

Abstract
LFA-1 (CD11a/CD18) is a member of the β2-integrin family of adhesion molecules important in leukocyte trafficking and activation. Although LFA-1 is thought to contribute to the development of experimental autoimmune encephalomyelitis (EAE) primarily through its functions on effector T cells, its importance on other leukocyte populations remains unexplored. To address this question, we performed both adoptive transfer EAE experiments involving CD11a-/- mice and trafficking studies using bioluminescent T cells expressing luciferase under the control of a CD2 promoter (T-lux cells). Transfer of encephalitogenic CD11a-/- T cells to wild type mice resulted in a significant reduction in overall EAE severity compared to control transfers. We also observed, using in vivo imaging techniques, that CD11a-/- T-lux cells readily infiltrated lymph nodes and the CNS of wild type recipients with kinetics comparable to CD11a+/+ transfers, although their overall numbers in these organs were reduced. Surprisingly, transfer of encephalitogenic wild type T cells to CD11a-/- mice induced a severe and sometimes fatal EAE disease course, associated with massive T cell infiltration and proliferation in the CNS. These data indicate that LFA-1 expression on leukocytes in recipient mice plays an important immunomodulatory role in EAE. Thus, LFA-1 acts as a key regulatory adhesion molecule during the development of EAE, serving both pro- and anti-inflammatory roles in disease pathogenesis.
Keywords: adhesion molecules, β2-integrins, T lymphocytes, bioluminescence, neuroimmunology
1. Introduction
Multiple sclerosis (MS) is an inflammatory CNS disease in which leukocytes, including T cells, migrate into the CNS to initiate tissue destruction. These autoreactive T cells are activated outside the CNS and migrate across the blood brain barrier (BBB) into the CNS parenchyma through the use of a well-established adhesion cascade that utilizes members of several adhesion molecule families and their ligands (reviewed in (Archelos et al., 1999; Engelhardt, 2006a; Engelhardt, 2006b; Ley et al., 2007; Luster et al., 2005). In experimental autoimmune encephalomyelitis (EAE), the animal model for MS, VLA-4 and several members of the β2-integrin family of adhesion molecules have been implicated as key components required for trafficking across the BBB and thereby contributing to disease development (Bullard et al., 2007a; Bullard et al., 2005; Laschinger et al., 2002; Wang et al., 2007; Yednock et al., 1992). The β2-integrin, LFA-1 (CD11a/CD18) is expressed on all leukocytes and has been extensively investigated in EAE and other MS model systems. However, the role of this adhesion molecule in EAE pathogenesis is controversial and conflicting results have been reported (Cannella et al., 1993; Gordon et al., 1995; Wang et al., 2007; Welsh et al., 1993; Willenborg et al., 1996). For example, Welsh et al. (Welsh et al., 1993) () found that treatment of mice with an anti-LFA-1 antibody resulted in a more severe inflammation and demyelination during EAE, while another study showed that CD11a-/- mice were significantly protected from disease (Wang). Although some of these different findings may result from technical differences in the induction or evaluation of EAE, they may also indicate that LFA-1 serves both pro- and anti-inflammatory roles during the initiation and progression of CNS inflammatory events.
In this report, we used both adoptive transfer and bioluminescent imaging techniques to further address the specific requirements for LFA-1 expression on both effector T cells and other leukocyte subsets during EAE development. We found that transfer of CD11a-/- MOG sensitized T cells to wild type recipients resulted in a significant, but incomplete attenuation of EAE, suggesting that LFA-1-independent adhesion mechanisms can also mediate effector T cell recruitment and activation. Surprisingly, the reciprocal transfer of wild type encephalitogenic T cells to CD11a-/- recipients resulted in exacerbated EAE and increased mortality. Using a recently developed T cell-specific transgenic luciferase reporter mouse (T-lux mice, (Azadniv et al., 2007)), we observed that both wild type and CD11a-/- MOG-sensitized bioluminescent T cells showed similar trafficking kinetics to the cervical lymph nodes, tail, brain and spinal cord in wild type recipients; however, the overall numbers of LFA-1 deficient T cells recruited to these organs was reduced. In contrast, wild type encephalitogenic T-lux cells showed a rapid accumulation in lymphoid organs and spinal cords following transfer into LFA-1 deficient mice. These results collectively demonstrate that LFA-1 expression on effector T cells, but not on other leukocyte subsets such as macrophages or dendritic cells, is required for the development of severe EAE. Furthermore, they indicate that this adhesion molecule also serves an additional regulatory role and acts to restrict effector T cell responses leading to CNS inflammation and demyelination.
2. Materials and Methods
Mice
LFA-1 deficient mice have been previously described (Ding et al., 1999). For all studies, we used CD11a-/- or CD11a +/- mice at an N16 backcross generation onto C57BL/6. The luciferase transgenic mouse line (T-lux), expressing firefly luciferase under the control of the human CD2 promoter, was generated in the C57BL/6 background as previously described (Azadniv et al., 2007). T-lux mice express luciferase activity in all CD3+ cells and bioluminescence generated by this enzyme is directly proportional to the number of cells expressing the gene, allowing real-time assessment of T cell proliferation and migration in vivo. For some experiments, LFA-1 deficient T-lux transgenic mice were used as sources of donor T cells and were generated by intercrossing the CD11a mutant and T-lux mice described above. Inbred T-lux transgenic or non-transgenic C57BL/6 mice were used as controls for all experiments. All studies were performed with approval from the UAB IACUC.
Induction of active and transferred EAE
For active EAE, control, CD11a−/− and CD11a+/− mice were immunized with MOG peptide35-55 (Biosynthesis, Lewisville, TX) as described (Bullard et al., 2007a). Onset and progression of EAE signs were monitored daily using a standard clinical scale ranging from 0 to 6 as follows: 0, asymptomatic; 1, loss of tail tone; 2, flaccid tail; 3, incomplete paralysis of one or two hind limbs; 4, complete hind limb paralysis; 5, moribund; and 6, dead. Only mice with a score of at least 2 (flaccid tail) for more than 2 consecutive days were judged to have onset of EAE. For each animal a cumulative disease index was calculated from the sum of the daily clinical scores observed between day 7 and day 30. To prepare encephalitogenic cells for adoptive transfer of EAE, mice were immunized as for active EAE, except they received only one PT injection. Spleens and lymph nodes were collected 8 days p.i., single cell suspensions were prepared, and RBCs were lysed. Cells (6 × 106 cells/ml) were cultured in RPMI 1640 medium (supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 IU/ml penicillin/streptomycin, and 2 × 10−5 M 2-ME, Invitrogen Life Technologies) with MOG35–55 (20 μg/ml) and IL-12 (30 ng/ml, R&D Systems) as previously described (Liu et al., 2006). Three days after initiation of culture, the cells were harvested, washed in PBS, and injected into recipient mice that were sublethally irradiated (500 rad) within 16 h before adoptive transfer. Onset and progression of EAE signs were monitored as for active EAE.
Imaging of live mice
Mice were subjected to bioluminescent imaging as previously described (Azadniv et al., 2007). Briefly, mice were anesthetized with isofluorane and placed in a light-tight chamber. The photographic (gray-scale) reference image was obtained at 10 minutes after D-luciferin injection; the bioluminescent image was collected immediately thereafter. Images were obtained with a CCD camera cooled to −120°C, using the IVIS Imaging System (Xenogen Corp., Alameda, CA) with the field of view set at 10 cm height. The photographic images were taken at a 0.2 second exposure, 8f/stop, 2 binning (resolution), and an open filter. The bioluminescent images used exposures ranging from 120 to 600 seconds, 1f/stop, 8 binning and open filter. The bioluminescent and gray-scale images were overlaid using Living Image software (Xenogen Corp.). Igor image analyses software (Wavemetrics, Lake Oswego, OR) was employed to obtain a pseudocolor image representing bioluminescence intensity (blue, least intense; red, most intense). The total counts were normalized to image acquisition. Ex vivo images were obtained by removal of brain and spinal cord from wild type to wild type and wild type to CD11a-/- transfers. Tissues were imaged at 3X magnification after treatment with the Luciferase Assay system (Promega).
Statistics
Statistical significance between controls, CD11a-/- and CD11a+/- mice for active and transferred EAE experiments was calculated using the Wilcoxon signed-rank test. Statistical significance between transferred EAE groups in the bioluminescence experiments was calculated using one way ANOVA and the Kruskal Wallis test.
3. Results
Transfer of MOG-sensitized T cells to CD11a-/- mice induces severe EAE
In our hands, active EAE development in CD11a-/- mice was significantly delayed and attenuated compared to control mice, similar to results reported by Wang and colleagues (Fig. 1A, Table 1; p=0.0001, Wilcoxon rank sign test) (Wang et al., 2007). However, in our studies using CD11a-/- mice, disease incidence was lower (60% vs. 80%) and disease onset was delayed (28 vs. 14 days) compared to the results reported by Wang and colleagues (Wang et al., 2007). In addition, we induced EAE in CD11a+/- mice and observed an intermediate but significantly different phenotype to that of CD11a-/- and wild type mice with respect to day of onset and overall severity (Fig. 1B, Table 1; p<0.001). These latter findings further implicate LFA-1-mediated interactions and functions in the development of EAE and suggest that the overall level of CD11a expression is critical in regulating both the onset and progression of CNS inflammation and demyelination in this model.
Figure 1.

The clinical course of active EAE is significantly attenuated in CD11a-/- and CD11a+/- mice compared to wild type mice. A, Active EAE was induced as described in Materials and Methods and signs of disease scored for 30 days. Results shown are the daily mean clinical score for wild type (n=17) and CD11a-/- mice (n=19) from three experiments. B, Active EAE was also induced in CD11a-/- mice. Results shown are the daily mean clinical score for wild-type (n=11) and CD11a-/- mice (n=19) from three experiments.
Table I.
EAE signs in wild-type mice, CD11a-/- and CD11a+/- mice.
| CDIA | Disease OnsetB | Disease IncidenceC | |
|---|---|---|---|
| Wild type n=17 |
41 | 17.5d | 100 |
| CD11a-/- n=19 |
8d | 28d | 63 |
| Wild type n=11 |
59 | 15.5d | 100 |
| CD11a+/- n=20 |
22 e | 22d | 95 |
Cumulative Disease Index is the mean of the sum of daily clinical scores observed between days 7 and 30.
Disease onset is defined as the first day of two consecutive days with a clinical score of two or more. Mice with no signs of disease are assigned a score of 30d.
Disease incidence is defined as the percent of mice that displayed any clinical signs of disease.
Significantly different from WT (p=0.0001, Wilcoxon signed rank test)
Significantly different from WT (p<0.001, Wilcoxon signed rank test)
We extended these studies by performing adoptive transferred EAE to determine the role of LFA-1 on effector T cells versus other leukocytes The course of EAE in wild-type mice receiving CD11a-/- encephalitogenic T cells was significantly attenuated (p=0.0039, Wilcoxon signed rank test) and slightly delayed in onset (13d vs. 11d; Fig. 2A, Table 2). These results suggest that LFA-1 expression on effector T cells is important for promoting severe disease. However, EAE development was not completely inhibited, suggesting that this adhesion molecule is not absolutely required for T cell activation and trafficking into the CNS in this model. To determine if absence of LFA-1 expression on monocyte/macrophages and other leukocytes would also result in attenuated disease, we induced EAE by transferring MOG-sensitized T cells from wild-type mice into CD11a-/- recipients (Fig. 2B, Table 2). Unexpectedly, the severity of EAE was significantly increased in these mice compared to control transfers (p=0.0039). All recipient mice reached a score of at least 5 and at later time points approximately 30% of the mice died (data not shown). This latter disease phenotype is markedly different from our previous studies using Mac-1-, p150,95-, and ICAM-1-deficient mice, where all combinations of transfers produced significantly attenuated disease and, in some cases, no disease at all (Bullard et al., 2007a; Bullard et al., 2005; Bullard et al., 2007b).
Figure 2.

The clinical course of adoptive transferred EAE is significantly attenuated on transfer of CD11a-/- MOG sensitized T cells to wild type recipients, but exacerbated on transfer of wild type or CD11a-/- MOG-sensitized T cells to CD11a-/- recipients. A, Transferred EAE was induced as described in Materials and Methods in wild-type mice (n=8) by injecting encephalitogenic T cells (2.5 × 107) derived from CD11a-/- mice with active EAE. As a control transferred EAE was induced in wild type mice (n=7) by injecting encephalitogenic T cells (2.5 × 107) derived from wild-type mice with active EAE. B, Transferred EAE was induced as in A, in wild type (n=7) and CD11a-/- mice (n=7) by injecting encephalitogenic T cells derived from wild-type mice with active EAE. Results shown are the daily mean clinical score from two separate experiments.
Table 2.
Transferred EAE signs in wild-type mice and CD11a-/- mice.
| CDIa | Disease Onsetb | Disease Incidencec | |
|---|---|---|---|
| WT > WT n=7 |
20 | 10.6d | 100 |
| CD11a-/- > WT n=8 |
9d | 13d | 100 |
| WT > WT n=7 |
20 | 10.6d | 100 |
| WT > CD11a-/- n=7 |
29e | 9.3d | 100 |
Cumulative Disease Index is the mean of the sum of daily clinical scores observed between days 7 and 30.
Disease onset is defined as the first day of two consecutive days with a clinical score of two or more.
Disease incidence is defined as the percent of mice that displayed any clinical signs of disease.
Significantly different from WT to WT (p=0.0039, Wilcoxon signed rank test)
Significantly different from WT to WT (p=0.0039, Wilcoxon signed rank test)
Bioluminescent imaging of T-lux wild type and CD11a-/- T cells during the course of disease in transferred EAE
Based on our findings of exacerbated EAE, we predicted that loss of LFA-1 expression in recipient mice might result in temporal changes in the trafficking, localization, or expansion of encephalitogenic T cells during the initiation and progression of EAE. To address this possibility, we performed transferred EAE using T cells isolated from T-lux mice (Azadniv et al., 2007). Transfer of wild-type MOG-sensitized T-lux cells resulted in rapid trafficking and preferential localization to cervical lymph nodes in wild type recipients as determined by in vivo (Fig. 3A) and ex vivo imaging (data not shown). By day 6, the cells had trafficked throughout the secondary lymphoid system and expanded dramatically. At the same time point, T cells were observed at the base of the tail and in the tail itself, suggesting that the sacral spinal cord is a major entry point for encephalitogenic T cells. T cell infiltration in the tail and lower spinal cord increased throughout the remaining time points and, by day 13, infiltration into the brain was also readily apparent in these mice (Fig. 3A). Ex vivo imaging of brain and spinal cord from these mice at day 20 demonstrated a marked infiltration of T cells (Fig. 3D). In contrast, antigen-specific CD11a-/- T-lux cells transferred to wild-type mice trafficked to lymph nodes, but proliferated poorly and substantially fewer cells were present in the CNS by day 13 (Fig. 3B). Ventral imaging of wild type T-lux cells transferred into CD11a-/- recipients revealed a significant accumulation of T cells throughout the secondary lymphoid system and massive expansion, most notably and in the cervical lymph nodes (Kruskal Wallis test, p=0.005) for the duration of the study (Fig. 3C). In this experiment, wild type T-lux cells were observed to localize in the tail with kinetics similar to those of wild type-to-wild type transfers, but these T cells appeared in the brain more rapidly, with large numbers of present in the brain on days 9 and 13 (Fig. 3C). In addition, a number of distinct T cell-containing lesions were visible in the spinal cord on day 13. Ex vivo imaging of brain and spinal cord from these mice at day 15 confirmed the whole body imaging results and revealed a particularly marked infiltration of T cells into the brain (Fig. 3D).
Figure 3.
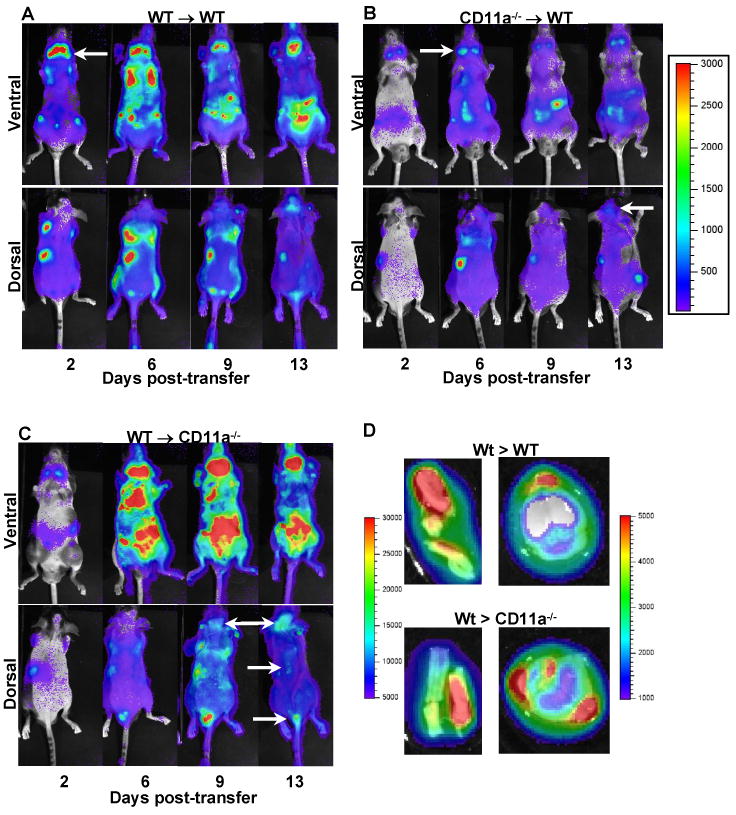
Bioluminescent imaging of wild type- or CD11a-/--T-lux T cells during the course of transferred EAE. Transferred EAE was induced as described in Materials and Methods, using MOG-sensitized T cells from wild type or CD11a-/- T-lux mice. The following combinations of transfers were performed: A, wild type to wild type (n=2), B, CD11a-/- to wild type (n=3) and C, wild type to CD11a-/- (n=4). Bioluminescent imaging was performed as described in Materials and Methods on days 2, 6, 9 and 13 post-transfer. Shown in each panel is representative ventral and dorsal imaging from one mouse at the indicated time points. Pseudo color scale for panels A-C is shown (100 min to 50,000 max) to emphasize spinal cord lesions or individual organ structures in which T cells have accumulated. Arrows indicate T cell accumulation in cervical nodes, brain or spinal cord. D, Ex vivo imaging of T-lux T cells in the brain and spinal cord of a mouse from a CD11a-/- mouse receiving wild type encephalitogenic T cells. At day 20 post-transfer, brain and spinal cord were removed, treated ex vivo with Luciferase assay system solution and imaged as described in Materials and Methods. Pseudo color scale for spinal cord images is shown (5,000 min to 30,000 max, left side of the images)) as well as for brain images (1,000 min to 5,000 max, to the right of the images).
We also quantified the infiltration of T-lux T cells in these adoptive transfer studies (Fig. 4). Total body ventral imaging of wild type mice receiving either wild type or CD11a-/- encephalitogenic T-lux T cells demonstrated marked expansion of these cells throughout the peripheral and mucosal lymph nodes before the onset of clinical signs of disease, peaking at day 9 for both sets of transfers. Wild type mice receiving CD11a-/- T-lux T encephalitogenic cells however had significantly reduced T cell expansion (Kruskal-Wallis test, p=0.0043) compared to the other two transfers groups (Fig. 4A). Similar results were obtained for whole body dorsal imaging of all three groups of mice, with significantly higher bioluminescent signal coming from the CD11a-/- mice receiving wild type encephalitogenic T cells primarily due to the marked accumulation of T cells in the brain and spinal cord (Fig. 4B, Kruskal-Wallis test, p=0.004). We also compared the bioluminescent signal in the cervical lymph nodes and brain and spinal cords (the latter two tissues as one group) between all three groups of transfers. The cervical lymph nodes are a major site encephalitogenic T cell accumulation during the course of EAE. We verified the identity of the cervical nodes, as opposed to other regional lymph nodes, by post-mortem imaging. T cell accumulation and proliferation in cervical nodes was readily detectable in all transfer groups, but was highest in wild type to CD11a-/- transfers (Fig. 4C). Not surprisingly, the bioluminescent signal was significantly higher in the brain and spinal cord in the wild type to CD11a-/- transfers (Kruskal-Wallis test, p=0.009) where T cell accumulation was readily apparent by imaging (Fig. 4D).
Figure 4.

Quantification of bioluminescence from cervical lymph nodes and brain and spinal cords during transferred EAE. Data were collected at days 2, 4, 6, 8, 9, 12, and 15 post-transfer from whole body ventral images (A), whole body dorsal images (B), cervical nodes (C) and the combination of brain and spinal cord (D) from all transferred EAE groups shown in Fig. 2. Results shown are the average of three to four mice per time point plotted as photons/sec versus days post transfer.
4. Discussion
Current paradigms describing the mechanisms of leukocyte trafficking in immune responses, including CNS autoimmune disease, highlight LFA-1 as a significant mediator of leukocyte adhesion to activated endothelial cells (reviewed in (Archelos et al., 1999; Engelhardt, 2006a; Ley et al., 2007; Luster et al., 2005; Smith et al., 2007; Steeber et al., 2005). Although we demonstrate here that LFA-1 is important for the development of severe EAE, we found that its expression is not absolutely required for effector T cell infiltration and activation in the CNS and in lymphoid organs, suggesting that redundancy exists among different integrin-mediated trafficking mechanisms in demyelinating disease. Mac-1, p150,95 and VLA-4 all likely substitute for LFA-1 in this setting, but other adhesion molecules may also contribute (Bullard et al., 2007a; Bullard et al., 2005; Yednock et al., 1992). Interestingly, both wild type and CD11a-/- encephalitogenic T cells were frequently seen at the base of the tail early in the course of EAE, before bioluminescent detection in the brain or spinal cord. Although it cannot be ruled out that the T cells found at the base of the tail first entered the CNS elsewhere, it may be that the selective recruit of effector T cells to this region occurs due to its unique anatomy, differential expression of adhesion molecules, or from myelin antigens selectively recruits effector T cells to the tail in this disease model. MOG-specific T cells also trafficked rapidly to cervical lymph nodes in all combinations of transfers we performed, with the most significant expansion observed in CD11a-/- recipient mice (Fig. 3).
An unexpected observation in our studies was that CD11a-/- recipient mice showed increased severity of EAE following transfer of wild type encephalitogenic T cells, suggesting that LFA-1 also plays a key role in inhibiting CNS inflammation and demyelination through its expression and activity on other non-effector T cell subsets. The findings contrast with our previous observations where only a mild transferred EAE phenotype was observed when wild type encephalitogenic T cells were transferred into CD11b- or CD11c mutant recipients, and highlight the unique roles of LFA-1 when compared to the other β2-integrin family members (Bullard et al., 2007a; Bullard et al., 2005). The increased severity of transferred EAE in CD11a-/- recipients could be due in part to altered T cell production of Th1/2 or Th17 cytokines. However, flow cytometric analysis of leukocytes isolated from lymph nodes and spinal cord did not show significant shifts in T cell production of several of the classic pro-inflammatory cytokines, including IFN-γ, TNF-α or IL-17 that might be expected to contribute to increased disease severity (data not shown). In contrast, Wang and colleagues reported elevated levels of IFN-γ and IL-17 produced by wild type T cells derived from inguinal lymph nodes during active EAE. The physiological significance of those results is unclear since these T cells were treated overnight with PMA and ionomycin and not derived from the spinal cord. Alternatively, a reduction in anti-inflammatory cytokine levels could also lead to changes in disease severity. Nevertheless, we did not observe a reduction in IL-10 levels, a well-established regulator of the autoreactive T cell repertoire in EAE and other autoimmune disease (data not shown)(Anderson et al., 2004; Kuchroo et al., 2002; Zhang et al., 2004). Future studies are necessary to examine additional cytokines to determine whether specific alterations occur and might contribute to disease exacerbation in this transferred EAE setting.
Little information is available regarding the role(s) of LFA-1 on non-effector T cell leukocyte subsets in demyelinating disease. Our data suggest that absence of LFA-1 on dendritic cells, neutrophils, γδ T cells and/or T regulatory cells, all of which contribute to EAE pathogenesis (Sospedra and Martin, 2005), may contribute to the disease phenotypes we report here. It is interesting to note that the wide-ranging effects of anti-CD11a antibody treatment on the course of EAE in previously published studies may have been due in part to differential inhibition of specific LFA-1-dependent functions. For example, in studies in which EAE was attenuated on anti-CD11a treatment (Gordon et al., 1995), T cell trafficking or dendritic cell function may have been altered. Furthermore, in studies in which EAE severity was increased on anti-CD11a treatment (Cannella et al., 1993; Welsh et al., 1993), the underlying mechanism may have been due in part to inhibition of T regulatory cells. Despite the inconsistent outcome in EAE studies, immunosuppressive therapies targeting LFA-1 in humans have shown beneficial effects in psoriasis and in preventing transplantation rejection (Barry and Kirby, 2004; Vincenti et al., 2007). Although our findings as well as others suggest that this adhesion molecule, like the α4-integrins, may also be an attractive therapeutic target for the treatment of MS, these results indicate that caution should be exercised with respect to specific inhibition of LFA-1.
Acknowledgments
The authors would like to acknowledge Graham Kennedy Spillman for encouragement and support and critical reading of the manuscript. The authors acknowledge the support of the Laboratory for Multi-Modality Imaging Assessment. This research was supported by NIH grants NS046032 (SRB and DCB), and AHA grant to DCB. Kari Dugger was supported by a supplement to NS046032 for promotion of diversity in health-related research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson AC, Reddy J, Nazareno R, Sobel RA, Nicholson LB, Kuchroo VK. IL-10 plays an important role in the homeostatic regulation of the autoreactive repertoire in naive mice. J Immunol. 2004;173:828–834. doi: 10.4049/jimmunol.173.2.828. [DOI] [PubMed] [Google Scholar]
- Archelos JJ, Previtali SC, Hartung HP. The role of integrins in immune-mediated diseases of the nervous system. Trends Neurosci. 1999;22:30–38. doi: 10.1016/s0166-2236(98)01287-9. [DOI] [PubMed] [Google Scholar]
- Azadniv M, Dugger K, Bowers WJ, Weaver CT, Crispe IN. Imaging CD8+ T cell dynamics in vivo using a transgenic luciferase reporter. Int Immunol. 2007;19:1165–1173. doi: 10.1093/intimm/dxm086. [DOI] [PubMed] [Google Scholar]
- Barry J, Kirby B. Novel biologic therapies for psoriasis. Expert Opin Biol Ther. 2004;4:975–987. doi: 10.1517/14712598.4.6.975. [DOI] [PubMed] [Google Scholar]
- Bullard DC, Hu X, Adams JE, Schoeb TR, Barnum SR. p150,95 (CD11c/CD18) expression is required for the development of experimental autoimmune encephalomyelitis. Amer J Path. 2007a;170:2001–2008. doi: 10.2353/ajpath.2007.061016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard DC, Hu X, Schoeb TR, Axtell RC, Raman C, Barnum SR. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:6327–6333. doi: 10.4049/jimmunol.175.10.6327. [DOI] [PubMed] [Google Scholar]
- Bullard DC, Hu X, Schoeb TR, Collins RG, Beaudet AL, Barnum SR. Intercellular adhesion molecule-1 expression is required on multiple cell types for the development of experimental autoimmune encephalomyelitis. J Immunol. 2007b;178:851–857. doi: 10.4049/jimmunol.178.2.851. [DOI] [PubMed] [Google Scholar]
- Cannella B, Cross AH, Raine CS. Anti-adhesion molecule therapy in experimental autoimmune encephalomyelitis. J Neuroimmunol. 1993;46:43–55. doi: 10.1016/0165-5728(93)90232-n. [DOI] [PubMed] [Google Scholar]
- Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163:5029–5038. [PubMed] [Google Scholar]
- Engelhardt B. Immune cell migration across the blood-brain barrier: molecular mechanisms and therapeutic targeting. Future Neurol. 2006a;1:47–56. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm. 2006b;113:477–485. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- Gordon EJ, Myers KJ, Dougherty JP, Rosen H, Ron Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;62:153–160. doi: 10.1016/0165-5728(95)00120-2. [DOI] [PubMed] [Google Scholar]
- Kuchroo VK, Anderson AC, Waldner H, Munder M, Bettelli E, Nicholson LB. T cell response in experimental autoimmune encephalomyelitis (EAE): role of self and cross-reactive antigens in shaping, tuning, and regulating the autopathogenic T cell repertoire. Annu Rev Immunol. 2002;20:101–123. doi: 10.1146/annurev.immunol.20.081701.141316. [DOI] [PubMed] [Google Scholar]
- Laschinger M, Vajkoczy P, Engelhardt B. Encephalitogenic T cells use LFA-1 for transendothelial migration but not during capture and initial adhesion strengthening in healthy spinal cord microvessels in vivo. Eur J Immunol. 2002;32:3598–3606. doi: 10.1002/1521-4141(200212)32:12<3598::AID-IMMU3598>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Liu L, Graham GJ, Damodaran A, Hu T, Lira SA, Sasse M, Canasto-Chibuque C, Cook DN, Ransohoff RM. Cutting edge: the silent chemokine receptor D6 is required for generating T cell responses that mediate experimental autoimmune encephalomyelitis. J Immunol. 2006;177:17–21. doi: 10.4049/jimmunol.177.1.17. [DOI] [PubMed] [Google Scholar]
- Luster AD, Alon R, von Andrain UH. Immune cell migration in inflammation: present and future therapeutics. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Smith A, Stanley P, Jones K, Svensson L, McDowall A, Hogg N. The role of the integrin LFA-1 in T-lymphocyte migration. Immunol Rev. 2007;218:135–146. doi: 10.1111/j.1600-065X.2007.00537.x. [DOI] [PubMed] [Google Scholar]
- Sospedra M, Martin R. Immunology of multiple sclerosis *. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- Steeber DA, Venturi GM, Tedder TF. A new twist to the leukocyte adhesion cascade: intimate cooperation is key. Trends Immunol. 2005;26:9–12. doi: 10.1016/j.it.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Vincenti F, Mendez R, Pescovitz M, Rajagopalan PR, Wilkinson AH, Butt K, Laskow D, Slakey DP, Lorber MI, Garg JP, Garovoy M. A phase I/II randomized open-label multicenter trial of efalizumab, a humanized anti-CD11a, anti-LFA-1 in renal transplantation. Am J Transplant. 2007;7:1770–1777. doi: 10.1111/j.1600-6143.2007.01845.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kai H, Chang F, Shibata K, Tahara-Hanaoka S, Honda S, Shibuya A, Shibuya K. A critical role of LFA-1 in the development of Th17 cells and induction of experimental autoimmune encephalomyelytis. Biochem Biophys Res Commun. 2007;353:857–862. doi: 10.1016/j.bbrc.2006.12.104. [DOI] [PubMed] [Google Scholar]
- Welsh CT, Rose JW, Hill KE, Townsend JJ. Augmentation of adoptively transferred experimental allergic encephalomyelitis by administration of a monoclonal antibody specific for LFA-1 alpha. J Neuroimmunol. 1993;43:161–167. doi: 10.1016/0165-5728(93)90087-f. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Staykova MA, Miyasaka M. Short term treatment with soluble neuroantigen and anti-CD11a (LFA-1) protects rats against autoimmune encephalomyelitis: treatment abrogates autoimmune disease but not autoimmunity. J Immunol. 1996;157:1973–1980. [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]