

Abstract
Considerable evidence indicates that CD4+ T cells are important in the pathogenesis of rheumatoid arthritis (RA), but the antigens recognized by these T cells in the joints of patients remain unclear. Previous studies have suggested that type II collagen (CII) and human cartilage gp39 (HCgp39) are among the most likely synovial antigens to be involved in T cell stimulation in RA. Furthermore, experiments have defined dominant peptide determinants of these antigens when presented by HLA-DR4, the most important RA-associated HLA type. We used fluorescent, soluble peptide–DR4 complexes (tetramers) to detect synovial CD4+ T cells reactive with CII and HCgp39 in DR4+ patients. The CII-DR4 complex bound in a specific manner to CII peptide-reactive T cell hybridomas, but did not stain a detectable fraction of synovial CD4+ cells. A background percentage of positive cells (<0.2%) was not greater in DR4 (DRB1*0401) patients compared with those without this disease-associated allele. Similar results were obtained with the gp39-DR4 complex for nearly all RA patients. In a small subset of DR4+ patients, however, the percentage of synovial CD4+ cells binding this complex was above background and could not be attributed to nonspecific binding. These studies demonstrate the potential for peptide–MHC class II tetramers to be used to track antigen-specific T cells in human autoimmune diseases. Together, the results also suggest that the major oligoclonal CD4+ T cell expansions present in RA joints are not specific for the dominant CII and HCgp39 determinants.
Rheumatoid arthritis (RA) is a disease of unknown etiology characterized by chronic inflammation in multiple joints (1). In a significant fraction of patients, this persistent synovitis leads to destruction of articular cartilage and surrounding structures and is a cause of significant morbidity. Considerable evidence suggests that CD4+ T cells are important in the pathogenesis of this disease (1–3). Importantly, the major genetic contribution to RA involves HLA class II alleles dominated by HLA-DRB1*0401 and *0404 in Caucasian populations, but also including other DR4 subtypes as well as *0101 (4). These alleles all share a sequence motif at positions 67–74 of the third hypervariable region of the DRβ chain, termed the shared epitope (5), which profoundly affects peptide binding and CD4+ T cell recognition (4). Synovial tissue and fluid from inflamed joints of RA patients usually contain large numbers of CD4+ T cells, including activated CD4+ T cells (1–3). Studies of synovial T cells also have demonstrated sets of related oligoclonal CD4+ T cell expansions in individual patients that express highly homologous T cell receptors (TCRs) (6). Based on these findings, it has been hypothesized that disease-associated HLA-DR molecules present arthritogenic cartilage antigens and cause stimulation and expansion of antigen-specific T cells in the joint. This T cell response then drives the inflammatory process.
The stimulating antigens in RA, however, are unknown. One proposed autoantigen is type II collagen (CII), the predominant protein in articular cartilage. Immunization with CII is known to induce arthritis in animals bearing particular major histocompatibility complex (MHC) class II molecules (7). In addition, there is evidence of enhanced humoral and cellular immunity to CII in RA in some studies (8–12). Recently, transgenic expression of DRB1*0401 and *0101 alleles was shown to confer susceptibility to collagen-induced arthritis in mice (13, 14). The major immunodominant determinant in both *0401 and *0101 transgenic mice was localized to peptides within residues 261–273 of human CII (13–15). Although other cartilage or synovial proteins have been proposed as candidate antigens in RA, DR4-binding epitopes have been defined only for human cartilage glycoprotein 39 (HCgp39) (16, 17). This protein is secreted by synovial cells and articular chondrocytes and is abundant in inflamed joints. Similar to CII, HCgp39 induces arthritis in mice after immunization (16). In addition, peripheral blood T cells from RA patients have been shown to respond to this protein (16). The predominant epitope recognized by T cells in DR4 patients was encompassed within residues 263–275. This epitope is identical to the dominant epitope defined in mice transgenic for DR*0401 after immunization with native HCgp39 (17).
Recently, multimeric peptide–MHC complexes (tetramers) have been developed that have the capacity to bind to antigen-specific T cells (18, 19). The ability to be stimulated by antigen is not required for an antigen-specific T cell to bind these tetramers. In this regard, studies using peptide–MHC class I tetramers have demonstrated that methods relying on the function of antigen-reactive T cells, such as limiting dilution analysis, greatly underestimate the size of the antigen-specific population in peripheral blood (18, 20–22). Here, we report an attempt to use peptide–MHC class II tetramers to identify antigen-reactive CD4+ T cells in the synovial fluid of patients with RA. The results show that patients with established RA do not have detectable frequencies of synovial fluid CD4+ T cells reactive with the dominant CII epitope. Although nearly all RA patients also have undetectable frequencies of T cells reactive with the dominant HCgp39 epitope, a few samples with levels above background were found.
Materials and Methods
Patients and HLA-DR Typing.
Patients were referred initially by rheumatologists on the basis of having RA and the availability of synovial fluid for analysis. Synovial fluid samples were obtained for clinical indications. All patients enrolled met the American College of Rheumatology criteria for the classification of RA (23) and were studied under the guidelines of the human subjects institutional review boards at the National Jewish Medical and Research Center (Denver). In general, patients with established severe disease and with evidence of active inflammatory arthritis were chosen for study. Mononuclear cells from peripheral blood and synovial fluid were isolated by Ficoll/Hypaque density gradient centrifugation (Pharmacia) and cryopreserved until analysis. HLA-DRB1 typing was performed by using standard molecular typing techniques at the University of Colorado Health Sciences Center Clinical Immunology and Histocompatibility Laboratory.
T Cell Hybridomas.
Mice transgenic for DRB1*0401 were immunized with human CII, and draining lymph node cells were used to generate CII-specific T cell hybridomas as described previously (14). Two hybridomas (DR4hCII-38.8 and DR4hCII-11.5) specific for the dominant determinant, CII(263–270), were used in the current study.
T cell hybridomas also were generated by transfection to express the TCR genes of clonal CD4+ expansions identified in RA synovial cells (6). The TCR α- and β-chain variable-region gene fragments were amplified from cellular RNA by reverse transcription–PCR with 5′ primers specific for the TCR gene leaders and antisense primers specific for junctional sequences of the clonal expansions. These primers also included restriction sites to permit insertion of the amplified fragments into a TCR β-chain expression vector, pHBAcPrMCβ2, adjacent to murine Cβ2 or a TCR α-chain expression vector, pSFFMCα, adjacent to murine Cα (E. Boen, A. Crownover, M. McIlhaney, A. Korman, and J.B., unpublished results). The chimeric TCR genes were introduced into the TCR-negative and human CD4-expressing T cell hybridoma 54ζ by electroporation as described (24). Transfectants were screened for surface expression of TCR and verified to be functional as determined by release of IL-2 after stimulation with an appropriate Vβ-selective superantigen presented by autologous Epstein–Barr virus-transformed B cells. All of the transfected hybridomas used in the present study also were tested for stimulation to CII(258–272) and HCgp39(263–275) peptides in the context of DR4 (*0401)-expressing antigen-presenting cells, and none responded (data not shown).
Preparation of MHC Class II Molecules with Covalent Peptides and Fluorescent, Soluble, Multivalent Peptide–MHC Complexes.
Soluble MHC class II molecules were prepared by using a baculovirus transfer vector containing separate cloning sites for expressing the class II α-chain under the control of the P10 promoter and the β-chain under the control of the polyhedrin promoter in the same virus (25). Portions of the HLA-DRA1 and HLA-DRB1*0401 genes encoding the extracellular domains were cloned in this vector as indicated in Fig. 1. Several modifications were made to the β-chain gene, also shown in Fig. 1. The natural signal peptide was replaced with that of the β-chain of the mouse IEk molecule. Sequence encoding any of three different DR4-binding peptides and a flexible linker containing a thrombin cleavage site was inserted between the signal peptide and the N terminus of the mature β-chain. The amino acids of the three peptides corresponded to CII (259–272), HCgp39 (263–275), and Borrelia burgdorferi outer surface protein A (OspA) (164–176), which encompass previously described dominant epitopes (14, 16, 17, 26). In addition, sequence encoding a peptide that could be biotinylated with the Escherichia coli enzyme BirA was attached to the 3′ end of the gene (19, 27). The complete constructions were incorporated into baculovirus by using the BaculoGold system (PharMingen), and high-titered virus was prepared.
Figure 1.

Expression of DR4 in baculovirus. The HLA-DRA1 and DRB1*0401 genes were cloned into a previously described baculovirus transfer vector (25). Modifications were made as described in the text to add covalently attached antigenic peptides (25) and a peptide tag for biotinylation by the enzyme BirA (19).
Soluble DR4 proteins containing the different peptides were purified from the supernatants of infected High5 insect cells (Invitrogen) by immunoaffinity chromatography using the anti-DRα mAb LB3.1 (American Type Culture Collection, Manassas, VA) and size-exclusion chromatography by using Superdex-200 (Pharmacia). The proteins were biotinylated with BirA (Avidity, Denver) and incorporated into saturated complexes with phycoerythrin (PE)-streptavidin (BioSource International, Camarillo, CA) as described previously (19).
Immunofluorescence Analysis for Peptide–DR4 Complex Binding.
The binding of multivalent peptide–DR4 complexes (tetramers) to T cell hybridomas was performed as described (19). Based on these previous studies, T cells usually were incubated with 20 μg/ml tetramer for 3 hr at 37°C. In some studies, concentrations as high as 40 μg/ml were studied. In an attempt to enhance peptide–MHC tetramer binding, separate samples also included a mAb directed to murine Cβ (H597; ref 28) at 2 μg/ml during the incubation period. In separate wells, biotinylated H597 was used to quantitate surface TCR expression of the T cell hybridomas and was detected with PE-avidin (Southern Biotechnology Associates).
The percentages of CD4+ T cells in peripheral blood or synovial fluid staining with peptide–DR4 complexes or anti-Vβ mAbs were determined by using three-color immunofluorescence analysis. CD4 expression was determined by using a PerCP-conjugated mAb to CD4 (Becton Dickinson). Monocytes/macrophages, the predominant source of nonspecific staining with peptide–DR4 complexes, were stained by using a fluorescein-conjugated mAb to CD14 (Ancell, Bayport, MN). Cells (0.5–1 × 106 in 50 μl of complete medium) were incubated with peptide–DR4 complexes at 20 μg/ml for 3 hr at 37°C, previously determined to be optimal conditions for specific binding to TCR (19). Separate aliquots of synovial cells were coincubated with peptide–MHC complexes and anti-CD3 (OKT3) used at 2 μg/ml. Cells subsequently were incubated with mAbs to CD4 and CD14 for 30 min at 4°C. Separate aliquots of all samples, in which the peptide–DR4 complex was omitted during the initial 3-hr incubation, also were stained with biotinylated mAb to TCR Vβ14 and/or Vβ17 followed by PE-avidin as described (6).
The viable mononuclear cell population was identified by using forward and 90° light-scatter patterns and analyzed for fluorescence intensity with a FACScan (Becton Dickinson). In general, the data are shown for viable cells that do not express CD14 and presented as the percentage of CD4+ cells staining with peptide–MHC or anti-Vβ. For most synovial samples, greater than 40 × 103 events were analyzed for each reagent combination.
Results
Specific Binding of Collagen Type II Peptide–DR4 Tetramers to T Cell Hybridomas.
Soluble DR4 (DRB1*0401) bearing a covalently attached peptide from either human CII, HCgp-39, or the outer surface protein A (Osp-A) of B. Burgdorferi were prepared. A biotinylated peptide tag on the DRβ-chain C terminus was included in the construct to allow tetramer formation at a later time. To test the binding and specificity of the covalent CII-DR4 complex, we used T cell hybridomas (DR4hCII-36.8 and 11.5) derived from DR4-transgenic mice after immunization with human CII (14). Previous studies determined that these hybridomas responded to the major immunodominant determinant, CII(263–270). In the presence of presenting cells bearing DR*0401/*0404, these hybridomas released large amounts of IL-2 in response to this peptide at concentrations as low as 1.0 μg/ml, whereas control T hybridomas (with TCRs derived from RA synovial CD4+ T cell expansions) showed no response in cultures with CII peptide concentrations as high as 100 μg/ml (data not shown). Incubation of the CII-specific hybridomas in wells precoated with avidin and bound with biotinylated CII-DR4 at concentrations as low as 20 ng/ml resulted in a dose-dependent stimulation and production of IL-2 (data not shown). This response was specific in that the same hybridomas did not release IL-2 in avidin-coated plates bound with biotinylated gp39-DR4 or OspA-DR4 at concentrations as high as 2,000 ng/ml. Control hybridomas failed to respond to any of the peptide–DR4 preparations.
Tetramers were prepared by binding PE-streptavidin to the biotinylated peptide–DR4 molecules. The same CII-reactive hybridomas then were stained with various concentrations of these complexes (Fig. 2). Specific staining was observed at 20 μg/ml, determined in previous studies to be near optimal for immunofluorescence staining with similarly designed tetramers (19), and doubling of this concentration resulted in only slightly more immunofluorescence intensity. Neither of the two CII-specific hybridomas showed any staining with the gp39-DR4 tetramer (Fig. 2) or OspA-DR4 tetramer (data not shown) used at equivalent concentrations. Of note, hybridoma DR4hCII-11.5, compared with 36.8, showed less intense staining with the CII-DR4 tetramer, but tetramer binding was enhanced significantly by coincubating with an anti-TCR mAb (H597) (Fig. 2). This enhancement of staining was specific in that no increase in binding was observed for the other peptide–DR4 tetramers. In separate studies, we noted that the addition of anti-CD3 mAb also resulted in enhancement of CII-DR4 tetramer binding (data not shown). This finding provided the rationale for coincubating RA synovial cells with tetramer and anti-CD3 mAb to try to enhance tetramer binding (see below). Fig. 2 also shows that two control T cell hybridomas, with TCRs derived from RA synovial CD4+ clonal expansions, failed to stain differentially with any of the tetramers tested, despite equivalent or higher TCR levels compared with the CII-specific hybridomas.
Figure 2.
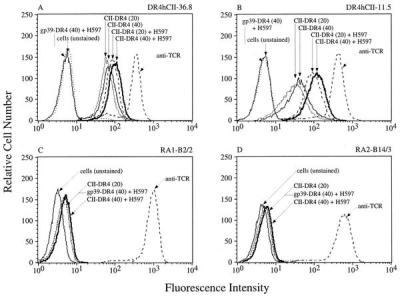
Staining of T cell hybridomas with the CII-DR4 and gp39-DR4 tetramers. Two CII-specific hybridomas are shown in A and B. Control T cell hybridomas in C and D were transfected to express TCRs of RA synovial clonal expansions and do not recognize CII (258–272) or HCgp39(263–275). All hybridomas were incubated with the indicated peptide–DR4 complexes at 20 μg/ml or 40 μg/ml with or without the anti-TCR mAb, H597. In separate wells, TCR expression was determined with biotinylated H597 followed by PE-avidin.
Search for CII and HCgp39 Peptide-Specific T Cells in RA Patients.
The clinical characteristics and HLA-DR types of the RA patients studied are shown in Table 1. In general, patients with established severe disease and with evidence of active inflammatory arthritis were chosen for study. The patients were divided into two groups based on the presence of a DRB1*0401 allele, without knowledge of any results of synovial fluid analyses. Four patients in group 1 harbored two RA-associated DR alleles, and four patients in group 2 carried single copies of RA-associated alleles that were not *0401 (Table 1). Probably related to their DR haplotypes (3, 4), patients in group 1 tended to have more severe disease with a higher frequency of positive rheumatoid factor tests, rheumatoid nodules, and joint destruction. Still, the two groups appeared to be reasonably well matched for these disease characteristics. Nearly all patients in both groups were on multiple medications including low-dose prednisone, nonsteroidal antiinflammatory drugs, methotrexate, as well as various other disease-modifying antirheumatic disease drugs.
Table 1.
Clinical characteristics of patients studied in relation to HLA-DR typing
| Characteristic | Group 1, *0401-positive (n = 10) | Group 2, *0401-negative (n = 10) |
|---|---|---|
| HLA-DRB1 typing | ||
| Allele 1 | All *0401 | No *0401 |
| Allele 2 | 2 *0101, 2 *0404 | 2 *0101, 1 *0404, 1 *0405 |
| Mean years of age (range) | 54 (35–73) | 56 (31–76) |
| Female/male | 6/4 | 5/5 |
| Mean years of disease (range) | 13.8 (5–28) | 10.9 (1.5–32) |
| Rheumatoid factor (present/absent) | 9/1 | 7/3 |
| Rheumatoid nodules (present/absent) | 6/4 | 3/7 |
| Joint destruction† (present/absent) | 10/0 | 7/3 |
Destruction refers to the presence of bone erosions on joint radiographs and/or deformities on physical examination.
Initial studies of peripheral blood cells from RA patients or healthy individuals did not show detectable frequencies of CD4+ T cells binding any of the tetramers (data not shown). We therefore focused our analyses on synovial fluid T cells, which seemed likely to have the highest frequency of cartilage antigen-specific cells in these patients. Fig. 3 shows a representative analysis of patients' cells after staining with the CII-DR4 and gp39-DR4 tetramers. Tetramer-staining cells essentially were undetectable in both synovial fluid samples, with detection limits in the range of 1 in 1,000 CD4+ T cells. Identical results were obtained when cells were coincubated with tetramer and anti-CD3 during the staining period (Fig. 3).
Figure 3.

Staining of RA synovial fluid cells with peptide–DR4 tetramers. Representative flow cytometric analyses are shown for synovial fluid cells from patients RA-6 (*0401/*1401) and RA-10 (*0101/*1601) stained with CII-DR4 (Top), gp39-DR4 (Middle), or anti-TCR Vβ17 (Bottom). For each histogram, CD14+ cells have been excluded, and the results are shown separately for the CD4+ and CD4− populations. Results are also shown for separate samples in which anti-CD3 was coincubated with tetramer. Approximately 50 × 103 cells were analyzed for each reagent combination. The gates used to determine positive cells are shown, and the percentages of cells staining with tetramer or anti-Vβ17 mAb are indicated in parentheses.
True peptide–DR4 tetramer binding T cells should be confined to the CD4+ T cell population. Therefore, the CD4− population provides an internal negative control for possible low frequency staining. In the example shown in Fig. 3, the frequency of CII-DR4- or gp39-DR4-positive cells was at least as great in the CD4− vs. CD4+ population, and this was true regardless of the threshold used to define tetramer-positive staining. For all *0401 patients (group 1), the mean percentage (±SE) of cells staining positive with the CII-DR4 tetramer in the CD4+ vs. CD4− population was 0.07 ± 0.02 vs. 0.17 ± 0.05, respectively (Fig. 4A). Furthermore, no individual in this group showed an increased frequency greater than 0.01% in the CD4+ vs. CD4− population. Comparison of gp39-DR4 tetramer staining in the CD4+ vs. CD4− populations of group 1 showed similar results overall (Fig. 4B). One sample from group 1 and two from group 2, however, showed an increase of positive cells in the CD4+ vs. CD4− population by greater than 0.2% (see below).
Figure 4.
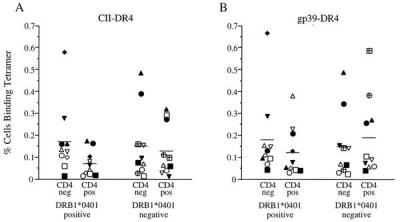
Percentages of synovial cells binding tetramer. The data are shown for CD4+ CD14− and CD4− CD14− cells staining with CII-DR4 (A) or gp39-DR4 (B) complexes. Results for RA patients in group 1 (*0401-positive) and group 2 (*0401-negative) are presented separately, and within each group, the same symbol is used for synovial cells obtained from an individual patient. Horizontal lines indicate the mean values for each group. Two individuals in group 2 (circle with plus sign; square with plus sign) carry non-*0401 DR4-susceptibility alleles (*0404 and *0405).
The results for all patients are shown in Fig. 4, in which synovial T cells from the two groups of patients are compared. Overall, no increased frequency of CD4+ cells binding the CII-DR4 tetramer was found in synovial fluid from the *0401 patients (mean percentage ± SE was 0.07 ± 0.02 vs. 0.13 ± 0.04 in group 1 vs. group 2) (Fig. 4A). Of note, the highest absolute percentage of CII-tetramer-positive cells in the CD4+ population of group 1 was 0.17%, a frequency of less than 1 in 500 CD4+ T cells. Higher values were noted in three *0401-negative patients, but binding appeared to be nonspecific. Thus, the only individual with significantly more staining in the CD4+ population was homozygous for DRB1*1501, an allele unrelated to *0401 or RA susceptibility. Anti-CD3 coincubation did not result in a significant increase in CII-DR4 tetramer-positive cells in any patient in the *0401 group (data not shown).
A similar low frequency of positive cells was detected in most of the synovial samples stained with the gp39-DR4 tetramer (Fig. 4B). The mean frequency of positive CD4+ cells was no greater in the *0401-positive compared with the *0401-negative group (0.12 ± 0.04 vs. 0.19 ± 0.06). The percentage of synovial CD4+ cells in one *0401-positive patient was noted to be 0.38%. Although a relatively low percentage, this value was greater than the percentage staining with the CII-DR4 tetramer (0.04%) in the same sample and greater than the percentage of positive cells in the CD4− population (0.15%). Interestingly, two *0401-negative patients also showed relatively high percentages of positive gp39-DR4 cells (Fig. 4B). DR typing revealed that one of these patients was *0404 and the other was *0405. Although these positive cells stained at low density, a differential increase was noted for gp39-DR4 vs. CII-DR4 tetramer staining and for CD4+ vs. CD4− populations in each patient (Fig. 4B).
Discussion
The rationale for using peptide–MHC class II tetramers to investigate the antigen specificity of synovial CD4+ T cells in RA is based on considerable evidence. For example, there is a strong association of certain DR4 alleles, such as *0401, with disease susceptibility and progression (3–5), and the leading hypotheses to explain these associations involve CD4+ T cell recognition of DR4-presented peptides. Experimental support for CII and HCgp39 as autoantigens in RA also has been provided, and it seemed most likely that CD4+ T cells reactive with these antigens would be enriched in synovium vs. peripheral blood. Importantly, despite the abundance of CD4+ T cells in synovial tissue and fluid, their responses to antigen have been difficult to study, at least in part, because of various functional defects (29). These functional deficiencies can be circumvented by the use of peptide–MHC tetramers, which bind to cells based on the level and affinity of expressed TCRs (19) and therefore independently of cellular function. Studies of peripheral blood CD8+ T cells in patients with various infectious diseases have shown that peptide–MHC class I tetramers identify a much larger number of antigen-specific cells compared with assays that rely on cell function (18, 20–22). Although peptide–MHC class II tetramers have not been used previously to enumerate CD4+ T cells in human autoimmune diseases such as RA, the OspA-DR4 complex described herein was used recently to identify Osp-A-reactive cells in the synovial fluid of patients with chronic, antibiotic-resistant Lyme arthritis (A. L. Meyer, C. Trollmo, F.C., P.M., A. C. Steere, D. A. Hafler, J.K., and B. T. Huber, unpublished results).
Studies in our laboratory and others' have demonstrated that RA synovial fluid contains multiple, expanded clonal CD4+ populations, which usually are not detectable in the peripheral blood of the same patients (ref. 6; reviewed in refs. 3 and 30). Although possibly enriched in certain TCR Vβ-expressing subsets, most subsets examined thus far harbor such expansions. Furthermore, a number of clonal expansions occupy more than 1% of the total synovial CD4+ cells, including several found in different individual DR4 patients studied in the current analysis (6). The expression of related TCRs among different clones and persistence of the same synovial T cell clones in individual patients suggest that a significant proportion of these T cells have been selected and stimulated in the joint by conventional peptide antigens. Results from the present analysis with peptide–DR4 tetramers strongly suggest that nearly all of these expanded synovial CD4+ clones are not specific for the dominant determinants of CII or HCgp39.
The CII-DR4 covalent complex bound in a specific manner to CII-specific T cell hybridomas. Using this complex, we should have been able to detect frequencies of tetramer-positive cells in synovial fluid or peripheral blood as low as 1 in 500 (0.2%). The ability to detect such a low frequency of positive cells was greatly aided by the exclusion of monocytes/macrophages in the immunofluorescence analysis. These phagocytic cells tended to nonspecifically bind and probably internalize the tetramers during the incubation. For the CII-DR4 tetramer, the frequencies of positive CD4+ cells were less than 0.2% for every DR4 patient studied. Importantly, even this low level appeared to reflect nonspecific binding in that staining was extremely low-density, the CD4− population contained as many positive cells as the CD4+ population, and values for DR4 patients were no greater than for those not expressing DR4. Still, it is possible that our analysis may have missed CII-reactive T cells in RA. Previous studies have shown that the intensity of binding with peptide–MHC class II tetramers is dependent on the affinity of TCR for peptide–MHC. If synovial CII-specific T cells were of uniformly low affinity, they might not have been detected. We tried to compensate for this possibility by coincubating the tetramers with anti-CD3, which appears to enhance tetramer binding to low-affinity T cell hybridomas.
We also may have failed to find CII-specific T cells because this study addressed RA patients with established severe disease. The shortest duration of disease for a DR4 patient in the present study was 5 years. It is possible that CII-specific T cells might be more apparent in patients closer to the onset of disease. Finally, despite the results obtained from DR4-transgenic mice and peptide–DR4-binding studies, it is possible that a totally separate CII epitope is recognized in RA patients.
Although similar results were obtained when staining synovial cells with the gp39-DR4 tetramer, our conclusions are more tentative for several reasons. First, we have not been able to test the covalent gp39-DR4 molecule or multivalent complex for activity with a set of T cells or hybridomas specific for this epitope. We also noted three synovial samples with slightly higher frequencies of tetramer-binding cells in the CD4+ population. Only one of these patients was in the group with *0401 alleles (matching the tetramer MHC molecule). The other individuals carried DR4 susceptibility alleles (*0404 and *0405), the only patients to do so in group 2. The rationale for originally dividing the patients as we did was based on studies showing that some T cell hybridomas specific for CII and HCgp39 and restricted by *0401 may not respond to the same peptide presented by *0404- or *0405-encoded molecules (14, 17). It seems likely, however, that these closely related DR4 molecules may cross-present peptides to a subset of antigen-specific CD4+ T cells originally restricted by a different DR4 allele. A higher frequency of tetramer-positive cells was noted in the CD4+ vs. CD4− populations in each of these individuals. In addition, the CII-DR4 tetramer staining of the same samples showed only background levels of binding. Still, it is emphasized that in these few samples, the frequency of positive cells and the staining density per cell were low. Sorting the tetramer-staining cells from synovial samples to develop T cell clones should determine whether they are indeed specific for this HCgp39 epitope.
Acknowledgments
These studies were supported in part by National Institutes of Health Grants AI-17134, AI-22295, and AI18785 and General Clinical Research Center Grant M01-RR-00051 from the Division of Research Resources.
Abbreviations
- RA
rheumatoid arthritis
- CII
human type II collagen
- HCgp39
human cartilage glycoprotein 39
- TCR
T cell receptor
- PE
phycoerythrin
References
- 1.Kavanaugh A F, Lipsky P E. In: Clinical Immunology: Principles and Practice. Rich R R, Fleisher T A, Schwartz B D, Shearer W T, Strober W, editors. St. Louis: Mosby; 1996. pp. 1093–1115. [Google Scholar]
- 2.Panayi G S, Lanchbury J S, Kingsley G H. Arthritis Rheum. 1992;35:729–735. doi: 10.1002/art.1780350702. [DOI] [PubMed] [Google Scholar]
- 3.Falta M T, Kotzin B L. In: T Cells in Arthritis. Miossec P, van den Berg W B, Firestein G S, editors. Basel: Birkhauser; 1998. pp. 201–231. [Google Scholar]
- 4.Nepom G T. Adv Immunol. 1998;68:315–332. doi: 10.1016/s0065-2776(08)60563-5. [DOI] [PubMed] [Google Scholar]
- 5.Gregersen P K, Silver J, Winchester R J. Arthritis Rheum. 1987;30:1205–1213. doi: 10.1002/art.1780301102. [DOI] [PubMed] [Google Scholar]
- 6.Striebich C C, Falta M T, Wang Y, Bill J, Kotzin B L. J Immunol. 1998;161:4428–4436. [PubMed] [Google Scholar]
- 7.Stuart J M, Townes A S, Kang A H. J Clin Invest. 1982;69:673–683. doi: 10.1172/JCI110495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee S, Luthra H S, Moore S B, O'Fallon W M. Clin Exp Rheumatol. 1988;6:373–380. [PubMed] [Google Scholar]
- 9.Stuart J M, Huffstutter E H, Townes A S, Kang A H. Arthritis Rheum. 1983;26:832–840. doi: 10.1002/art.1780260703. [DOI] [PubMed] [Google Scholar]
- 10.Londei M, Savill C M, Verhoef A, Brennan F, Leech Z A, Duance V, Maini R N, Feldmann M. Proc Natl Acad Sci USA. 1989;86:636–640. doi: 10.1073/pnas.86.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snowden N, Reynolds I, Morgan K, Holt L. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 12.Kim H-Y, Kim W-U, Cho M-L, Lee S K, Youn J, Kim S-I, Yoo W-H, Park J-H, Min J-K, Lee S-H, Park S-H, Cho C-S. Arthritis Rheum. 1999;42:2085–2093. doi: 10.1002/1529-0131(199910)42:10<2085::AID-ANR8>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 13.Rosloniec E F, Brand D D, Myers L K, Whittington K B, Gumanovskaya M, Zaller D M, Woods A, Altmann D M, Stuart J M, Kang A H. J Exp Med. 1997;185:1113–1122. doi: 10.1084/jem.185.6.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosloniec E F, Brand D D, Myers L K, Esaki Y, Whittington K B, Zaller D M, Woods A, Stuart J M, Kang A H. J Immunol. 1998;160:2573–2578. [PubMed] [Google Scholar]
- 15.Fugger L, Rothbard J B, Sonderstrup-McDevitt G. Eur J Immunol. 1996;26:928–933. doi: 10.1002/eji.1830260431. [DOI] [PubMed] [Google Scholar]
- 16.Verheijden G F, Rijnders A W, Bos E, Coenen-de Roo C J, van Staveren C J, Miltenburg A M, Meijerink J H, Elewaut D, de Keyser F, Veys E, Boots A M. Arthritis Rheum. 1997;40:1115–1125. doi: 10.1002/art.1780400616. [DOI] [PubMed] [Google Scholar]
- 17.Cope A P, Patel S D, Hall F, Congia M, Hubers H A, Verheijden G F, Boots A M H, Menon R, Trucco M, Rijnders A W, Sonderstrup G. Arthritis Rheum. 1999;42:1497–1507. doi: 10.1002/1529-0131(199907)42:7<1497::AID-ANR25>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Altman J D, Moss P A H, Goulder P J R, Barouch D H, McHeyzer-Williams M G, Bell J I, McMichael A J, Davis M M. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 19.Crawford F, Kozono H, White J, Marrack P, Kappler J. Immunity. 1998;8:675–682. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 20.Callan M F, Tan L, Annels N, Ogg G S, Wilson J D, O'Callaghan C A, Steven N, McMichael A J, Rickinson A B. J Exp Med. 1998;187:1395–1402. doi: 10.1084/jem.187.9.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieganowska K, Hollsberg P, Buckle G J, Lim D G, Greten T F, Schneck J, Altman J D, Jacobson S, Ledis S L, Hanchard B, et al. J Immunol. 1999;162:1765–1771. [PubMed] [Google Scholar]
- 22.Tan L C, Gudgeon N, Annels N E, Hansasuta P, O'Callaghan C A, Rowland-Jones S, McMichael A J, Rickinson A B, Callan M F. J Immunol. 1999;162:1827–1835. [PubMed] [Google Scholar]
- 23.Arnett F C, Edworthy S M, Bloch D A, McShane D J, Fries J F, Cooper N S, Healey L A, Kaplan S R, Liang M H, Luthra H S, et al. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 24.Vollmer J, Weltzien H U, Moulon C. J Immunol. 1999;163:2723–2731. [PubMed] [Google Scholar]
- 25.Kozono H, White J, Clements J, Marrack P, Kappler J. Nature (London) 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- 26.Gross D M, Forsthuber T, Tary-Lehmann M, Etling C, Ito K, Nagy Z A, Field J A, Steere A C, Huber B T. Science. 1998;281:703–706. doi: 10.1126/science.281.5377.703. [DOI] [PubMed] [Google Scholar]
- 27.Schatz P J. Bio/Technology. 1993;11:1138–1143. doi: 10.1038/nbt1093-1138. [DOI] [PubMed] [Google Scholar]
- 28.Kubo R T, Born W, Kappler J W, Marrack P, Pigeon M. J Immunol. 1989;142:2736–2642. [PubMed] [Google Scholar]
- 29.Maurice M M, Lankester A C, Bezemer A C, Geertsma M F, Tak P P, Breedveld F C, van Lier R A, Verweij C L. J Immunol. 1997;159:2973–2978. [PubMed] [Google Scholar]
- 30.Struyk L, Hawes G E, Chatila M K, Breedveld F C, Kurnick J T, van den Elsen P J. Arthritis Rheum. 1995;38:577–589. doi: 10.1002/art.1780380502. [DOI] [PubMed] [Google Scholar]