

Abstract
Inflammatory destruction of insulin-producing β cells in the pancreatic islets is the hallmark of insulin-dependent diabetes mellitus, a spontaneous autoimmune disease of non-obese diabetic mice resembling human juvenile (type I) diabetes. Histochemical analysis of diabetic pancreata revealed that mononuclear cells infiltrating the islets and causing autoimmune insulitis, as well as local islet cells, express the CD44 receptor; hyaluronic acid, the principal ligand of CD44, is detected in the islet periphery and islet endothelium. Injection of anti-CD44 mAb 1 hr before cell transfer of diabetogenic splenocytes and subsequently on alternate days for 4 weeks induced considerable resistance to diabetes in recipient mice, reflected by reduced insulitis. Contact sensitivity to oxazolone was not influenced by this treatment. A similar antidiabetic effect was observed even when the anti-CD44 mAb administration was initiated at the time of disease onset: i.e., 4–7 weeks after cell transfer. Administration of the enzyme hyaluronidase also induced appreciable resistance to insulin-dependent diabetes mellitus, suggesting that the CD44–hyaluronic acid interaction is involved in the development of the disease. These findings demonstrate that CD44-positive inflammatory cells may be a potential therapeutic target in insulin-dependent diabetes.
The inflammatory cascade in affected organs of autoimmune diseases is a complex process that involves triggering of the immunological response, release of chemokines, cytokines, and toxic agents (e.g., reactive oxygen) by the activated cells, stimulation of endothelial cells, up-regulation of cell surface adhesion molecules, transendothelial cell migration, and a shift in the Th1/Th2 balance in favor of the Th1 cells (1). Hence, the destructive autoimmune inflammatory process depends substantially (but not exclusively) on cell migration and cell interaction with matrix components of target organs. The destruction of pancreatic islet β-cells in insulin-dependent diabetes mellitus (IDDM) by invading leukocytes and the consequent deterioration of the insulin-dependent glucose homeostasis is an excellent example of such an autoimmune process (2, 3), although neither the nature of the triggering self-antigen nor the molecules associated with its recognition and presentation have been unequivocally identified.
Whereas the function of selectins and integrins in supporting inflammatory cell migration and lodgment has been well established (4), the role of cell surface CD44 has only recently attracted attention (5). Alternative splicing and/or posttranslation modifications generate many CD44 isoforms. The large array of CD44 isoforms is mainly attributable to the insertion of amino acid sequences, encoded by different combinations of 10 variant exons, into a membrane proximal position of the extracellular domain. Transcripts in which these variant exons are spliced out encode the most common and widely expressed 85-kDa isoform, known as standard CD44. The expression of CD44 isoforms containing sequences encoded by the variant exons (CD44v) is tightly regulated. Expression of CD44v isoforms is restricted to epithelial cells and cells undergoing activation or differentiation, as well as some progressor tumors. CD44 isoforms are involved in a wide variety of cellular processes, such as cell–cell and cell–matrix interactions, cell trafficking, presentation of cytokines and growth factors to traveling cells, and the uptake and intracellular degradation of hyauloronic acid (HA), the principal ligand of this receptor. In addition to HA, other tissue and cell components (e.g., collagen, fibronectin, laminin, serglycin, and osteopontin) can interact with CD44 (reviewed in refs. 5 and 6).
Although the triggering mechanism of IDDM remains elusive, it is clear that the entire process depends on the migration of inflammatory cells into the pancreatic islets and their interaction with their matrix. Because CD44 is associated with both activities (5), we decided to study its involvement in experimental IDDM in non-obese diabetic (NOD) mice by exploring the anti-diabetogenic effect of mAbs directed against CD44 constant epitopes. To this end, we resorted to the well established (7) NOD mouse transfer model, using the same experimental protocol we had previously adopted to arrest CD44-positive lymphoma dissemination (8, 9) and to inhibit the development of collagen-induced arthritis (10). Because of the consistency of the model and the high incidence of involved animals, we decided to use this model, rather than the spontaneous one, to address the major question related to this study: do CD44 and its major ligand, HA, associate functionally with autoimmune insulitis leading to IDDM, and can they serve as potential therapeutic targets? Here we show that, in this animal model, CD44 and HA targeting by specific antibody and enzyme, respectively, confer appreciable resistance to diabetes.
Materials and Methods
Mice.
Male and female NOD mice or BALB/c mice obtained from The Jackson Laboratory or Harlan Laboratories (Jerusalem) were maintained in a specific pathogen-free animal facility at 21°C and supplied with acidified water (pH 2.7) and mouse chow ad libitum.
mAbs and Enzymes.
The following rat anti-mouse mAbs were used: the anti-mouse CD44 constant region (rat, IgG2b), obtained from hybridoma IM7.8.1 (American Type Culture Collection, TIB-235) (11); the anti-mouse CD44 constant region (rat, IgG2b), obtained from hybridoma KM81 (American Type Culture Collection, TIB-241) (12); the anti-mouse CD44 constant region (rat, IgG2a), obtained from hybridoma IRAWB14.4 (provided by P. W. Kincade, Oklahoma Medical Research Foundation) (13). The anti-mouse cell surface Ig idiotype (rat, IgG2b) obtained from hybridoma 4D2 (provided by J. Haimovich, Tel Aviv University) (14) or rat IgG2b Ig (PharMingen) were used as an isotype-matched control mAb, to exclude Fc-mediated effects. The hybridomas were injected into the peritoneal cavity of nude mice, and the mAbs were purified from the ascitic fluid by protein G-Sepharose chromatography, as described in our previous study (15). Heparinase (H-2519) and hyaluronidase (H-3757) were obtained from Sigma.
Experimental Design.
Diabetic [confirmed by glucosuria (see below)] female donor NOD mice, aged 15–20 weeks, were killed, and suspensions of spleen leukocytes were washed twice in RPMI 1640 medium containing 10% fetal calf serum. A quantity of 25 × 106 leukocytes was injected i.v. into each irradiated (550 cGy) male recipient mouse, aged 8–10 weeks. A number of recipient mice were injected with PBS whereas the remaining animals were injected with 150 μg of IM7.8.1 anti-CD44 or control mAbs per mouse. A 200-μl volume of PBS or mAbs (diluted in PBS) was injected i.v. into the recipient mice 1 hr before cell transfer and then i.p. every other day for 4 weeks (a total of 12 injections of 150 μg of mAb per mouse). To determine the ability of anti-CD44 mAb to inhibit IDDM after its onset (i.e., prediabetes), NOD recipient mice were checked twice weekly for the development of prediabetic activity by using the intraperitoneal glucose tolerance test (IPGTT) (see below). Any mouse exhibiting a positive IPGTT (usually 4–7 weeks after cell transfer) was randomly assigned to one of two groups given IM7.8.1 anti-CD44 mAb or isotype-matched control mAb (4D2), according to the above protocol. To monitor the development of diabetes, the percentage of disease-free animals (glucosuria negative) was recorded over time. Each experiment was repeated at least two or three times. The statistical significance of the findings was determined (unless otherwise indicated) by the Generalized Gehan test (Breslow) (16). To avoid presentation bias, repeated experiments were pooled (unless otherwise indicated), and the statistical analysis was adjusted to account for interexperimental variations (16).
Assessment of Diabetes by Glucose Determination.
Diabetes was monitored by testing urinary glucose with a Teststrip (Medi-Test, Combi 9, Macherey-Nagel, Düren, Germany) twice weekly and was considered positive after the appearance of glucosuria in at least two determinations. The IPGTT was performed as follows: blood was drawn from the paraorbital plexus at 0 min and 60 min after an i.p. injection of glucose (1 g/kg body weight). Plasma glucose levels were determined (as glucose mmol/liter) with a Glucose Analyzer 2 (Beckman Instruments). A glucose level above 15 mmol/liter at the 60 min time point was considered a positive IPGTT.
Histopathology.
Pancreatic tissue was fixed in 10% buffered formalin and was embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin. Sections containing a total of more than 17 islets from each pancreas were screened and scored by an uninformed observer according to the following criteria: 0, no cell infiltration; 1, periinsulitis and cell infiltration involving up to 20% of islet area; 2, cell-infiltration involving up to 50% of islet area; 3, cell infiltration involving up to 75% of islet area; and 4, cell infiltration involving 90–100% of islet area.
CD44 and HA Histochemistry.
CD44 and HA histochemistry of pancreatic sections were performed as described in ref. 17, except that normal rabbit, rather than goat, serum and rabbit, rather than goat, anti-rat IgG secondary antibody were used. The slides were counterstained with hematoxylin and were mounted with permount.
Delayed Type Hypersensitivity (DTH) Assessments.
Solutions of 2% and 0.5% 4-ethoxymethylene-2-phenyl-oxazolin-5-one (oxazolone, E-0753, Sigma) were prepared by dissolving 200 and 50 mg, respectively, of oxazolone in 8 ml of acetone and 2 ml of mineral oil (Kodak). The abdomen of each mouse was painted with 100 μl of 2% oxazolone. On day 6, the right ear was painted with 10 μl of 0.5% oxazolone, and differences (Δ) between right and left ear thickness, indicating DTH development, were determined by microcaliper.
Results
CD44 and HA Histochemical Analysis of Diabetic Pancreatic Islets.
Diabetes was induced in sublethally irradiated male NOD mice by transfer of splenocytes isolated from diabetic female NOD mice. After 20–50 days, the recipient mice developed diabetes, as indicated by the appearance of glucose in the urine. Immunohistochemical analysis of pancreatic sections prepared from the diabetic recipient mice revealed that mononuclear cells (MNCs), mostly lymphocytes, infiltrating the islets of Langerhans, as well as the islet cells themselves, were intensively stained after incubation with KM81 (Fig. 1 A and B) or IM7.8.1 (results not shown) anti-CD44 mAbs and the indicator antibody. Similar sections did not react with the same concentrations of rat isotype-matched (IgG2b) Ig (Fig. 1C) or rat isotype-matched irrelevant mAb (4D2) (results not shown). The pancreatic islets of normal BALB/c mice failed to be stained by the same concentration of the diabetic islet-interacting anti-CD44 mAb and the indicator antibody (Fig. 1D). An entirely different reaction pattern was revealed with 9A4 anti-CD44v6 and 10D1 anti-CD44v4 mAbs (provided by J. Moll, Karlsruhe Research Center, Karlsruhe, Germany). The anti-CD44v6 mAb intensively stained small groups of epithelial islet cells, but not the infiltrating MNCs. Identical concentrations of anti-CD44v4 mAb, displaying the same isotype (rat, IgG1), did not stain the islets of diabetic recipient mice. Neither of the two antibodies stained the pancreatic islets of BALB/c mice (results not shown).
Figure 1.
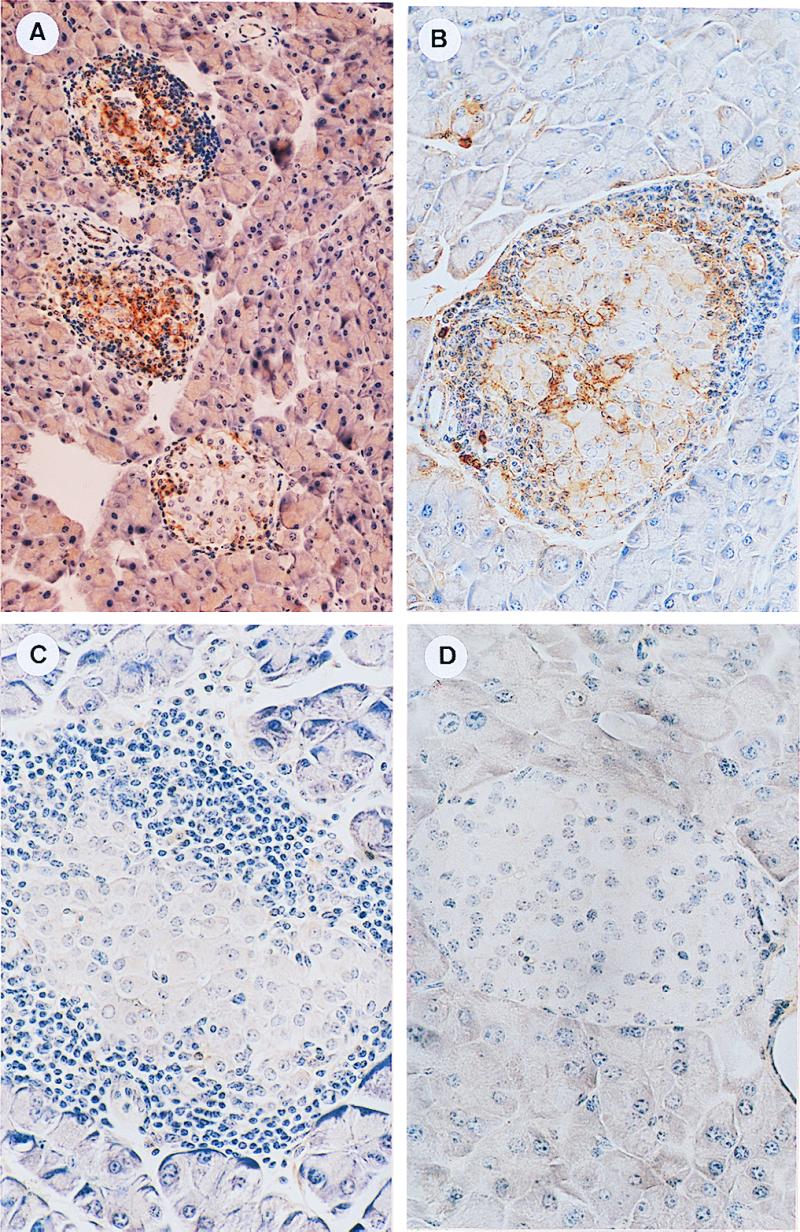
CD44 immunohistochemical analysis of pancreatic islets. Sections of pancreas derived from diabetic recipient NOD mice (A, B, and C) or normal BALB/c mice (D) were incubated with KM81 anti-CD44 mAb (A, B, and D) or with the same concentration (5 μg/ml) of isotype-matched control Ig (IgG2b) (C). Bound antibodies were identified with biotinylated rabbit anti-rat IgG (7.5 μg/ml), using the avidin-biotin horseradish peroxidase detection system. CD44 is present on infiltrating MNCs and local cells of diabetogenic islets (A and B) but not on normal islet cells of BALB/c mice (D). CD44 staining of diabetogenic islets is not observed after their preincubation with isotype-matched control Ig (C). (A, ×176; B–D, ×352.)
The HA content of the diabetic islets of recipient NOD mice was analyzed with biotinylated HA binding protein (bHABP) (17) and the indicator streptavidin-alkaline phosphatase. Hyaluronate staining was detected along the connective tissue surrounding the islets (Fig. 2A) and in a few small blood vessels penetrating the islets (not shown in this figure). HA staining was prevented by preincubating the pancreatic sections with hyaluronidase (17) (Fig. 2B), proving the specificity of the reaction. In contrast, HA was barely detectable after staining with bHABP in sections of pancreatic islets derived from normal BALB/c mice (Fig. 2C), suggesting that the inflammatory cascade induces the expression of hyaluronate.
Figure 2.
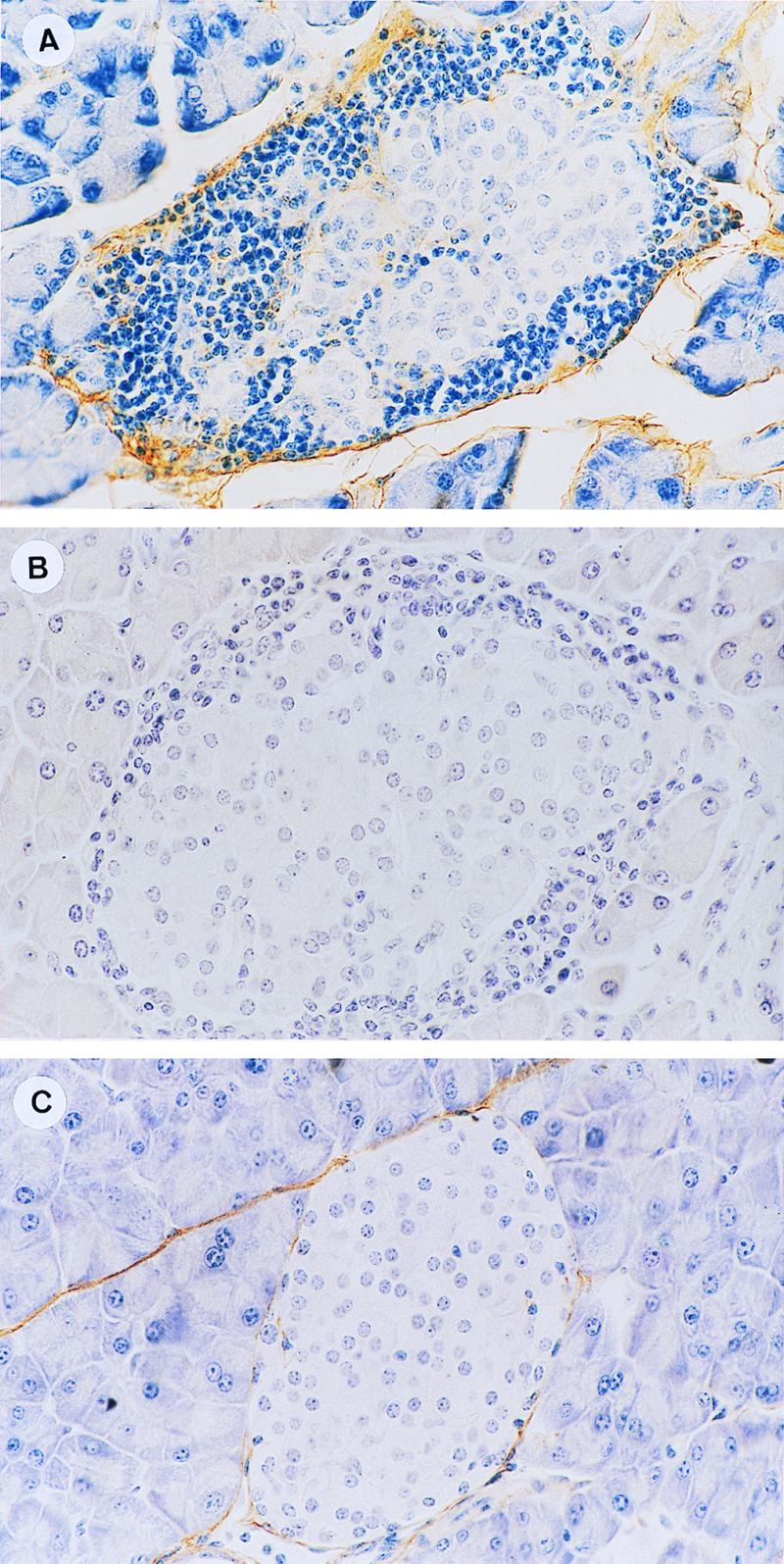
HA histochemical analysis of pancreatic islets. Sections from diabetic recipients (A and B) or normal BALB/c mice (C), pretreated [100 relative turbidity reducing units (rTRU)/ml] (B) or not pretreated (A and C) with hyaluronidase, were incubated with bHABP (2.5 μg/ml). Bound HA was identified by using the avidin-biotin peroxidase detection system. HA was detected in the connective tissue surrounding the islets (A) whereas normal islets (C) were only marginally stained by the same concentration of bHABP. Preincubation with hyaluronidase prevented the bHABP staining of the diabetogenic islets (B). (×352.)
The above results imply that both CD44 and HA are up-regulated in the pancreatic islets of the diabetic recipient NOD mice, suggesting that cell surface CD44 and matrix HA support the migration of destructive inflammatory cells into the islets. Therefore, we tested the ability of anti-CD44 mAb and the enzyme hyaluronidase to interfere with this event, thereby protecting the insulin-producing β cells and, consequently, sustaining glucose homeostasis.
The Anti-Diabetogenic Effect of Anti-CD44 mAb.
The anti-diabetogenic effect of the anti-CD44 mAb, which recognizes a constant epitope on the CD44 receptor, was tested in recipient-irradiated male NOD mice infused with splenocytes of female diabetic NOD mice. The data pooled from 10 similar experiments are presented in Fig. 3. The recipient mice were injected with PBS, anti-CD44 mAbs, or control mAb, using the protocol described in Materials and Methods. Diabetes development over time was monitored by recording the appearance of glucose in the urine. Within 55 days, 90% of the recipient mice treated with PBS or with isotype-matched (IgG2b) control mAb (4D2) developed diabetes. In contrast, within the same period, 55% of the mice injected with IM 7.8.1 anti-CD44 mAb were diabetes-free, and close to 50% remained free of disease at day 75: i.e., 47 days after termination of antibody administration. On day 100 after transfer (or 72 days after termination of antibody administration), 12 of 46 mice injected with anti-CD44 mAb (26%) did not show any signs of diabetes. At that time, only 2 of the 93 control mice (≈2%) were free of diabetes. Similarly to the control groups, all mice injected with IRAWB14.4 anti-CD44 mAb developed diabetes within 55 days after cell transfer, although some delay in disease appearance was observed.
Figure 3.

The anti-diabetogenic effect of anti-CD44 mAb. Male NOD mice were injected with IM7.8.1 anti-CD44 mAb, isotype-matched control mAb (4D2), IRAWB14.4 anti-CD44 mAb, or PBS, 1 hr before splenocyte transfer from diabetic NOD female mice. The mice were then injected with the same reagents on alternate days for the next 4 weeks, as described in the experimental protocol. Development of diabetes was monitored by glucosuria determination. The data pooled from 10 similar experiments are shown. The Breslow statistical analysis (p) was used to compare the PBS group with all of the other groups. NS, not significant.
To evaluate the anti-diabetogenic effect of anti-CD44 mAb on the active disease, the standard injection protocol was delayed by several weeks to include prediabetic recipients with positive IPGTT. Fig. 4 reveals that 45% of the mice injected with anti-CD44 mAb did not develop diabetes for at least 100 days after the detection of prediabetes by IPGTT (or 72 days after termination of antibody administration) whereas all mice injected with 4D2 mAb were diabetic on day 100. This experiment demonstrates that anti-CD44 mAb induces considerable resistance to diabetes even when the recipients are treated at an early stage of detectable disease.
Figure 4.

Inhibition of prediabetes onset by anti-CD44 mAb. Recipient NOD female mice with onset of active prediabetes, as determined by IPGTT 4 to 7 weeks after cell transfer, were randomly assigned to two groups. One group was injected with IM7.8.1 anti-CD44 mAb, and the other with isotype-matched control mAb (4D2) on alternate days for 4 weeks. Development of diabetes was monitored by glucosuria determination. Data were pooled from two similar experiments. The Breslow statistical analysis (p) was used to compare the 4D2 group with the IM7 group. Note that the IPGTT positive prediabetic mice were still glucosuria negative at the beginning of the experiment. Therefore, they were categorized as “diabetes-free mice.”
To demonstrate the association between the inhibition of diabetes and blockage of islet inflammation attributable to anti-CD44 mAb treatment, we scored islet cell infiltration in experimental recipient mice injected with this antibody and in control mice injected with the 4D2 isotype-matched control antibody, using the standard protocol described in Fig. 3. Recipient mice were killed 56 days after cell transfer, when the anti-diabetogenic effect of anti-CD44 antibody was revealed by the glucose test. The degree of inflammation in each screened pancreatic islet was histopathologically analyzed and scored by a noninformed observer (see Materials and Methods and legend to Table 1). The average inflammation score for 139 islets from four mice injected with control mAb was 2.4 whereas the average score of 156 islets from four mice injected with anti-CD44 mAb was 0.7 (Table 1), implying 3.4× less intensive islet cell infiltration. Examples of histological sections of pancreatic islets obtained from a different experiment, showing results similar to those listed in Table 1, are presented in Fig. 5. The islets of the control recipient mice are heavily infiltrated by MNCs (Fig. 5 A–C; insulitis score close to 3) whereas those of the anti-CD44 mAb-injected recipients display only a small focal infiltrate at the periphery, especially around blood vessels (Fig. 5 D–F; insulitis score of up to 1).
Table 1.
Insulitis inhibition by anti-CD44 mAb
| Injected mAb | Mouse | No. of scored islets | Average score/islet |
|---|---|---|---|
| 4D2 | 1 | 17 | 3.0 |
| 2 | 24 | 1.5 | |
| 3 | 58 | 2.9 | |
| 4 | 40 | 2.2 | |
| Total | 139 | 9.6 | |
| Mean = 2.40 ± 0.35 | |||
| IM7.8.1 | 5 | 40 | 0.7 |
| 6 | 26 | 0.8 | |
| 7 | 51 | 0.5 | |
| 8 | 39 | 0.9 | |
| Total | 156 | 2.9 | |
| Mean = 0.72 ± 0.08 |
Male NOD mice were injected with anti-CD44 mAb or isotype-matched control mAb (4D2), using the protocol described in Fig. 3. Insulitis was graded on a scale of 0–4, 56 days after cell transfer: i.e., 28 days after termination of antibody administration, as described in Material and Methods. At this time, the anti-diabetogenic effect of the anti-CD44 mAb was clearly observed by the glucose test. The number of scored islets per mouse and the average insulitis score per mouse, as well as the total score and mean ± SEM for each group, are shown. P < 0.03, by the Mann–Whitney U test.
Figure 5.

Histological analysis of pancreatic islets derived from control recipients (A–C) and from the corresponding recipients treated with anti-CD44 mAb (D–F). Pancreatic tissue from recipient mice (as described in the legend to Table 1) were fixed in 10% buffered formalin, embedded in paraffin, and 5-μm sections were stained with hematoxylin and eosin. The pancreatic endocrine islets of the control recipients are heavily infiltrated by MNCs (A–C) whereas those of the anti-CD44 mAb-injected mice display a small focal infiltrate at the periphery (D–F). (×158.)
Anti-CD44 mAb Injection Does Not Influence Normal Immune Responses of Recipient NOD Mice.
The finding that anti-CD44 mAb induced appreciable resistance to diabetes in recipient NOD mice raised the question of whether normal immunological responses were simultaneously affected by the antibody. For in vivo testing, we chose the delayed type hypersensitivity reaction, because of its resemblance to the inflammatory responses in autoimmune diseases. Recipient NOD mice were injected with 150 μg/mouse IM 7.8.1 anti-CD44 mAb or with the same amount of isotype-matched control mAb (4D2), using our standard protocol. When the anti-diabetogenic effect of anti-CD44 mAb was already evident (Fig. 6), the DTH response to oxazolone was assessed by measuring the difference in thickness (Δ) (in mm × 10−2) between the hapten-painted right ear and nonpainted left ear. Recipient mice given IM 7.8.1 anti-CD44 mAb had a Δ of 7.5 ± 1.3 (n = 6), and animals injected with isotype-matched control mAb (4D2) had a Δ of 5.8 ± 0.6 (n = 7), which is statistically insignificant (P = 0.45 by the Mann–Whitney U test). On day 31 after cell transfer, all of the experimental animals were killed, and the proliferative response of their splenocytes to stimulation with Con A was measured by 3H-thymidine incorporation. In this and in an additional independent experiment, no difference was observed in the mitogen-induced proliferation response (results not shown). These results were confirmed by an additional experiment in which injection of anti-CD44 mAb induced, in contrast to control mAb (4D2), anti-diabetogenic activity whereas the oxazolone DTH response was not influenced. The average of Δ units (mm × 10−2) in recipient mice injected with IM7.8.1 anti-CD44 mAb was 14.3 ± 1.2 (n = 5) versus 16.3 ± 1.62 (n = 7) in the control mice (P = 0.9 by the Mann–Whitney U test). Collectively, these findings imply that, under these experimental conditions, the anti-diabetogenic dose of anti-CD44 mAb did not influence cellular immune responses such as DTH and mitogen-induced T lymphocyte proliferation.
Figure 6.

The anti-diabetogenic dose of anti-CD44 mAb does not influence DTH development. Male NOD mice were injected with IM7.8.1 anti-CD44 mAb or isotype-matched control mAb (4D2) 1 hr before splenocyte transfer from diabetic female NOD mice, and then every other day for an additional 4 weeks. On day 20 after cell transfer, when the anti-CD44 mAb anti-diabetogenic effect was evident, all recipients were sensitized with oxazolone, which was applied to their abdomen. After 6 days, they were repainted in the right ear. The difference in thickness between the right and the left ear (Δ) was determined by microcaliper 1 day later. Development of diabetes was monitored by glucosuria determination. Statistical analysis was carried out as described in Fig. 4. The difference in DTH (Δ ear thickness) between the two groups is statistically insignificant according to the Mann–Whitney U test.
The Anti-Diabetogenic Effect of Hyaluronidase.
Because the interaction between cell surface CD44 and HA may be critical to the development of the diabetogenic inflammatory cascade, we tested whether degradation of HA by hyaluronidase would ameliorate disease pathology. More than 90% of recipient mice injected 1 hr before cell transfer and then every other day for 4 weeks with PBS or with 20 units of the control enzyme heparinase developed diabetes within 60 days. In contrast, recipients injected with 20 units of hyaluronidase on the same days generated considerable resistance to the disease (P = 0.003, hyaluronidase group vs. heparinase group) (Fig. 7). Note that the heparinase and the hyaluronidase had similar specific activities.
Figure 7.

The anti-diabetogenic effect of hyaluronidase. Recipient NOD mice were injected 1 hr before cell transfer and then every other day for 4 weeks with PBS or 20 units of heparinase (Hep'ase) or hyaluronidase (H'ase). Development of diabetes was monitored by glucosuria. Data were pooled from two similar experiments.
Discussion
CD44 molecules play a critical role in the development of inflammatory responses, leading to physiological eradication of the invading microorganisms (18) or of the pathological manifestations of autoimmunity (10, 19–22). We have shown here that both the CD44 receptor and HA, its principal ligand, are involved in the development of IDDM in NOD mice, as deduced from the anti-diabetogenic effects of anti-CD44 mAb and the enzyme hyaluronidase, respectively.
Immunohistochemical staining showed that the pancreatic islets of diabetic NOD mice contain infiltrating lymphocytes expressing CD44 (ref. 23 and Fig. 1). Using the reverse transcriptase–PCR (24) or immunohistochemical analysis (Fig. 1), it further was demonstrated that splenocytes and local cells of pancreatic islets from diabetic NOD mice (but not from BALB/c mice) display enhanced expression of CD44 isoforms, including CD44 isoform(s) containing v6 epitopes. Furthermore, we showed by histochemical analysis (Fig. 2) that, in contrast to normal pancreatic islets, the islet periphery of a diabetogenic pancreas, as well as its endothelium, contain HA, the principal ligand of CD44. These findings suggest that the inflammatory cascade in the diabetogenic pancreas is associated with the up-regulation of both CD44 and hyaluronan. Indeed, it was demonstrated that the proinflammatory cytokines tumor necrosis factor-α and IL-1β, as well as bacterial lipopolysaccharide, induce expression of HA in endothelial cell lines and on endothelial cells in primary culture (25). The association between cell migration-dependent activities, such as inflammation or malignant metastasis, and interactions between cell surface CD44 and HA has been well documented. For example, it was demonstrated that the CD44-HA interaction allows cell migration into staphylococcal enterotoxin B-induced inflamed sites, as interference with this interaction by anti-CD44 mAbs or by the enzyme hyaluronidase inhibited the inflammatory process (18). Lymphocytes capable of CD44/HA-dependent rolling interactions were found within inflamed tonsils and in the peripheral blood of patients from a pediatric rheumatology clinic (26). We previously reported that injection of anti-CD44 mAb or hyaluronidase prevents LB T-cell lymphoma invasion of lymph nodes (8, 9). In addition, it has been documented that treatment with anti-CD44 mAb induces resistance to experimental autoimmune diseases such as collagen-induced arthritis (10, 19, 20), trinitrobenzenesulfonic acid-induced colitis (21), and experimental allergic encephalomyelitis (22), all of which are characterized by destructive inflammation of the target organs. Hence, CD44 can be included in the inventory of prodiabetic targets such as the adhesive molecules l-selectin, α4,β7, lymphocyte function-associated antigen-1, mucosal addressin cell adhesion molecule-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 or the cytokines IFN-γ and IL-6, the proinflammatory effects of all of which are manifested by the inhibitory activity of the corresponding mAbs (7, 27). Like l-selectin (4), the CD44 receptor enables primary cell attachment to and rolling on the endothelium (25). It is possible, however, that CD44 is preferentially (but not exclusively) expressed on cells migrating to potential inflammatory sites (e.g., pancreatic islets or joints) whereas l-selectins are presented on cells infiltrating peripheral lymph nodes (4).
What can be learned from this study regarding the mechanism of CD44-dependent inflammatory islet invasion and CD44 targeting in the experimental model of IDDM? (i) Anti-CD44 mAb can induce resistance to IDDM when injected in the prediabetic phase (Fig. 4). This finding shows that the relatively late effector phase of the disease is susceptible to CD44 targeting. Although this information should encourage the clinical application of this experimental approach, we still do not know whether also the induction phase is sensitive to the CD44 targeting procedure. However, because the inflammatory process in the pancreatic islet up-regulates local HA expression, we must assume that the major anti-diabetogenic effect of anti-CD44 antibody interferes with the perturbation of the inflammatory cascade by blocking the HA receptor of newcomer MNCs. (ii) Suppression of diabetes progression by anti-CD44 mAb is correlated with the inhibition of MNCs infiltration into the pancreatic islets, although periislet inflammation around the blood vessels is frequently observed. These findings suggest that the antibody protects the insulin-producing β cells by interfering with the penetration of the inflammatory cells into the islets or with their localization inside the islets. (iii) Whereas the diabetic inflammatory cascade is susceptible to anti-CD44 mAb, the generation of DTH in the same recipient mice and the mitogen-induced proliferative response of their splenocytes are resistant to the inhibitory effect, although both cell activities are CD44-dependent (28, 29). These results imply that the CD44 of inflammatory cells may be more sensitive to targeting by antibody than the CD44 of cells involved in normal immune responses, an observation that may be useful in the clinical setting. (iv) Because both the blocking of cell surface CD44 and the degradation of HA by hyaluronidase impeded diabetes development, it is likely that the interaction between inflammatory cell CD44 and the HA of the islet endothelium and/or islet matrix enables destructive invasion of the islet. (v) The specific epitope targeted by the anti-CD44 mAbs is critical to their anti-diabetogenic effects, as reflected by the marginal activity of IRAWB14, in contrast to that of IM7.8.1. Interestingly, in the collagen-induced arthritis autoimmune model, the disease was further enhanced after IRAWB14 treatment (30). Because IRAWB14 augments the binding of HA to CD44 (13), these findings are hardly surprising.
When considering therapeutic targeting, the diversity of the CD44 molecule, because of its variable region, generated by alternative splicing, has an advantage over other proinflammatory molecules (e.g., l-selectins, integrins, mucosal addressin cell adhesion molecule-1, vascular cell adhesion molecule-1, tumor necrosis factor-α, IFN-γ, and IL-6), which have a much more restricted structure. In this context, CD44 is not “just another” proinflammatory therapeutic target, but it has a unique and independent significance. Using anti-CD44 mAbs directed against a variant CD44 epitope, rather than against a constant epitope, we may be able exclusively or preferentially to target the destructive inflammatory cells, sparing normal cells that express different CD44 isoforms committed to physiological functions. In conclusion, the molecular flexibility of the CD44-receptor provides us with an excellent opportunity to target pathological CD44, while leaving normal CD44 undamaged. This concept is now under study.
Acknowledgments
We thank Mr. Herzel Harrison (Rohnert Park, CA) for his continuously generous support. We also thank Dr. Alexandra Mahler for her editorial assistance and Ms. Sharon Saunders for typing the manuscript. This work was supported by the Szydlowsky Foundation.
Abbreviations
- IDDM
insulin-dependent diabetes mellitus
- CD44v
variant CD44
- HA
hyaluronic acid
- NOD
non-obese diabetic
- IPGTT
intraperitoneal glucose tolerance test
- bHABP
biotinylated HA binding protein
- MNCs
mononuclear cells
- DTH
delayed type hypersensitivity
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Van Noort J M, Amor S. Int Rev Cytol. 1998;178:127–206. doi: 10.1016/s0074-7696(08)62137-3. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch A, Suarez-Pinzon W L. Biochem Pharmacol. 1998;55:1139–1149. doi: 10.1016/s0006-2952(97)00492-9. [DOI] [PubMed] [Google Scholar]
- 3.Wegmann D R. Curr Opin Immunol. 1996;8:860–864. doi: 10.1016/s0952-7915(96)80016-1. [DOI] [PubMed] [Google Scholar]
- 4.Springer T A. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 5.Naor D, Vogt Sionov R, Ish-Shalom D. Adv Cancer Res. 1997;71:241–319. doi: 10.1016/s0065-230x(08)60101-3. [DOI] [PubMed] [Google Scholar]
- 6.Lesley J, Hyman R, Kincade P W. Adv Immunol. 1993;54:271–335. doi: 10.1016/s0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 7.Michie S A, Sytwu H-K, McDevitt J O, Yang X-D. Curr Top Microbiol Immunol. 1998;231:65–83. doi: 10.1007/978-3-642-71987-5_5. [DOI] [PubMed] [Google Scholar]
- 8.Zahalka M A, Naor D. Int Immunol. 1994;6:917–924. doi: 10.1093/intimm/6.6.917. [DOI] [PubMed] [Google Scholar]
- 9.Zahalka M A, Okon E, Gosslar U, Holzmann B, Naor D. J Immunol. 1995;154:5345–5355. [PubMed] [Google Scholar]
- 10.Nedvetzki S, Walmsley M, Alpert E, Williams R O, Feldmann M, Naor D. J Autoimmun. 1999;13:39–47. doi: 10.1006/jaut.1999.0294. [DOI] [PubMed] [Google Scholar]
- 11.Trowbridge I S, Lesley J, Schulte R, Hyman R, Trotter J. Immunogenetics. 1982;15:299–312. doi: 10.1007/BF00364338. [DOI] [PubMed] [Google Scholar]
- 12.Miyake K, Medina K I, Hayashi S-I, Ono S, Hamaoka T, Kincade P W. J Exp Med. 1990;171:477–488. doi: 10.1084/jem.171.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lesley J, He Q, Miyake K, Hamann A, Hyman R, Kincade P W. J Exp Med. 1992;175:257–266. doi: 10.1084/jem.175.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maloney D G, Kaminski M S, Burowski D, Haimovich J, Levy R. Hybridoma. 1985;4:191–209. doi: 10.1089/hyb.1985.4.191. [DOI] [PubMed] [Google Scholar]
- 15.Zahalka M A, Okon E, Naor D. J Immunol. 1993;150:4466–4477. [PubMed] [Google Scholar]
- 16.Kalbfleisch J D, Prentice R L. The Statistical Analysis of Failture Time Data. New York: Wiley; 1980. pp. 16–19. [Google Scholar]
- 17.Gakunga P, Frost G, Shuster S, Gunha G, Formby B, Stern R. Development (Cambridge, UK) 1997;124:3987–3997. doi: 10.1242/dev.124.20.3987. [DOI] [PubMed] [Google Scholar]
- 18.DeGrendele H C, Estess P, Siegelman M H. Science. 1997;278:672–675. doi: 10.1126/science.278.5338.672. [DOI] [PubMed] [Google Scholar]
- 19.Mikecz K, Brennan F R, Kim J H, Glant T T. Nat Med. 1995;1:558–563. doi: 10.1038/nm0695-558. [DOI] [PubMed] [Google Scholar]
- 20.Verdrengh M, Holmdahl R, Tarkowski A. Scand J Immunol. 1995;42:353–358. doi: 10.1111/j.1365-3083.1995.tb03667.x. [DOI] [PubMed] [Google Scholar]
- 21.Wittig B, Schwärzler C, Föhr N, Günthert U, Zöller M. J Immunol. 1998;161:1069–1073. [PubMed] [Google Scholar]
- 22.Brocke S, Piercy C, Steinman L, Weissman I L, Veromaa T. Proc Natl Acad Sci USA. 1999;96:6896–6901. doi: 10.1073/pnas.96.12.6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faveeuw C, Gagnerault M-C, Lepault F. J Immunol. 1994;152:5969–5978. [PubMed] [Google Scholar]
- 24.Milde K F, Alonso M, Kong S S, Alejandro R, Mintz D H, Pastori R L. Diabetes. 1996;45:718–724. doi: 10.2337/diab.45.6.718. [DOI] [PubMed] [Google Scholar]
- 25.Mohamadzadeh M, DeGrendele H, Arizpe H, Estess P, Siegelman M. J Clin Invest. 1998;101:97–108. doi: 10.1172/JCI1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Estess P, DeGrendele H C, Pascual V, Siegelman M H. J Clin Invest. 1998;102:1173–1182. doi: 10.1172/JCI4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campbell I L, Kay T W H, Oxbrow L, Harrison L C. J Clin Invest. 1991;87:739–742. doi: 10.1172/JCI115055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camp R L, Scheynius A, Johansson C, Puré E. J Exp Med. 1993;178:497–507. doi: 10.1084/jem.178.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moll J, Schmidt A, van der Putten H, Plug R, Ponta H, Herrlich P, Zöller M. J Immunol. 1996;156:2085–2094. [PubMed] [Google Scholar]
- 30.Mikecz K, Dennis K, Shi M, Kim J H. Arthritis Rheum. 1999;42:659–668. doi: 10.1002/1529-0131(199904)42:4<659::AID-ANR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]