

Abstract
Heat shock protein (hsp) 90 is an ATP-dependent molecular chaperone, which maintains the active conformation of client oncoproteins in cancer cells. An isoform, hsp90α, promotes extra-cellular maturation of matrix metalloproteinase (MMP)-2 involved in tumor invasion and metastasis. Knockdown of histone deacetylase (HDAC) 6, which deacetylates lysine residues in hsp90, induces reversible hyper-acetylation and attenuates ATP binding and chaperone function of hsp90. Here, utilizing mass spectrometry, we identified seven lysine residues in hsp90α that are hyper-acetylated, following treatment of eukaryotic cells with a pan-HDAC inhibitor that also inhibits HDAC6. Depending on the specific lysine residue in the middle domain involved, while acetylation affects ATP, co-chaperone and client protein binding to hsp90α, acetylation of all seven lysines increased the binding of hsp90α to 17-allyl-amino-demethoxy geldanamycin (17-AAG). Notably, following treatment with the pan-HDAC inhibitor panobinostat (LBH589), the extra-cellular hsp90α was hyper-acetylated and it bound to MMP-2, which was associated with increased in vitro tumor cell invasiveness. Treatment with anti-acetylated hsp90α antibody inhibited in vitro invasion by tumor cells. Thus, reversible hyper-acetylation modulates the intra- and extra-cellular chaperone function of hsp90, and targeting extra-cellular hyper-acetylated hsp90α may undermine tumor invasion and metastasis.
Introduction
Heat shock protein 90 is a constitutively and ubiquitously expressed, ATP-dependent molecular chaperone (1). It exerts an essential role in proper folding and in maintaining the active conformation, intracellular disposition and proteolytic turnover of a large number of the pro-growth and pro-survival substrate client oncoproteins in cancer cells (1). Therefore, hsp90 has emerged as a promising target in cancer therapy (2). Activation of client proteins by hsp90-based chaperone machine involves an ordered association with several co-chaperones, e.g., p23, cdc37 and Aha-1, linked to the ATPase cycle of hsp90, which may also direct client protein specificity (3-5). Hsp90 exists predominantly as a homodimer, with transient association between N-terminal domains, thus functioning as a dimeric ‘molecular clamp’ (6). Each hsp90 monomer is modular with three well-defined domains. These include the N-terminal nucleotide-binding domain (NTD), a middle domain (MD) that completes the ATPase site and binds to client proteins, as well as the C-terminal dimerization domain (CTD) (7, 8). ATP binding and hydrolysis triggers conformational change in the hsp90 homodimer, which is crucial for its binding to the co-chaperones, as well as for its interaction with various client oncoproteins in the cancer cells (1, 9, 10). The hsp90 chaperone cycle includes a) an open, apo, nucleotide-free conformation in which each of the three domains in each monomer presents hydrophobic surface to the large inter-domain cleft, a conformation most optimal for client protein binding; b) an ATP bound intermediate state and c) a closed ADP bound state (8). There are two isoforms of hsp90, i.e., hsp90α and hsp90β, which are encoded by two separate genes (11, 12). Only hsp90α has been described to be extra-cellular, where it serves as a molecular chaperone and activates matrix metalloproteinase (MMP)-2 (11, 12).
In addition to co-chaperone association as well as ATP binding and hydrolysis, post-translational modifications such as hyper-phosphorylation (13-15), S-nitrosylation and reversible hyper-acetylation have also been shown to regulate the chaperone function of hsp90 (16-18). Several serine-threonine phosphorylation sites have been identified in hsp90. Although hyper-phosphorylation negatively regulates hsp90 chaperone function, the role of site-specific phosphorylation in modulating hsp90 function has yet to be fully elucidated. Lysine (K) acetylation is a reversible modification mediated by opposing actions of acetyltransferases (HATs) and deacetylases (HDACs) in which an acetyl group is covalently linked to lysine residues of target proteins (19). Following treatment with a variety of pan-histone deacetylase inhibitors (HDIs), including the hydroxamic acid analogues vorinostat, LAQ824 and panobinostat (LBH589), or following siRNA mediated knockdown of HDAC6, reversible hyper-acetylation of hsp90 has been documented (17, 18). Overall, hyper-acetylation of hsp90 was shown to inhibit the ATP, co-chaperone p23 and client protein binding to hsp90, directing the client proteins to polyubiquitylation and proteasomal degradation (18). In a recent report, Scroggins et al identified the K294 in the MD of hsp90α as a discrete acetylation site (20). They also determined that the acetylation status of K294 is a strong determinant of client protein and co-chaperone binding to hsp90α. Although they noted that hsp90 is acetylated at more than one site, identification of these sites or their functional significance was not determined (20). In the present study, we determined the identity and functional significance of the domain-specific seven lysine residues that are hyper-acetylated, following treatment with pan-HDAC inhibitors that also inhibit HDAC6. Remarkably, hyper-acetylated hsp90α was extra-cellular and acted as a chaperone for MMP-2, which promoted in vitro invasion by breast cancer cells. Our findings also demonstrate that treatment with anti-acetyl lysine-69 hsp90α antibody markedly inhibits the invasiveness of breast cancer cells.
Materials and Methods
Cell lines, antibodies and plasmids
HEK293T, MDA-MB-468, MDA-MB-231 and T47D cells were all purchased from American Tissue Culture Collection (Manassas, VA). HEK293T and MDA-MB-468 cells were maintained in Dulbecco' modified Eagle's medium (DMEM); T47D and MDA-MB-231 cells were maintained in RPMI medium containing 10% FBS. The following antibodies used were purchased from commercial sources: anti-CHIP (Abcam, Cambridge, MA), anti-hsp40 (SPA-450, StressGen, Victoria, BC, Canada), anti-hsp90α (SPA-840, StressGen), anti-hsp90α (polyclonal, GeneTex, San Antonio, TX) anti-p23 (Alexis Biochemicals, San Diego, CA, 804-023-R100), anti-acetyl-lysine (monoclonal) and anti-AKT (Cell Signaling, Beverly, MA), anti-Acetyl lysine (polyclonal, Upstate-Millipore), anti-HA.11 (Monoclonal, Covance, Berkeley, CA), anti-cRaf and anti-MMP2 (Santa Cruz Biotech., Santa Cruz, CA), anti-FLAG (M2 monoclonal and F polyclonal), ANTI-FLAG® M2 agarose, anti-β-actin (Sigma, St. Louis, MO) and agarose conjugates (Upstate, Lake Placid, NY). Plasmids expressing FLAG (F)-hsp90 and HA-p300 have previously been described (21-22). Hsp90 mutants were generated by site-directed mutagenesis using the QuikChange kit from Stratagene (Cedar Creek, TX) (23). All mutants were verified by sequencing at the DNA Core Lab of MCG. The sequence of mutagenesis primers is provided in Table 1.
Table 1. Sequence of mutagenesis primers.
| Mutation site | Sequence of primer |
|---|---|
| K69Q | 5-GAA AGC TTG ACA GAT CCC AGT CAA TTA GAC TCT GGG A |
| K69R | 5-GAA AGC TTG ACA GAT CCC AGT AGA TTA GAC TCT GGG A |
| K100Q | 5-GAT ACT GGA ATT GGA ATG ACC CAG GCT GAC TTG ATC |
| K100R | 5-GAT ACT GGA ATT GGA ATG ACC AGG GCT GAC TTG ATC |
| K292Q | 5-TCG ATC AAG AAG AGC TCA ACC AAA CAA AGC CCA TCT G |
| K292R | 5-TCG ATC AAG AAG AGC TCA ACA GAA CAA AGC CCA TCT G |
| K327Q | 5-TGG GAA GAT CAC TTG GCA GTG CAG CAT TTT TCA GTT G |
| K327R | 5-TGG GAA GAT CAC TTG GCA GTG AGG CAT TTT TCA GTT G |
| K478Q | 5-GTG ATG AGA TGG TTT CTC TCC AGG ACT ACT GCA CCA G |
| K478R | 5-GTG ATG AGA TGG TTT CTC TCA GGG ACT ACT GCA CCA G |
| K546Q | 5-GAA GAC TTT AGT GTC AGT CAC CCA AGA AGG CCT GGA ACT |
| K546R | 5-GAA GAC TTT AGT GTC AGT CAC CAG AGA AGG CCT GGA ACT |
| K558 | 5-TCC AGA GGA TGA AGA AGA GCA AAA GAA GCA GGA AG |
| K558R | 5-TCC AGA GGA TGA AGA AGA GAG AAA GAA GCA GGA AG |
Acetylated-K69 hsp90α antibody
Affinity-purified polyclonal antibody against Ac-K69-hsp90α was generated by Alpha Diagnostic (San Antonio, TX) based on the synthetic 12 amino acid peptide flanking K69 (acetylated) ETLTDPSKLDSGK. Affinity-purified antibody was checked by performing an ELISA, using free peptide containing acetylated lysine. The antibody specifically recognized acetylated peptide but not non-acetylated peptide dotted on nitrocellulose membrane (data not shown). The antibody also recognized increase in hsp90α acetylation following HDAC inhibtor-treatment and untreated acetylated hsp90α (see Figure 6B).
Figure 6. Anti-Ac-K69 antibody inhibits hsp90α-dependent in vitro invasion by breast cancer cells.
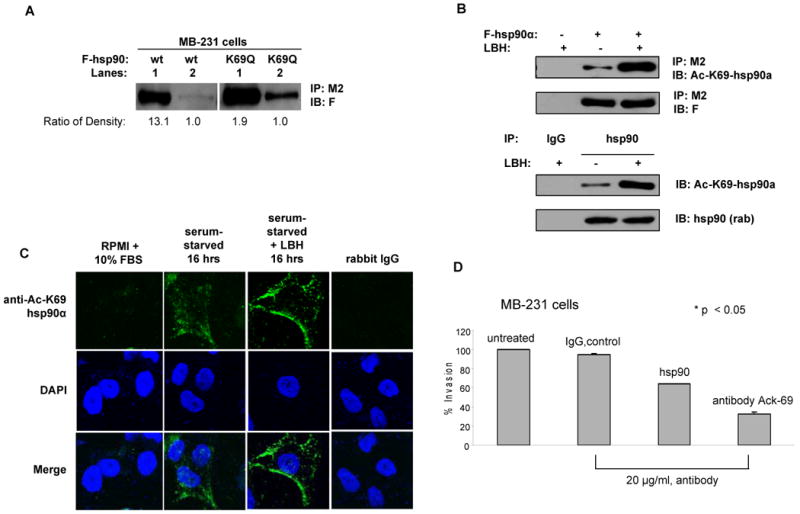
(A) K/Q substitution at K69 promotes extracellular location of hsp90α. MB-231 cells transfected with either F-hsp90 or K69Q mutant were cultured under serum-free condition for 24 hours. Total extracellular and intracellular hsp90α were immunoprecipitated with anti-M2 conjugated beads and immunoblotted with anti-F antibody. The intensity of the bands was quantified using ImageQuant 5.2 software, and the ratio for intracellular to extracellular hsp90 is shown below the panel. Lane 1 is intracellular, and Lane 2 is extracellular. (B) Anti-Ac-K69 hsp90α antibody specifically recognizes acetylated form of both exogenous and endogenous hsp90α expressed in MB-231 cells. MB-231 cells were transfected with F-hsp90α followed by the treatment with LBH589. Immunoprecipitates of F-hsp90α with anti-M2 conjugated beads were immunoblotted with anti-K69-hsp90α antibody for the acetylation status and anti-F antibody for F-hsp90α expression. Cells transfected with empty vector followed the treatment with LBH589 served as control for specificity. Acetylation of endogenous hsp90α induced by LBH589 was also detected with anti-AcK69-hsp90α antibody (Lower panel). Immunoprecipitates of endogenous hsp90α with anti-hsp90α antibody from cell lysates of MB-231 cells treated with or without LBH589 were immunoblotted with either anti-AcK69-hsp90α or anti-hsp90α (rabbit) antibody. IgG served as the control for specificity of the immunoprecipitates. (C) LBH589 induces surface localization of acetylated hsp90α in MB-231 cells. Serum-starved MB-231 cells were treated with 40 nM LBH589 for 16 hours, followed by staining with anti-AcK69 hsp90α antibody and confocal microscopy. Cell cultured in RPMI with 10 % FBS and cells stained with rabbit IgG served as controls. (D) Inhibition of in vitro invasion by MB-231 cells by anti-AcK69 hsp90α antibody. Serum-starved MB-231 cells treated with 20 μg/mL anti-hsp90α or anti-AcK69 hsp90α antibody were used for determining in vitro matrigel invasion. Untreated cells, or cells treated with IgG were used as controls. Columns, average results of three independent experiments; bars, SD. *, p < 0.05.
Transfections, immunoprecipitations and immunoblots
Following culture in the plates for 24 hours, cells were transfected by Lipofectamine Plus following the protocol provided by Invitrogen (Carlsbad, CA). Transfected cells were cultured in full-medium containing drugs dissolved in DMSO or vehicle for 24 hours. Cellular extracts were prepared by directly adding lysis buffer (25 mM Tris-phosphate, pH 7.8, 2 mM DTT, 2 mM 1,2-diaminocyclohexane-N′,N′,N′,N′-tetraacetic acid, 10% glycerol, 0.2% Triton X-100) to the cells on ice. For immuno-precipitations (IP), 2 ×106 cells in 100 mm dishes were transfected and/or treated as described above. Cellular extracts were prepared and immunoprecipitation performed as described (24). For immunoblotting, cellular extracts or immunoprecipitates were separated on SDS-PAGE, transferred to a nitrocellulose membrane, probed with antibodies, and visualized with enhanced chemiluminescence, as described (24).
Purification of acetylated hsp90α
Using anti-FLAG M2 affinity beads, F-tagged hsp90α protein was affinity captured from F-hsp90α transfected-HEK 293 cells that had been treated with 100 nM LBH589 (Novartis Pharmaceuticals Inc., East Hanover, NJ). This was followed by immunoprecipitation of acetylated F-hsp90α using acetyl lysine agarose beads. The immunoprecipitated proteins were resolved by 8% SDS-PAGE gel and visualized using Coomassie Blue stain.
Mass spectrometric determination of hsp90α acetylation sites
For comprehensive detection of the acetylation sites, MS in combination with MS/MS was utilized (25). First, gel slices from SDS PAGE separation of cell lysates were subjected to in-gel tryptic digestion to create peptides whose mass can be searched against public data bases. For peptide detection, we used an Applied Biosystems 4700 Proteomics Analyzer with Mascot (Matrix Science) protein search engine. The sample was loaded using α-cyano-4-hydroxycinnamic acid (CHCA) into the instrument according to manufacturer's instructions and run in a data dependent MS plus MS/MS mode. This allows for the documentation of the peptides creating a protein fingerprint for protein identification as well as documentation of the peptides for further analysis through post-source fragmentation. Once the peptides were detected, the instrument was told to go back to each sample and fragment the individual top 20 peptides creating fragments of different lengths that can be re-assembled by the computer to predict, in combination with the identification from the fingerprint data, the sequence of the peptide. The sequence data, in turn was used to bolster the protein identification call. The program allows for certain user input modifications that will allow for missed cleavages as a result of enzyme inefficiency as well as blocked sites. The software also allows for input of potential modifications that might add additional mass to a peptide allowing the search engine to call the peptide in either its modified or unmodified state and with or without missed cleavages resulting from any modifications or enzyme inefficiencies. All of this information was amassed and the call for identification and of the potential modification at a certain site was statistically calculated and the highest probability calls are reported.
ATP-Sepharose binding Assay
Hsp90α in 200 μg of cell lysates was affinity-precipitated using KinaseBind™γ-phosphate-linked ATP resin (Innova Biosciences) at 4°C for 4 hours. After washing three or four times with the lysis buffer, the resin was pelleted and SDS/PAGE analysis was performed (18).
Biotinylated-geldanamycin (B-GA) binding assay
Biotinylated (B)-GA binding to hsp90 was assessed as described previously (26). Briefly, cell-lysates were incubated with or without 17-AAG (Developmental Therapeutics Branch of CTEP/NCI/NIH) for 1 hour at 4°C, and then incubated with B-GA to displace 17-AAG from hsp90 for another 1h. GM-bound hsp90 was captured by biotin-GA linked to streptavidin Mutein Matrix (Roche Diagnostics, Indianapolis, IN) for 1 h at 4°C. The unbound supernatant was removed and the beads were washed three times with lysis buffer. The precipitates were immunoblotted for hsp90.
Isolation of hsp90α on cell surface
MDA-MB-231 cells transfected with hsp90α expression constructs cultured in RPMI-1640 (without serum) were harvested by centrifugation (1000g, 10 min at 4°C) and washed twice with ice-cold PBS. Proteins on the cell surface were labeled following the protocol supplied by the manufacturer (PIERCE, Rockford, IL). Briefly, cells were resuspended in EZ-Link® Sulfo-NHS-SS-Biotin (0.25 mg/mL) at 4°C for 30 min. The biotinylation reaction was terminated by addition of Quenching Solution. Following biotinylation, the cells were washed 3 times with TBS, pelleted by centrifugation and solubilized in Lysis buffer. The cell lysates were further disrupted by brief sonication. Solubilized biotinylated membrane proteins from same amount of cell lyastes were captured using Immobilized NeutrAvidin Gel, extensively washed with Wash Buffer and re-suspended in 2X SDS sample buffer prior to immunoblot analysis.
In vitro invasion assays
In vitro invasion assay were performed, as previously described (12, 27) by using the Cultrex® Cell Invasion Assay kit (RandD Systems, Minneapolis, MN). In brief, serum-starved cells in 50 μL serum-free medium with or without antibody or IgG were placed in the top chamber and allowed to invade for 24 hours. The lower chambers (assay chamber) were filled with 10% FBS medium. After incubation, migrated cells on the upper chamber of the membrane were dissociated with cell dissociation solution containing Calcein AM at 37°C for 1 hour and read the bottom plate at 485 nm excitation and 520 nm emission.
In vitro protein acetylation assay
Protein acetylation assays were performed as described previously (28). Briefly, reactions (30 μL) were carried out at 30°C for 1hour with M2 beads bound FLAG-hsp90α (25 μL M2 beads mixed with 10μl of in vitro translated FLAG-hsp90α and washed three times with HAT buffer) and 50 ng of p300 protein (upstate) in HAT buffer (50 mM Tris-HCl, pH 8.0, 10% glycerol, 1mM DTT, 1mM PMSF, 0.1 mM EDTA and 50 nM acetyl-CoA (sigma). After three time wash with HAT buffer, the samples were then subjected to western blot analysis with anti-acetyl-lysine antibody.
Confocal microscopy
MDA-MB-231 cells were cultured in a chamber slide in RPMI medium with 10% FBS or under serum-free conditions with or without 40 nM LBH589 for 16 hours and stained with anti-acetyl (Ac)-K69 antibody. Briefly, after 16 hours incubation, cells were washed with PBS and fixed with 4% paraformaldehyde for 10 minutes. Following this, the slides were blocked with 3% BSA for 30 minutes and incubated with primary antibody at a dilution of 1:100 in blocking buffer for 2 hours. Following three washes with PBS the slides were incubated in Alexa Fluor 488 anti-rabbit secondary antibody (Molecular probes, Invitrogen) for one hour at 1:3000 dilution. After three washes with PBS, the cells were counterstained with DAPI using Vectashield mountant with DAPI and imaged using Zeiss LSM510 confocal microscope.
Results
Hyperacetylation of hsp90α involves p300 as the acetyl-transferase
We first confirmed that hsp90 is acetylated both in vitro and in vivo, as well as determined the HAT responsible for inducing hyperacetylation of hsp90α. In HEK293 cells with ectopic expression of FLAG (F)-hsp90α, treatment with the pan-HDAC inhibitors LBH589 (Figure 1A) or vorinostat (Figure 1B) resulted in hyper-acetylation of F-hsp90α. To determine whether p300 acts as the HAT for hsp90α, following incubation with p300 and acetyl-CoA, in vitro translated hsp90α was analyzed for its acetylation status by Western analysis with anti-AcK antibody. As shown in Figure 1C, p300 was necessary for the acetylation of hsp90α in the in vitro assay. Immunoprecipitated F-hsp90 from LBH589-treated cells was used as the positive control. In HEK293 cells, it was determined that p300 can be co-immunoprecipitated with hsp90α (Supplemental Figure 1A). Co-incubation with p300 was discovered to dose-dependently stimulate the acetylation of hsp90α (Figure 1D). This was not seen with PCAF (data not shown). However, knock-down of p300 using siRNA only partially decreased LBH589 induced acetylation of hsp90 (Supplemental Figure 1B). Therefore, p300 appeared not to be the sole but one of the HATs both for the in vitro and in vivo acetylation of hsp90α.
Figure 1. Treatment with pan-HDAC inhibitor induces acetylation of hsp90α, and p300 is the HAT for hsp90α.
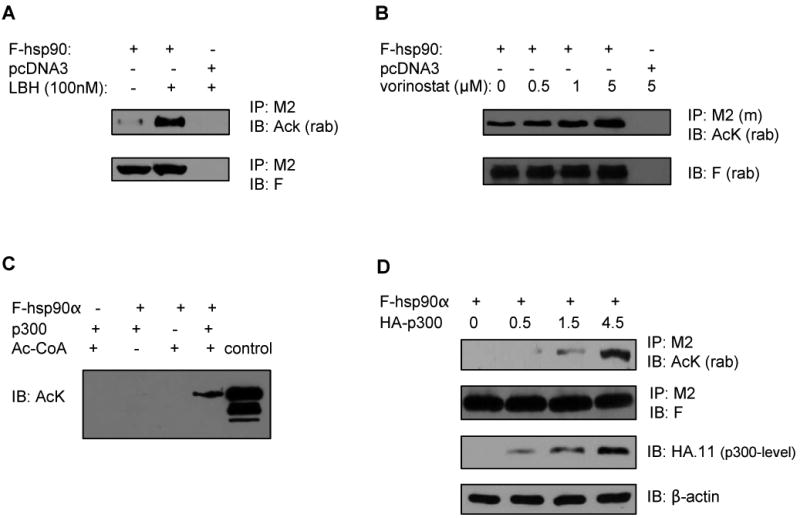
(A, B) HEK293 cells were transfected with either FLAG (F)-tagged hsp90α (3 μg) or empty vector pcDNA3 and treated with the indicated concentration of LBH589 (LBH) (A) or vorinostat (B) for 24 hours. Immunoprecipitates (IP) with anti-F (M2) antibody-conjugated beads were immunoblotted (IB) with either anti-rabbit acetyl lysine (AcK) or anti-rabbit FLAG (F) antibody. (C) p300 acts as a histone acetyl-transferase for hsp90α in vitro. The in vitro translated hsp90α was incubated with the combination of recombinant p300 and acetyl (Ac)-CoA followed by immunoblotting with anti-AcK antibody. The lane labeled ‘control’ contained LBH-induced acetylated F-hsp90α (D) p300 promotes acetylation of hsp90 in a dose dependent manner. HEK293 cells were co-transfected with F-hsp90α and the indicated amount of HA-tagged p300 (μg). Immunoprecipitates with anti-M2 antibody were immunoblotted with either anti-AcK or anti-F antibody. Immunoblots with anti-HA.11 or β-actin antibody served as loading control for p300 levels.
Identity and functional significance of lysine residues in hsp90α hyper-acetylated by pan-HDAC inhibitors. Next, we determined the identity of the acetylated lysine residues in hsp90α induced by HDAC inhibitor LBH589. HEK293 cells transfected with F-hsp90α, were treated with 100 nM LBH589, and the acetylated F-hsp90α was affinity immunopurified using anti-FLAG conjugated M2 agarose, followed by agarose beads bearing immobilized anti-AcK antibody. The enriched acetylated hsp90α was analyzed by nano-HPLC/MS/MS in a mass spectrometer. Seven acetylated lysine residues were identified in hsp90α: K69, K100, K292, K327, K478, K546 and K558. Figure 2A shows a three dimensional space-filling molecular structural model of hsp90α, which shows that all of the identified lysine residues that are acetylated reside on the surface and thus accessible for modification.
Figure 2. The individual K/R or K/Q substitutions do not affect the overall acetylation level of hsp90 but affect the ability of hsp90α to bind ATP, co-chaperones and client proteins.
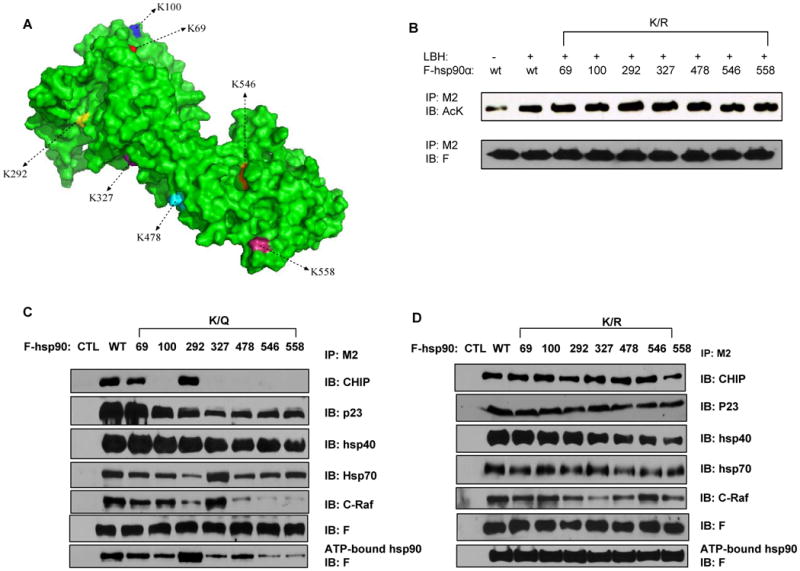
(A) The space-filling molecular structure model of hsp90. The seven lysine residues are shown in different colors. (B) Transfectants of F-hsp90α, with or without K/R substitutions, were treated with or without 100 nM of LBH. Following this, immunoprecipitates with M2 antibody were immunoblotted with either anti-AcK or anti-F antibody. (C, D) Transfectants of F-hsp90α with K/Q (C) but not K/R substitutions (D) affect ATP binding of hsp90α. Precipitates from the mixture of cell lysates containing hsp90α and ATP-sepharose were analyzed with anti-F antibody. Also, following transfections of F-hsp90α, with or without K/Q or K/R substitutions, immunoprecipitates with M2 antibody were immunoblotted with anti-CHIP, anti-p23, anti-hsp40, anti-hsp70, anti-c-Raf or anti-F antibody.
To assess the impact of acetylation at the various lysine residues on hsp90α function, point mutations were introduced to create acetylation-deficient (lysine to arginine, K/R) and acetylation-mimetic (lysine to glutamine, K/Q) mutants on the FLAG (F)-tagged hsp90α. First, we determined whether any of the K/R point mutations affects the overall hyperacetylation of F-hsp90α induced by either the co-transfected p300 or by treatment with LBH589. None of the individual K/R mutants showed any change in the hyperacetylation induced by either the co-transfected p300 (Supplemental Figure 2) or by treatment with LBH589 (Figure 2B). These mutants were also analyzed for their ability to bind ATP, as well as for binding to co-chaperones, client proteins and the biotinylated (B) GA (geldanamycin) (26). Although none of the K/R mutants compromised the ATP binding of F-hsp90α (Figure 2D), all but one of the acetylation-mimetic mutants (K/Q) showed decreased binding to ATP (Figure 2C). The exception was the K292Q mutant, which demonstrated increased binding to ATP (Figure 2C). The significance of this is unclear, although K292 is in the hinge region at the beginning of the middle domain (MD) of hsp90α, a region which is well conserved from yeast to human hsp90 (20, 29). Increased acetylation of hsp90 by a pan-HDAC inhibitor or HDAC6 siRNA had been shown to inhibit the binding of hsp90 with co-chaperones, e.g., p23, and client proteins (18). Here, we found that acetylation-mimetic mutants (K/Q) of the lysine residues in the middle domain, i.e., K100, K292, K327, K478, K546 and K558, displayed decreased binding with the co-chaperones p23 and to a lesser extent hsp40 (Figure 2C). While binding of K/Q mutants at K69, K100, K327, K478, K546 and K558 to CHIP was decreased, binding of K/Q mutant at K292 was not affected (Figure 2C). Additionally, binding of K/R mutants was similar to the binding of WT hsp90α to CHIP (Figure 2D). Acetylation-mimetic mutants of hsp90α also showed disrupted binding to hsp70 and with its client protein c-Raf, except with the K327Q mutant (Figure 2C), Binding of hsp70 and c-Raf to K/R mutants appeared to be not significantly altered (Figure 2D), In contrast, notably, the acetylation-mimetic K/Q mutants showed increased binding to B-GA (Figure 3A). This is consistent with the observation that acetylation of the endogenous hsp90α due to treatment with LBH589 was associated with increase in B-GA binding of hsp90α in MDA-MB-468 cells (Figure 3B). LBH589-induced hsp90 acetylation in MDA-MB-468 cells also promoted 17-AAG binding to the acetylated endogenous hsp90, since B-GA binding to hsp90 was reduced by 17-AAG treatment more in those cells exposed to LBH589 as compared to those that were unexposed (Figure 3C and 3D uppermost panel). Also, following treatment with LBH589, each K/R mutant of hsp90α showed increased binding to B-GA which could not be displaced by co-treatment with 17-AAG, suggesting decreased binding of K/R mutants to 17-AAG (Figure 3D).
Figure 3. LBH589 (LBH)-induced acetylation increases B-GA and 17-AAG binding to hsp90α.
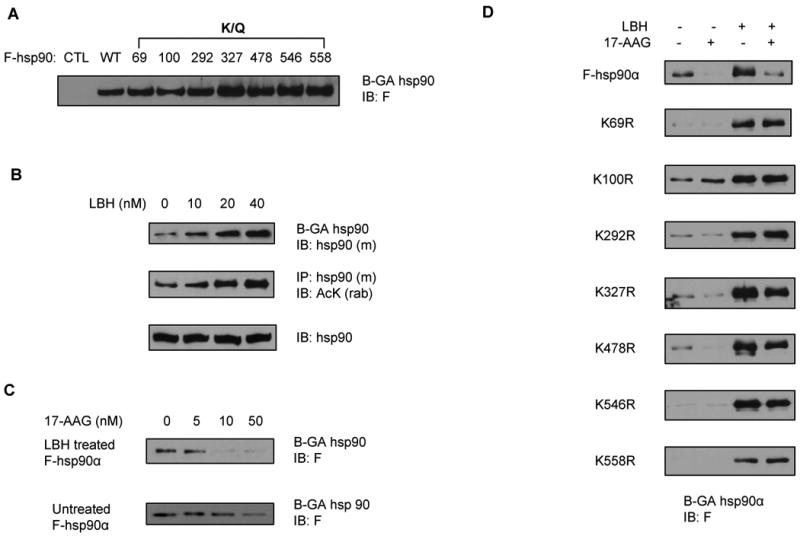
(A) Individual K/Q substitutions increase hsp90α binding to biotinylated (B)-GA. Cell lysates from Figure 2D above were also incubated with B-GA followed by streptavidin coated agarose beads and eluted proteins were analyzed with anti-F antibody. (B) LBH589 increases hsp90 binding to GA and induces hsp90 acetylation dose-dependently. MB-468 cells cultured in DMEM medium containing 10 % FBS were treated with the indicated concentration of LBH589 for 16 hours. Following this, cell lysates were incubated with B-GA followed by streptavidin coated agarose beads and eluted proteins were analyzed with anti-hsp90α antibody. Acetylation and expression level of endogenous hsp90 were detected with anti-AcK and anti-hsp90 antibody, respectively. (C) LBH589 treatment preferentially increases hsp90α binding to 17-AAG. MB-468 cells ectopically expressing F-hsp90α were treated with 100 nM of LBH for 16 hours. Following this, equal amount of cell lysates were incubated with the indicated doses of 17-AAG for 30 min at 4°C, followed by incubation with B-GA and streptavidin-coated agarose beads. Precipitates from streptavidin coated beads were analyzed with anti-F antibody. (D) individual K/R substitution disrupted the affinity of LBH589-treated hsp90α for 17-AAG. Following treatment with either vehicle or 100 nM of LBH, cell lysates from HEK293 cells expressing F-hsp90 or K/R substitutions were incubated with vehicle or 50 nM of 17-AAG followed by incubation with B-GA and streptavidin coated agarose beads. Precipitates from streptavidin coated beads were analyzed with anti-F antibody.
Role of lysine hyperacetylation in extra-cellular location of hsp90α
Previous reports have shown that the inducible isoform hsp90α, but not hsp90β, can be secreted and found on the surface of cancer cells, although it lacks the classical signal sequence (12, 30). Serum-starvation, hypoxia, high concentration of glucose, as well as oxidative stress have all been shown to promote the export and extra-cellular location of hsp90α (30-32). Consistent with these reports, our findings demonstrate that serum-starvation of breast cancer T47D cells promoted secretion and extra-cellular localization of the endogenous hsp90α (Figure 4A), as well as of the ectopically expressed F-hsp90α (Figure 4B). Notably, under both circumstances, hsp90α was acetylated (Figure 4A and 4B). Notably, treatment with LBH589, in a dose-dependent manner, also promoted and increased the export and extra-cellular localization of hyper-acetylated hsp90α from T47D cells into the culture medium (Supplemental Figure 3A). These data indicate that hyper-acetylation stimulates the extracellular export of hsp90α. To further verify this, we determined the export and extra-cellular location of acetylation mimetic (K/Q) or acetylation-resistant (K/R) mutants transfected into T47D cells that were cultured under serum-free condition. As shown, K69Q, K100Q and K558Q substitutions promoted the export and extra-cellular location of hsp90α, while K/R substitutions at the same residues markedly reduced the export and extra-cellular location of hsp90α (Figure 4C). Co-treatment of cells transfected with K/R mutants with LBH589 increased the export and extra-cellular location of K/R mutants of hsp90α to a variable extent, with K100R and K558R mutants showing less export than the other mutants (Supplemental Figure 3B). Immunoblot analysis of the solubilized biotinylated membrane proteins also showed that acetylation-mimetic mutants K69Q, K100Q, K327Q and K558Q exist more on the cell surface compared to the wild-type hsp90 (Figure 4D).
Figure 4. Acetylation-dependent extra-cellular localization of hsp90α.
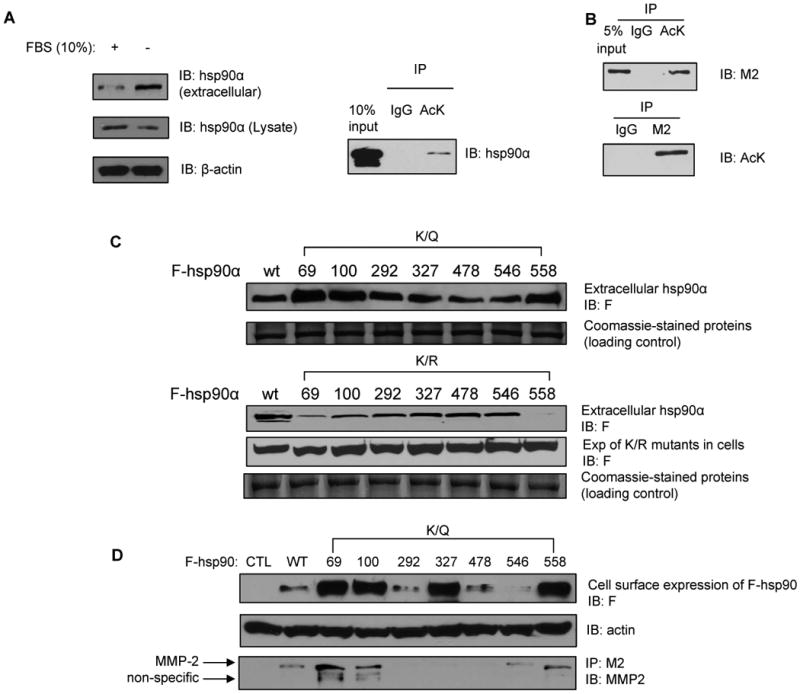
(A) Serum-starvation of T47D breast cancer cells promotes extra-cellular localization of hsp90α. Concentrated extra-cellular medium or cell lysates from T47D cells, which were either serum-starved or cultured in 10 % FBS, were immunoblotted with anti-hsp90α antibody. β-actin served as a loading control. (A, right panel and B) Under starvation, both endogenous (4A, right panel) and exogenous hsp90α (B) are secreted from T47D cells in the acetylated form. Extra-cellular hsp90α was immunoprecipitated with anti-AcK antibody and immunoblotted with either anti-hsp90α or M2 antibody. (C) In serum-starved T47D cells, K/R substitutions at K69, K100 and K558 decrease, while K/Q substitutions increase the level of extracellular hsp90α. Supernatants of serum starved T47D cells transfected with the indicated F-tagged hsp90α mutant constructs were concentrated and immunoblotted with anti-F antibody. Coomassie-stained non-specific proteins served as the loading control. (D) K/Q substitution affects hsp90 expression on cell surface. MB-231 cells transfected with the indicated constructs were cultured under serum-free condition for 24 hours and followed by the labeling of surface protein with biotinylation. Biotinylated hsp90 on cell surface was detected with anti-F antibody. Biotinylated actin on cell surface served as loading control. Supernatants of serum-starved MB-231 cells transfected with the indicated F-tagged K/Q hsp90α mutant constructs were also concentrated and immunoprecipitates with anti-M2 conjugated beads were immunoblotted with anti-MMP-2 antibody.
Extracellular hyperacetylated hsp90α binds MMP-2 and promotes tumor cell invasion
In cancer cells, extra-cellular hsp90α was shown to act as a chaperone and assist in the maturation of the matrix metalloproteinase (MMP)-2 to its active form (12). Consistent with this, our findings show that K/Q mutants expressed in MB-231 cells, especially K69Q, K100Q and K558Q, which are preferentially extra-cellular under serum-free culture conditions, bind MMP-2 in the extracellular medium (Figure 4D). Overall, these data indicate that acetylation promotes not only the extra-cellular location of hsp90α but also facilitates its chaperone association with MMP-2. Next, we determined whether following treatment with sub-lethal concentrations of pan-HDAC-inhibitor increased extra-cellular levels of acetylated hsp90α increases invasiveness of cancer cells. Figure 5A shows that treatment with LBH589 dose dependently increased matrigel invasion by MB-231 cells. This was associated with increased extra-cellular binding of acetylated hsp90α with MMP-2 (Figure 5B). Similar increase in the invasiveness of MB-468 cells was observed following treatment with LBH589 or SAHA (Figure 5C). We next determined whether increased extra-cellular location of K69Q, K100Q or K558Q mutant promotes in vitro invasiveness of breast cancer cells. For this, we created stable transfectants of MB-468 cells expressing the wild-type hsp90α, or the K/Q or K/R mutants of hsp90α at the residues K69, K100 and K558, and their in vitro invasiveness in the matrigel-based assay was evaluated. As compared to the MB-468 cells expressing the wild-type hsp90α, stable transfectants of MB-468 cells with K/Q but not K/R mutants at K69, K100 and K558 demonstrated increased in vitro invasiveness (Figure 5D).
Figure 5. Acetylation of hsp90α promotes in vitro invasion by breast cancer cells.
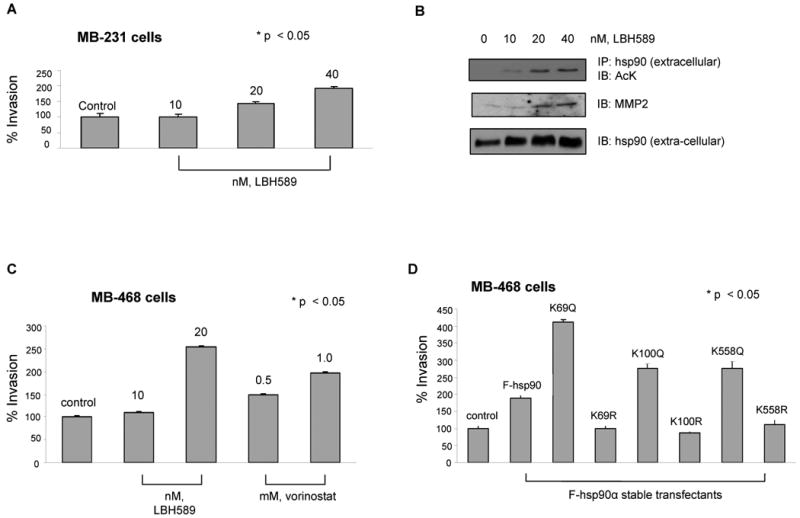
(A and B) LBH promotes in vitro invasion of MB-231 cells, associated with extra-cellular location and binding of acetylated hsp90α to MMP-2. (A) MB-231 cells were treated with the indicated concentrations of LBH589 for 16 hours. Following this, an aliquot of the cells was used for determining in vitro matrigel invasion (see text for details) Columns, average results of three independent experiments; bars, SD. *, p < 0.05. (B) Alternatively, supernatant of serum starved LBH treated MDA-MB-231 cells were concentrated and immunoprecipitates with anti-hsp90α antibody were immunoblotted with anti-AcK, anti-MMP-2 or hsp90α antibody. (C) HDAC inhibitors promote in vitro invasion by MB-468 breast cancer cells. Serum-starved MB-468 cells were used for determining in vitro matrigel invasion in the indicated concentrations of LBH589 or vorinostat and then evaluated for in vitro invasion. Columns, average results of three independent experiments; bars, SD. *, p < 0.05. (D) Stable transfection of K/Q but not K/R substituted mutants promote in vitro invasion by MB-468 cells. Serum-starved MB-468 cells expressing either F-hsp90α, or K/Q or K/R substituted mutants were used for determining in vitro matrigel invasion. Columns, average results of three independent experiments; bars, SD. *, p < 0.05.
Treatment with anti-AcK hsp90α antibody inhibits in vitro invasion by breast cancer cells
The findings above raised the possibility that treatment with an antibody to acetylated (Ac) hsp90α would inhibit binding of hsp90α to MMP-2 that is involved in tumor invasiveness. Therefore, the anti-Ac- hsp90α would thereby inhibit invasiveness of breast cancer cells. To verify this, first we compared the extra-cellular location of the ectopically expressed K69Q mutant versus the wild-type hsp90α in the breast cancer MB-231 cells. As shown, compared to the location of the wild-type hsp90α, which is mostly intracellular (extra-cellular to intracellular ratio of 13.1), relatively more of the K69Q mutant hsp90α was extracellular (extra-cellular to intracellular ratio of 1.9) (Figure 6A). Next, a polyclonal antibody was generated against the acetylated K69-containing peptide of hsp90α (anti-Ac-K69). The specificity of the antibody was confirmed by determining its ability to detect the increase in the acetylation of the ecotopically expressed (Figure 6B, upper panel) or endogenous hsp90α (Figure 6B, lower panel), following treatment with the HDAC inhibitor LBH589. As shown, following treatment of cells with LBH589, the anti-Ac-K69 hsp90α antibody recognized the increase in the acetylated hsp90α (Figure 6B). Importantly, the anti-Ac-K69 hsp90α antibody selectively recognized acetylated hsp90α (Supplemental Figure 4A). In contrast, the commercially available polyclonal anti-hsp90α antibody non-specifically recognized both the acetylated and un-acetylated hsp90α, without showing specific increase in the epitope detection following treatment of the cells with serum-starved condition or with LBH589 (Supplemental Figure 4B). Therefore, this raised the possibility that the anti-Ac-K69 antibody might be more selective in attenuating the role of Ac-hsp90α in the invasiveness of cancer cells. Figure 6C demonstrates that, as compared to the control IgG-treated or untreated MB-231 cells, serum-starved MB-231 cells demonstrated surface location of the acetylated hsp90α when stained with the anti-Ac-K69 antibody (Figure 6C). Additionally, treatment of serum-starved MB-231 cells with LBH589 led to increased levels of the membrane associated acetylated hsp90α, as detected by the anti-Ac-K69 antibody. Next, we compared the effect of anti-hsp90α with anti-Ac-K69 hsp90α antibody on the in vitro invasiveness of MB-231 breast cancer cells. Figure 6D clearly demonstrates that, while the control IgG had no significant effect and the commercially available polyclonal anti-hsp90α antibody only modestly inhibited the in vitro invasiveness of MB-231 cells, treatment with the anti-Ac-K69 hsp90α markedly inhibited the matrigel invasion by MB-231 cells (Figure 6D). Thus acetylation of K69 in hsp90α may play an important role in extra-cellular location and chaperone association of hsp90α with MMP-2. Additionally, exposure to anti-Ac-K69 hsp90α antibody could inhibit invasion of breast cancer cells.
Discussion
Recently, the hsp90 ortholog in the yeast was shown to physically or genetically interact with at least 10% of the yeast proteome (33). By analogy, hsp90 is increasingly recognized as a chaperone and master regulator for the key cell signaling networks or cellular transcription in human cells, especially in the transformed cells (34). Hyperacetylation of hsp90 resulting from inhibition of HDAC6 has been shown to inhibit the ATP, co-chaperone p23 and client protein binding of hsp90. This directs the client proteins, including several oncoprotein signaling kinases and nuclear hormone receptors to polyubiquitylation and proteasomal degradation (17, 18, 35-37). Although it is likely that not all of the known hsp90 client proteins would be uniformly affected (38), post translational modifications of hsp90 such as phosphorylation and acetylation influence the chaperone function of hsp90, thereby affecting the signaling networks that govern the malignant phenotype and response to therapy of cancer cells (39). By utilizing mass spectrometry on hsp90α isolated from HDAC inhibitor-treated cells, we could document hyperacetylation of surface-residing seven lysine residues in the N-terminus and the middle domain of hsp90α. Unlike a previous report, in our study, under the experimental conditions we employed for mass spectrometry studies, K294 was not found to be acetylated in hsp90α (20). Most likely this is so because different tryptic digestion protocols and different HDAC inhibitors were utilized in the two studies. Therefore, there may have been preservation and detection of K294 acetylation in the previous report and not of the other acetylated lysine residues identified in our study (20). In the previous report, authors had conjectured that lysine residues other than K294 may also be involved in the acetylation/deacetylation modification (20). Although other HATs may be involved, in the present studies we also determined that p300 is one of the HATs involved in acetylating hsp90α.
In our studies, all but one of the acetylation-mimetic mutants showed decreased binding to ATP. The exception was the K292Q mutant, which demonstrated increased binding to ATP. Most K/Q mutants of the lysine residues in the middle domain, i.e., K292, K478, K546 and K558, displayed decreased binding to p23, hsp40, hsp70, as well as with the client protein c-Raf. Binding to CHIP, which is a C-terminal interacting protein of hsp70 (40), was also disrupted. Conversely, the K/Q mutants showed increased binding to B-GA. This suggests that charge neutralization at the lysine residues in the middle domain of hsp90α decreases the ATP and co-chaperone binding but increases GA binding. Consistent with this, treatment with LBH589, which dose-dependently induced hyper-acetylation of hsp90, also increased the B-GA binding to hsp90α in a dose dependent manner. In a previous report, we demonstrated that co-treatment with a pan-HDAC inhibitor enhances antileukemia effects of 17-AAG with greater depletion of hsp90 client proteins in the leukemia cells (41). This strongly suggested that hyperacetylation of hsp90 could augment 17-AAG binding to hsp90 and inhibition of its chaperone function, resulting in superior antileukemia activity of the combination of HDAC inhibitor and 17-AAG. Indeed, in LBH589-treated cells 17-AAG was able to more efficiently displace B-GA, indicating increased affinity of the hyperacetylated hsp90α for binding to 17-AAG. However, for unclear reasons, this was not observed in LBH589-treated K/R mutants of hsp90α. Overall, these observations create a rationale to determine the in vivo activity of the combination of HA-HDI and 17-AAG against human leukemia cells.
Recently, hsp90 was shown to be present on the surface of metastatic melanoma cells (42). It is the inducible isoform hsp90α, not hsp90β, which is secreted and present on the surface of cancer cells, (12). Serum-starvation of breast cancer cells promoted secretion and extra-cellular localization of the endogenous or ectopically expressed hsp90α, which was notably hyper-acetylated under these circumstances. LBH589 treatment also promoted the export and extra-cellular localization of hyper-acetylated hsp90α into the culture medium. Although it lacks the classical signal sequence, the hyper-acetylated hsp90α is extra-cellular. Our data also show that K69Q, K100Q and K558Q substitutions promote the export and extra-cellular location of hsp90α, while K/R substitutions at the same residues markedly reduce the export and extra-cellular location of hsp90α. To a variable extent, treatment with LBH589 was also able to induce the export and extra-cellular location of the K/R mutants of hsp90α. Thus, acetylation status of hsp90α at specific sites may influence the extra-cellular localization of hsp90α. While hsp70, p23, cdc37 and Hip (hsp70 interacting protein) have also been detected in the extra-cellular environment of tumor cells, their precise function in this location has not been elucidated (30, 43). Whether ATP binding and hydrolysis is required for the extra-cellular chaperone function of hsp90α remains to be determined.
Extra-cellular hsp90α acts as a chaperone and assists in the maturation of the matrix metalloproteinase (MMP)-2 to its active form (12). Our data also show that acetylation promotes not only the extra-cellular location of hsp90α but also facilitates its chaperone association with MMP-2. Acetylation-mimetic mutants K69Q, K100Q and K558Q exist more on the cell surface compared to the wild-type hsp90α. Therefore, HDAC inhibitor mediated hyperacetylation of the endogenous hsp90α at these residues most likely promotes binding and maturation of MMP-2. This is supported by the observation that K/Q mutants, especially at K69, K100 and K558 residues, bind MMP-2 in the extra-cellular medium. Eustace et al had clearly documented that the presence of hsp90α on the cell surface was critical for the in vitro invasiveness of HT-1080 fibrosarcoma cells (12-30). We further elucidate that sub-lethal concentrations of LBH589, which was associated with increased extra-cellular binding of acetylated hsp90α with MMP-2, increased invasiveness of breast cancer cells. Increased extra-cellular localization and chaperone association of K69Q, K100Q and K558Q with MMP-2 also promoted in vitro invasiveness by breast cancer cells. Previous studies have shown that treatment with extra-cellularly-restricted GA beads, designed to inhibit extra-cellular chaperone function of hsp90α, decreased active MMP-2 levels and inhibited invasiveness of cancer cells by 80% (12). Here, we generated a novel antibody against the acetylated K69 containing hsp90α peptide. This anti-Ac-K69 hsp90α antibody specifically recognizes the acetylated and not the un-acetylated endogenous hsp90α on the cell surface. Exposure to this antibody markedly inhibited the in vitro matrigel invasion by breast cancer cells, and this effect was superior to the inhibition observed with the commercially available anti-hsp90α antibody. This evidence supports that acetylation of K69 plays an important role in extra-cellular chaperone association of hsp90α with MMP-2, which may regulate invasion by breast cancer cells.
Serum-starvation, hypoxia, high concentrations of glucose and oxidative stress have all been shown to promote the export and extra-cellular location of hsp90α (13, 16, 32). Under cellular stress, the intracellular signaling events result in phosphorylation or hyperacetylation of hsp90α. Under serum- and nutrient-depleted, hypoxia-stressed environment, in the established primaries, hsp90α is likely to be hyper-acetylated and extra-cellular. Boyault et al have recently demonstrated that cellular stress and increased levels of misfolded polyubiquitylated proteins in cancer cells triggers the dissociation of a repressive HDAC6/HSF1 (heat shock factor 1)/hsp90 complex and a subsequent HSF1 activation, which induces the expression of major cellular chaperones (44). Dissociation of the complex also induces hsp90α acetylation, which would export hsp90α to the cell surface. Extra-cellularly, it would bind and promote the maturation of MMP-2, thereby facilitating invasion and metastasis. Parenthetically, activation of HSF1 and heat shock response by proteotoxic stress was identified as a potent modifier of tumorigenesis and required for tumor initiation and maintenance in cancer cells, highlighting a non-oncogene addictive stress phenotype of cancer cells (45, 46). A recent report demonstrated that in a multivariate analysis, the level of hsp90 expression was an independent prognostic factor of survival in patients with breast cancer (47). Our findings raise the possibility that, due to intra-tumoral stress in the primary breast cancers, increased expression, hyperacetylation and extracellular location of hsp90α promotes MMP maturation, increased tumor invasion and metastasis. This may be responsible for the overall negative impact of high hsp90α expression on the survival in breast cancer patients. Therefore, perhaps it is the expression of acetylated hsp90α which is the important determinant of metastases and overall prognosis in breast cancer. As its corollary, it would be important to determine whether the combination of LBH589 and anti-AcK69 hsp90α antibody will inhibit in vivo invasion and metastasis by breast cancer cells.
Supplementary Material
Abbreviations
- 17-AAG
17-allyl-amino-demethoxy geldanamycin
- CHCA
α-cyano-4-hydroxycinnamic acid
- CTD
C-terminal dimerization domain
- GA
geldanamycin
- HATs
histone acetyltransferases
- HDAC
histone deacetylase
- HDIs
pan-histone deacetylase inhibitors
- IP
immunoprecipitation
- K
Lysine
- LBH589
panobinostat
- MD
middle domain
- MMP
matrix metalloproteinase
- NTD
N-terminal nucleotide-binding domain
Footnotes
Author contributions: KB directed this research and was mainly responsible for manuscript preparation. YY helped in planning, and performed the experiments. JS and YT helped in the molecular modeling. WF and RR assisted in experiments. PA provided reagents and helped in directing the experimental plan. JN helped in the performance of the proteomic analyses.
References
- 1.Whitesell L, Lindquist SL. Hsp90 and the chaperoning of cancer. Nature Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 2.Isaacs J, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–17. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 3.Panaretou B, Siligardi G, Meyer P, et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone Aha1. Molec Cell. 2002;10:1307–18. doi: 10.1016/s1097-2765(02)00785-2. [DOI] [PubMed] [Google Scholar]
- 4.Morishima Y, Kanelakis KC, Murphy PJ, et al. The hsp90 cochaperone p23 is the limiting component of the multiprotein hsp90/hsp70-based chaperone system in vivo where it acts to stabilize the client protein hsp90 complex. J Biol Chem. 2003;278:48754–63. doi: 10.1074/jbc.M309814200. [DOI] [PubMed] [Google Scholar]
- 5.Roe S, Ali MM, Meyer P, et al. The mechanism of hsp90 regulation by the protein kinase-specific cochaperone p50cdc37. Cell. 2004;116:87–98. doi: 10.1016/s0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 6.Prodromou C, Panaretou B, Chohan S, et al. The ATPase cycle of hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000;19:4383–92. doi: 10.1093/emboj/19.16.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali MM, Roe SM, Vaughan CK, et al. Crystal structure of an hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–17. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shiau AK, Harris SF, Southworth DR, Agard DA. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell. 2006;127:329–40. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 9.Panaretou B, Prodromou C, Roe SM, et al. ATP binding and hydrolysis are essential to the function of the hsp90 molecular chaperone in vivo. EMBO J. 1998;17:4829–36. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grenert JP, Johnson BD, Toft DO. The importance of ATP binding and hydrolysis by hsp90 in formation and function of protein heterocomplexes. J Biol Chem. 1999;274:17525–33. doi: 10.1074/jbc.274.25.17525. [DOI] [PubMed] [Google Scholar]
- 11.Passarino G, Cavalleri GL, Stecconi R, et al. Molecular variation of human hsp90alpha and hsp90beta genes in Caucasians. Hum Mutat. 2003;21:554–5. doi: 10.1002/humu.9141. [DOI] [PubMed] [Google Scholar]
- 12.Eustace BK, Sakurai T, Stewart JK, et al. Functional proteomic screens reveal an essential extra-cellular role for hsp90α in cancer cell invasiveness. Nat Cell Biol. 2004;6:507–14. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- 13.Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007;282:9364–71. doi: 10.1074/jbc.M608985200. [DOI] [PubMed] [Google Scholar]
- 14.Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone hsp90. Embo J. 2006;25:367–76. doi: 10.1038/sj.emboj.7600930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A. Tyrosine phosphorylation of hsp90 within the P2X(7) receptor complex negatively regulates P2X(7) receptors. J Biol Chem. 2003;278:37344–51. doi: 10.1074/jbc.M301508200. [DOI] [PubMed] [Google Scholar]
- 16.Martínez-Ruiz A, Villanueva L, González de Orduña C, et al. S-nitrosylation of hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci USA. 2005;102:8525–30. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 18.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90 - a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 19.Minucci S, Pelicci P. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 20.Scroggins BT, Robzyk K, Wang D, et al. An acetylation site in the middle domain of hsp90 regulates chaperone function. Mol Cell. 2007;25:151–9. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koga F, Xu W, Karpova TS, McNally JG, Baron R, Neckers L. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc Natl Acad Sci USA. 2006;103:11318–22. doi: 10.1073/pnas.0604705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao BX, Chen HZ, Lei NZ, et al. P53 mediates the negative regulation of MDM2 by orphan receptor TR3. EMBO J. 2006;25:5703–15. doi: 10.1038/sj.emboj.7601435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiskus W, Pranpat M, Bali P, et al. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl expressing human leukemia cells. Blood. 2006;108:645–52. doi: 10.1182/blood-2005-11-4639. [DOI] [PubMed] [Google Scholar]
- 24.Chen Z, Lee FY, Bhalla KN, Wu J. Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825. Mol Pharmacol. 2006;69:1527–33. doi: 10.1124/mol.105.020172. [DOI] [PubMed] [Google Scholar]
- 25.Shkriabai N, Patil SS, Hess S, et al. Identification of an inhibitor-binding site to HIV-1 integrase with affinity acetylation and mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:6894–9. doi: 10.1073/pnas.0400873101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of hsp90 confers tumor selectivity on hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 27.Imamura F, Horai T, Mukai M, Shinkai K, Akedo H. Potentiation of invasive capacity of rat ascites hepatoma-cells by adriamycin. Cancer Res. 1990;50:2018–21. [PubMed] [Google Scholar]
- 28.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 29.Meyer P, Prodromou C, Hu B, et al. Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell. 2003;11:647–58. doi: 10.1016/s1097-2765(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 30.Eustace B, Jay D. Extracellular roles for the molecular chaperone, hsp90. Cell Cycle. 2004;3:1098–1100. [PubMed] [Google Scholar]
- 31.Liao DF, Jin ZG, Baas AS, et al. Purification and identification of secreted oxidative stress-induced factors from vascular smooth muscle cells. J Biol Chem. 2000;275:189–96. doi: 10.1074/jbc.275.1.189. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Li Y, Guan S, et al. Extra-cellular heat shock protein-90 alpha: linking hypoxia to skin cell motility and wound healing. Embo J. 2007;26:1221–33. doi: 10.1038/sj.emboj.7601579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao R, Davey M, Hsu YC, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–27. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 34.Neckers LJ. Heat shock protein 90: the cancer chaperone. Biosci. 2007;32:517–30. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Meng S, Wang H, et al. Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor LAQ824. Mol Cancer Ther. 2005;4:1311–19. doi: 10.1158/1535-7163.MCT-04-0287. [DOI] [PubMed] [Google Scholar]
- 36.Fiskus W, Ren Y, Mohaptra A, et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res. 2007;13:4882–90. doi: 10.1158/1078-0432.CCR-06-3093. [DOI] [PubMed] [Google Scholar]
- 37.Edwards A, Li J, Atadja P, Bhalla K, Haura E. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor dependent lung cancer cells. Mol Cancer Ther. 2007;6:1400–5. doi: 10.1158/1535-7163.MCT-06-0761. [DOI] [PubMed] [Google Scholar]
- 38.Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26:5468–76. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- 39.Aoyagi S, Archer TK. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Bio. 2005;15:565–7. doi: 10.1016/j.tcb.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99:12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.George P, Bali P, Annavarapu S, et al. Combination of histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 42.Becker B, Multhoff G, Farkas B, et al. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp Dermatol. 2004;13:27–32. doi: 10.1111/j.0906-6705.2004.00114.x. [DOI] [PubMed] [Google Scholar]
- 43.Shin BK, Wang H, Yim AM, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 44.Boyault C, Zhang Y, Fritah S, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–18. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–8. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, Kluger HM. High hsp90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67:2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.