]
- 17.Tanner K, Reichling DB, Levine JD. Nociceptor hyper-responsiveness during vincristine-induced painful peripheral neuropathy in the rat. J Neurosci. 1998;18:6480–6491. doi: 10.1523/JNEUROSCI.18-16-06480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang MS, Wu Y, Culver D, Glass JD. Pathogenesis of axonal degeneration: parallels between Wallerian degeneration and vincristine neuropathy. J Neuropathol Exp Neurol. 2000;59:599–606. doi: 10.1093/jnen/59.7.599. [DOI] [PubMed] [Google Scholar]
- 19.Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]