

Abstract
Angiotensin-converting enzyme (ACE) inhibition induces glomerular repair in the Munich Wistar Frömter (MWF) rat, a model of spontaneous glomerular injury. In this study, we investigated whether this effect is related to changes in glomerular cell number, particularly of podocytes, which are progressively lost with age. MWF rats with advanced nephropathy were studied at both 40 weeks and after 20 weeks of observation either with or without treatment with the ACE inhibitor lisinopril. Forty-week-old Wistar rats were used as controls. In untreated MWF rats, proteinuria, hypertension, glomerulosclerosis, and renal function worsened, while lisinopril induced regression of both functional and structural changes. Despite glomerular hypercellularity in untreated MWF rats, the number of endothelial cells per glomerulus did not change, and podocyte number even decreased. ACE inhibition halted the progressive increase in glomerular cell number and enhanced endothelial cell volume density. Surprisingly, lisinopril not only halted age-related podocyte loss but also increased the number of glomerular podocytes above baseline, which was associated with an increased number of proliferating Wilms tumor 1-positive cells, loss of cyclin-dependent kinase inhibitor p27 expression, and increased number of parietal podocytes. These data indicate that ACE inhibition restructures glomerular capillary, primarily by restoring the podocyte population in this model of glomerular injury. Increased parietal podocyte number in lisinopril-treated MWF rats suggests that the remodeling of Bowman’s capsule epithelial cells contributes to this effect.
Clinical studies have documented that single or multidrug antiproteinuric treatments based on angiotensin II blockade can stabilize, or even reverse, renal disease progression in both patients with diabetic and non-diabetic nephropathies even in advanced stages of the disease.1,2,3,4,5 Actually, regression of proteinuria and glomerulosclerosis by angiotensin converting enzyme (ACE) inhibition or angiotensin II type 1 receptor (AT1R) blockade has also been documented in experimental models of progressive nephropathies, such as puromycin aminonucleoside,6 chronic nitric oxide synthase inhibition,7 renal mass ablation,8,9,10 aging,11 and the Munich Wistar Frömter (MWF) rat.12,13,14 In the latter study, combined treatment with an ACE inhibitor and an AT1R blocker given from 25 to 40 weeks of age completely reversed proteinuria, and halted progressive glomerulosclerosis, particularly in glomeruli with mild sclerotic lesions.13 Recently three-dimensional reconstruction of the capillary tuft by serial section analysis allowed us to document the effects of administration of a high dose of an ACE inhibitor starting at 50 weeks of age, when rats had a more advanced nephropathy. This treatment not only remarkably reduced sclerosis volume in most glomeruli, but also increased the volume of the glomerular tuft occupied by intact capillary by up to 40%, indicating consistent glomerular tuft repair.14 So far, the therapeutic effect of angiotensin II blockade has been mainly attributed to its capability to control extracellular matrix deposition. Inhibition of collagen synthesis,7 transforming growth factor-β,14 and plasminogen activator inhibitor-1 expression10,11 were indeed proposed as possible mechanisms responsible for sclerosis regression. However, the possibility that ACE inhibitors or AT1R antagonists can modulate glomerular cell survival and repair is intriguing and not well explored yet.
Podocyte loss has been recognized as a causal factor for renal disease progression. A recent study performed in transgenic rats for human diphtheria toxin receptor has clearly documented a strict correlation between the extent of podocyte depletion, obtained by titrating the administration of the corresponding toxin, and defined stages of glomerular damage ranging from transient proteinuria to progressive decline of renal function.15 We have recently reported in the male MWF rat that podocyte number is progressively reduced with age, and this may importantly contribute to glomerular permselective defect, proteinuria, and renal scarring on the long-term.16 This evidence prompted us to characterize changes in resident glomerular cells and infiltrating/inflammatory cells during the development of sclerotic lesions in progressive proteinuric nephropathy in male MWF rats, and to investigate which, among glomerular components, is the key player for glomerular capillary restructuring and repair induced by ACE inhibition therapy.
Materials and Methods
Study Design
Twenty-nine male MWF rats from our colony17 and six Wistar rats (Charles River S.p.A, Calco, Italy) were used in this study. MWF rats were divided into three groups. Group 1 (n = 10) consisting of untreated animals, was studied at 40 weeks of age, at which time we previously documented about 50% podocyte loss associated with massive proteinuria and glomerulosclerosis16 (MWF 40W). Group 2 (n = 10) was left untreated and followed from 40 to 60 weeks of age (MWF 60W), whereas rats of group 3 (n = 9) were treated with a high dose of ACE inhibitor lisinopril in drinking water from 40 to 60 weeks of age (MWF+LIS 60W). The dose of lisinopril was progressively increased from 80 to 100 mg/L from 50 to 60 weeks to provide a better control of systolic blood pressure. A group of Wistar rats was studied at 40 weeks of age and used as control (Wistar 40W). Systolic blood pressure, urinary protein excretion, and serum creatinine were periodically measured during the observation period by conventional methods.14,16
All animals were maintained in a temperature-controlled room regulated with a 12-hour light/dark cycle, and they had free access to water and food (standard rat chow containing 18.5% protein by weight). Animal care and treatment were conducted in conformity with the institutional guidelines that are in compliance with national and international laws and policies (EEC Council Directive 86/609, OJL 358, 1987; DL n116, G.U., suppl. 40, 18/2/1992, Circolare No.8, G.U., 14/7/1994; Guide for the Care and Use of Laboratory Animals, National Research Council, 1996). At sacrifice, both kidneys were perfused under anesthesia with PBS and processed as previously described in detail.16
In additional experiments, two groups of untreated or lisinopril treated (from 40 to 60 weeks) MWF rats (n = 2 each) were injected with 5-bromo-2′-deoxyuridine (BrdU, Sigma Aldrich, St Louis, MO, i.p. 50 mg/kg dissolved in saline) for 5 days (the last administration 2 hours before the sacrifice). In addition the thymidine analog was also given in the drinking water for the same period of time to obtain a final dose of 100 mg/kg. Kidney specimens were fixed in paraformaldehyde 4% and processed for immunohistochemistry.
Immunohistochemistry
The identification and count of resident and infiltrating cells per glomerulus, proliferating cells, and parietal podocytes, and the evaluation of cyclin-dependent kinase inhibitor expression was performed on kidney sections by single or multiple immunostaining for the following antigens: Wilms’s tumor 1 (WT1), a podocyte-specific nuclear antigen18; RECA-1, a cell surface antigen expressed by all rat endothelial cells (ECs)19; Thy1.1, a mesangial cell (MC) antigen20; ED1, an antigen present in the rat monocytes and macrophages; Ki-67, a nuclear antigen expressed during all active phases of the cell cycle (G1 to M)21; the protein gene product 9.5, an ubiquitin C-terminal hydrolase found to localize in parietal epithelial cells (PECs) of the Bowman’s capsule in normal rat22; p27, a cyclin-dependent kinase inhibitor. Cell nuclei were labeled by 4′, 6-diamidine-2′-phenylindole dihydrochloride (DAPI) and the glomerular tuft by the wheat germ agglutinin (WGA). All of the immunofluorescence experiments were performed on sections (3 μm) from paraformaldehyde-lysine-periodate-fixed kidney specimens unless otherwise specified. Antibody incubations were performed at room temperature unless otherwise specified. After antigen unmasking (10 mmol/L citrate buffer, pH 6.0 for 20 minutes at 100°C) and blocking of non-specific sites (1% bovine serum albumin), sections were incubated with a mouse monoclonal antibody (moAb) to Ki-67 (dil 1:100 in PBS, Novacastra Laboratories Ltd, Newcastle, UK) overnight at 4°C, followed by Cy5-conjugated F(ab′)2 fragment donkey anti-mouse IgG (affinity purified 10 μg/ml in PBS, Jackson Immunoresearch Laboratories Inc., West Grove, PA) for 1 hour. After washing, sections were further incubated with a rabbit polyclonal antibody to WT1 (WT1 [C-19] 4 μg/ml in PBS, Santa Cruz Biotechnology, Santa Cruz, CA) for 3 hours, followed by Cy3-conjugated goat anti-rabbit IgG (12.5 μg/ml in PBS, Jackson Immunoresearch) for 1 hour, and finally with the fluorescein isothiocyanate (FITC)-conjugated WGA (12.5 μg/ml in PBS, Vector Laboratories Inc., Burlingame, CA) for 15 minutes at room temperature and with the nuclear stain DAPI (Sigma Aldrich, St Louis, MO) for 20 minutes at room temperature. At least 30 glomerular sections per animal were randomly acquired by confocal laser scanning microscope (LS510 Meta, Zeiss, Jena, Germany) and examined by an observer unaware of the identity of samples. For each glomerulus separate images for each marker were acquired and digitally merged. Glomerular cells were identified as DAPI-positive cells and counted, and the number of cells per glomerulus was estimated by morphometric analysis. By comparing superimposed images of WT1 and DAPI with those of Ki-67 plus DAPI, we identified and counted the number of cells positive for WT1, Ki-67, or both. Proliferating cells were identified as DAPI-positive cells expressing also Ki-67. Their percentage, in respect to the total cell number, was calculated to estimate the number of proliferating cells per glomerulus. Glomerular podocytes were identified as WT1-positive cells and counted as previously described.16 Finally, the percentage of WT1-positive cells also expressing Ki-67 was calculated, to estimate the number of proliferating WT1-positive cells per glomerulus.
To validate the method for identification of proliferating WT1-positive glomerular cells, we verified whether WT1-positive cells expressing Ki-67 were also stained for BrdU. Paraformaldehyde-fixed kidney sections (5 μm) were pretreated for BrdU immunostaining by DNA denaturation (2M/L HCl for 15 minutes at 37°C) followed by 10 minutes in 0.1 M/L borate buffer (pH 8.5). After blocking, sections were incubated with WT1 (C-19) antibody overnight at 4°C followed by Cy3-conjugated goat anti-rabbit IgG (12.5 μg/ml in PBS) for 1 hour. Then sections were incubated with a moAb to Ki-67 (dil 1:100 in PBS) for 3 hours followed by Cy5-conjugated F(ab′)2 fragment donkey anti-mouse IgG (10 μg/ml in PBS) for 1 hour and finally with a FITC-conjugated moAb to BrdU (dil 1:4 in PBS, Roche Diagnostics Co, IN) for 1 hour and DAPI. For each glomerulus separate images of WT1, Ki-67, BrdU, and DAPI were obtained and in the merged image the simultaneous expression of Ki-67 and BrdU was evaluated in WT1-positive cells.
To evaluate the expression of p27 in podocytes, kidney sections were incubated with goat polyclonal antibody to p27 (3 μg/ml in PBS, Santa Cruz) overnight at 4°C followed by FITC-conjugated donkey anti-goat IgG (25 μg/ml in PBS, Jackson) for 1 hour. Then sections were incubated with WT1 (C-19) antibody (4 μg/ml in PBS) for 3 hours followed by Cy5-conjugated goat anti-rabbit IgG (7.5 μg/ml in PBS, Jackson) for 1 hour and finally with rhodamine WGA (Vector) and DAPI. At least 30 glomeruli per animal were acquired and the percentage of WT1-positive cells expressing p27 was calculated.
To evaluate glomerular expression of the glial cell line-derived neutrophic factor (GDNF), kidney sections were incubated with a moAb raised against amino acids 78 to 134, representing the mature GDNF of human origin (4 μg/ml in PBS, Santa Cruz) overnight at 4°C, followed by Cy3-conjugated F(ab′)2 fragment donkey anti-mouse IgG (6.5 μg/ml in PBS, Jackson) for 1 hour and finally with FITC-WGA (Vector) and DAPI. From 40 to 60 glomeruli per section were randomly chosen and blindly examined.
To estimate the presence of parietal podocytes, kidney sections were incubated overnight at 4°C with a moAb to WT1 (WT1 [F-6] 4 μg/ml in PBS, Santa Cruz) followed by Cy3-conjugated F(ab′)2 fragment donkey anti-mouse IgG (6.5 μg/ml in PBS, Jackson) for 1 hour and then with rabbit anti-human protein gene product 9.5 antibody (1:200, AbD Serotec, Kidlington, Oxford, UK) for 3 hours, followed by Cy5-conjugated goat anti-rabbit IgG (7.5 μg/ml in PBS) for 1 hour. Finally FITC-WGA and DAPI were added. PECs in Bowman’s capsule were identified by co-staining with DAPI and protein gene product, and by morphology (WGA staining of Bowman’s capsule). Parietal podocytes were identified as PEC expressing WT1. The number of PECs and parietal podocytes was determined using nuclear count in at least 20 glomeruli for each animal, and the length of Bowman’s capsule was also measured. Then the parietal podocyte/PEC number ratio and PEC number/Bowman’s capsule length ratio were computed.
To investigate the presence of inflammatory cells infiltrating the glomerular tuft, kidney sections were incubated with a moAb to ED1 (dil 1:100, Chemicon, Temecula, CA) for 1 hour, followed by Cy3-conjugated F(ab′)2 fragment donkey anti-mouse IgG (6.5 μg/ml in PBS) for 1 hour and then with FITC-WGA. At least 30 glomeruli for each animal were acquired and ED1-positive cells counted.
To estimate glomerular ECs, sections were incubated with a mouse anti-rat RECA-1 moAb (10 μg/ml Serotec) for 1 hour, followed by Cy5-conjugated F(ab′)2 fragment donkey anti-mouse IgG (10 μg/ml in PBS) for 1 hour, and then with FITC-WGA and DAPI. Glomerular cells were identified by counting DAPI-positive nuclei surrounded by RECA-1 staining in at least 30 glomeruli per animal. Another series of sections was used to estimate the volume density (Vv) of glomerular ECs using 15 glomerular sections per animal.
To estimate the number of glomerular MCs, sections were incubated with a mouse anti-rat Thy1.1 moAb (dil 1:100, AbD Serotec) for 1 hour, followed by Cy3-conjugated F(ab′)2 fragment donkey anti-mouse IgG (6.5 μg/ml in PBS, Jackson) for 1 hour, and then with FITC-WGA and DAPI. Glomerular MCs were identified by counting DAPI-positive nuclei surrounded by Thy1.1 staining in at least 30 glomeruli per animal.
Morphometric Analysis
The extent of glomerular sclerotic lesions was estimated on a single kidney cortical section (3.5 μm in thickness) stained with periodic acid-Schiff examined by light microscopy (Zeiss, Jena, Germany) using computer-based morphometric analysis. For each kidney section, about 50 glomeruli were systematically digitized (glomeruli were consecutively encountered moving the microscope stage with an S-shape path) using a ×40 objective, and digital images were acquired and processed with interactive tools (Image J, http://rsb.info.nih.gov/ij) to outline separately the outer polygon of the glomerular capillary tuft and, if present, the sclerotic region of the glomerular tuft. The percentage of glomerular volume occupied by sclerotic changes was then calculated as the percent ratio of the sclerotic surface area over the total area of capillary tuft, based on the principle that area density is equivalent to volume density.23
Estimation of mean glomerular volume (VG) was performed on digital images of cortical tissue either stained with periodic acid-Schiff or labeled with fluorescein-WGA as previously described.16 The latter was used for the calculation of the mean number of podocytes per glomerulus. VG was calculated using the formula
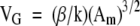 |
where Am is the mean glomerular cross-sectional area, k = 1.01 is a size distribution coefficient, and β = 1.38 is the shape coefficient for glomeruli that are assumed to be spherical.23
The mean number of resident and infiltrating cells per glomerular tuft was calculated as the product of cell volume density (Nv) and (VG),16 as previously described in details. Briefly, Nv was calculated as NV = NA / D̄ , where NA, the cell nuclear profile area density, is the ratio between the number of cell nuclear profiles and the measured glomerular capillary tuft area. D̄ is the average diameter of cell nuclei that we estimated from major and minor axis of nuclear sections, measured on digital images assuming ellipsoidal shape of cell nuclei. This technique was used to estimate average number of infiltrating cells per glomerulus, total number of resident cells, podocytes, ECs, and MCs. To further verify the precision of podocyte estimation, we used serial sections of an entire glomerular capillary and calculated the actual number of podocytes using high-resolution digitized images. Briefly, kidney tissue was obtained from a normal Wistar rat (40 weeks of age). Serial sections (1 μm thick) of a whole glomerular tuft were cut from an epon resin embedded sample (more than 200 sections in total) and sequentially collected. Digital micrographs of toluidine blue stained sections were acquired, from the beginning to the end of the Bowman’s capsule (141 sections), by optical microscopy (at 100x). Images were digitally rotated and translated using image analysis software developed in house to generate a stack of aligned images containing the entire capillary tuft. In each image of the stack the nuclei of podocytes were identified and marked with a colored circle on all images, as reported in supplemental Figure S1 (see http://ajp.amjpathol.org) for a representative section. The image stack was then digitally processed for automatic segmentation of colored volumes and counting, each volume corresponding to a podocyte nucleus. There is a complete agreement of the estimation of the podocyte number with the two techniques (supplemental Figure S1, see http://ajp.amjpathol.org).
The volume of capillary tuft occupied by ECs was determined on the basis of area density. To this purpose, digital images of capillary tuft outline were thresholded on green color for WGA staining to identify the area occupied by capillary cells and matrix and on red color to identify RECA-1 staining. Then the ratio between red and green pixels was calculated as the percentage of tuft cellular volume occupied by ECs.
Transmission Electron Microscopy of Visceral Podocytes and PECs
For transmission electron microscopy analysis, ultrathin sections (60 to 100 nm) were cut, from cortical kidney tissue samples embedded in epon resin, by an ultramicrotome (LKB, Bromme, Sweden), collected on copper grids, and stained with uranyl acetate and lead citrate. Ultrastructural analysis was performed by transmission electron microscopy (Morgagni 268D, Philips, Eindhoven, The Netherlands).
Statistical Analysis
Results are expressed as mean ± SD. Comparisons were made by analysis of variance and Bonferroni/Dunn post hoc analysis using the computer software Stat View (Abacus Concept Inc., Berkeley, CA). Statistical significance was defined as P < 0.05.
Results
Effect of ACE-Inhibition on Renal Function and Structure
As expected,14 male MWF rats developed massive proteinuria, hypertension, and impairment of renal function with age. As reported in Table 1, treatment with the ACE inhibitor lisinopril, besides controlling systolic blood pressure, reverted proteinuria and completely prevented the rise in serum creatinine. ACE inhibition also significantly (P < 0.01) reduced kidney weight as compared with untreated MWF rats (right kidney averaged 1.41 ± 0.16, 1.43 ± 0.10, and 1.04 ± 0.11 g in untreated rats at 40 and 60 weeks, and in lisinopril-treated rats at 60 weeks, respectively).
Table 1.
Effect of ACE Inhibition on Blood Pressure and Kidney Functional Parameters
| Age (weeks) | MWF | MWF+LIS | Wistar |
|---|---|---|---|
| SBP (mmHg) | |||
| 40 | 171 ± 4 | 170 ± 8*† | 130 ± 7‡ |
| 50 | 173 ± 5 | 138 ± 5*† | |
| 60 | 181 ± 7 | 130 ± 26 | |
| UPE (mg/24 hours) | |||
| 40 | 551 ± 72 | 632 ± 149 | 20 ± 4‡§ |
| 50 | 678 ± 53 | 197 ± 74*† | |
| 60 | 914 ± 236†¶ | 228 ± 91*† | |
| Screa (mg/dl) | |||
| 40 | 0.76 ± 0.20 | 0.77 ± 0.19 | 0.53 ± 0.1 |
| 50 | 0.97 ± 0.27 | 0.86 ± 0.11 | |
| 60 | 1.77 ± 0.77∥ | 0.91 ± 0.06 |
Data are mean ± SD.
P < 0.01 vs untreated MWF rats.
P < 0.01 vs 40 weeks of age within the same group.
P < 0.01 vs untreated MWF group and lisinopril-treated MWF rats at 40 weeks.
P < 0.05 vs lisinopril-treated MWF rats at 60 weeks.
P < 0.01 vs 50 weeks of age within the same group.
P < 0.01 vs 40 and 50 weeks of age within the same group, lisinopril-treated MWF group and Wistar rats.
SBP, systolic blood pressure; UPE, urinary protein excretion; Screa, serum creatinine; LIS, lisinopril.
The results of morphometrical analysis of glomerular structural changes at light microscopy are reported in Table 2. Mean VG was markedly higher in untreated MWF rats than in age-matched Wistar rats, and did not significantly change with age. In MWF rats given lisinopril, VG was significantly lower (P < 0.05) than in untreated animals. In untreated MWF rats at 40 weeks, 87 ± 7% of glomeruli showed capillary areas occupied by sclerosis accounting for more than 35% of the tuft volume. At 60 weeks almost all glomeruli were sclerotic and the fraction of glomerular volume with sclerotic changes further increased in MWF rats up to about 60%. Treatment with lisinopril significantly reduced the incidence of glomerulosclerosis and regressed glomerular lesions, sclerosis volume averaging less than 25% (P < 0.05 vs. untreated MWF rats at 40 weeks).
Table 2.
Glomerular Volume and Incidence and Extent of Glomerular Lesions
| MWF 40W | MWF 60W | MWF+LIS 60W | Wistar | |
|---|---|---|---|---|
| VG (μm3 x 106) | 1.75 ± 0.29 | 1.85 ± 0.25 | 1.40 ± 0.25*† | 0.70 ± 0.07‡ |
| Glomeruli affected by sclerosis (%) | 87 ± 7 | 99 ± 2** | 85 ± 8† | 4 ± 4‡ |
| Sclerosis Volume (%) | 35.8 ± 10.0 | 62.0 ± 9.8** | 24.8 ± 4.7*† | 0.3 ± 0.4‡ |
Data are mean ± SD.
P < 0.05,
P < 0.01 vs untreated MWF rats at 40 weeks.
P < 0.01 vs untreated MWF rats at 60 weeks.
P < 0.01 vs all MWF groups.
Abbreviations: VG, mean glomerular volume; LIS, lisinopril.
Glomerular volume expansion in MWF rats might reflect glomerular cell hypertrophy, or hypercellularity, or a combination of both. We then estimated, by histomorphometric analysis, the mean number of resident and infiltrating cells per glomerulus. As shown in Figure 1A, the mean number of cells per glomerulus in untreated MWF rats at 40 weeks was significantly higher (>60% higher, P < 0.01) than that measured in age-matched Wistar rats, and further increased at 60 weeks. Increased cellularity occurred in both non-sclerotic and sclerotic areas of the glomerular capillary tuft. In lisinopril-treated rats, mean glomerular cell number was significantly lower (P < 0.01) than in untreated MWF rats at the same age. Estimation of infiltrating/inflammatory cells within glomerular tufts, by ED1 immunostaining, allowed for documenting the presence of more infiltrating cells in MWF rats than in Wistar animals (Figure 1B). Mean number of ED1-positive cells per glomerulus was significantly increased by age in MWF rats, and was reduced by ACE inhibitor, but in any case it accounted only for <1% of total number of cells within the capillary tuft. We then estimated the number of proliferating cells in the capillary tuft by immunostaining. As shown in Figure 1C, the mean number of cells per glomerulus expressing the proliferation marker Ki-67 was significantly higher in MWF rats than in Wistar rats and was not significantly affected by age and treatment. In average proliferating cells were < 4% of total glomerular cells.
Figure 1.

Effect of ACE inhibition on resident and infiltrating cells per glomerulus. Estimation of the mean number of (A) cells per glomerulus, (B) infiltrating/inflammatory cells per glomerulus, and (C) proliferating cells per glomerulus in Wistar rats and in untreated or lisinopril-treated MWF rats. Results are mean ± SD. *P < 0.05 and **P < 0.01 vs. untreated MWF rats at 40 weeks; °P < 0.05 and °°P < 0.01 vs. untreated MWF rats at 60 weeks; §P < 0.01 vs. all MWF groups and #P < 0.01 vs. untreated MWF rats at 60 weeks.
Effect of ACE Inhibition on Visceral and Parietal Podocytes
Since glomerular cell number was importantly affected by the renal disease in MWF and, in particular, since changes in podocyte number contributed to disease progression,16 we investigated the effect of ACE inhibition treatment on the number of glomerular podocytes. As reported in Figure 2A, in 40-week-old untreated MWF rats podocyte number per glomerulus was lower than in age-matched Wistar rats (averaging 109 ± 14 vs. 159 ± 11 cell/glomerulus, respectively, P < 0.01) and further decreased at 60 weeks (58 ± 18 cell/glomerulus, P < 0.01 vs. MWF rats at 40 weeks and Wistar rats). Loss of podocyte number with age positively correlated with the extent of glomerular lesions (Figure 2B). At variance, lisinopril treatment increased the podocyte number per glomerulus above baseline, reaching values comparable to Wistar rats (144 ± 22 cell/glomerulus, P < 0.01 vs. untreated MWF rats, Figure 2A). Consistently, higher number of podocytes induced by ACE inhibition significantly correlated with lower glomerular volume occupied by sclerosis (Figure 2B).
Figure 2.

Effect of ACE inhibition on visceral and parietal podocytes. Estimation of (A) the mean number of podocytes per glomerulus. B: Statistically significant correlation, by linear regression analysis, between podocyte number/glomerulus and mean percentage of glomerular capillary occupied by sclerosis. C: WT1+Ki-67+ cells per glomerulus, (D) p27-positive podocytes, (E) PEC number/Bowman’s capsule length ratio, and (F) parietal podocytes/PEC ratio in Wistar rats and in untreated or lisinopril-treated MWF rats. Results are mean ± SD. *P < 0.05 and **P < 0.01 vs. untreated MWF rats at 40 weeks; °°P < 0.01 vs. untreated MWF rats at 60 weeks; ¶P < 0.01 vs. untreated MWF rats and #P < 0.05 and ##P < 0.01 vs. lisinopril-treated MWF rats at 60 weeks.
To investigate whether restoration of glomerular podocytes was associated to cell proliferation, we estimated the number of cells showing nuclear co-localization of WT1 and Ki-67 (see representative images in Figure 3A–C). As shown in Figure 2C, in Wistar rats, a very low number of WT1-positive cells also expressed Ki-67 (0.2 cells/glomerulus) and a mild increase was found in untreated MWF rats accounting for <5% of total WT1-positive cells, both at 40 and 60 weeks of age. At variance, a statistically significant higher number of WT1+ Ki-67+ cells (>11% in average, P < 0.01) was observed in lisinopril-treated animals, as compared with untreated MWF rats at both ages.
Figure 3.

Immunohistochemistry of proliferating WT1-positive cells. A–C: Representative confocal images of a glomerulus from a lisinopril treated MWF rat showing labeling for podocytes (WT1-positive nuclei in red), for cell nuclei (DAPI in blue), for cell proliferation (Ki-67-positive nuclei in pseudocolor, white), and for glomerular capillary tuft (WGA in green). A: Superimposed images of WT1 and DAPI reveal pink staining of podocyte nuclei. B and C: Some cells show nuclear colocalization of WT1 and Ki-67 (arrows). Cell nuclei positive only for Ki-67 identify proliferating cells, which are not podocytes (arrow heads). D–F: Representative images of a glomerulus from a lisinopril-treated MWF rat, which also received Brdu for 5 days before the sacrifice (see Materials and Methods for details). Positive nuclear staining for Brdu (in green, F) is detected in a WT1-positive cell (D) expressing ki67 (E). Final magnification = original ×400.
To confirm that our results were not affected by experimental artifacts, we compared, in WT1-positive cells, nuclear staining for the thymidine analog BrdU, which labels cells into the S-phase of the cell cycle, and the expression of Ki-67. As shown by representative images in Figure 3, D–F, in line with similar comparisons reported in the literature,24 in a series of glomerular sections, we verified that almost all cells expressing Ki-67 and WT1 were also stained for BrdU.
Since cell cycle progression and proliferation is favored by reduction in cyclin-dependent kinase inhibitors including p27,25,26 we investigated the glomerular podocyte expression of p27 in the four animal groups. In Wistar rats p27 was expressed in up to 73% of WT1-positive cells (Figure 2D) and a comparable percentage of podocytes expressed p27 in untreated MWF rats at 40 and 60 weeks. At variance, in lisinopril-treated rats the percentage of p27-expressing podocytes was significantly lower (about 41%) than in untreated animals at baseline.
It has been shown that PECs of the Bowman’s capsule may express a podocyte phenotype,27,28 and these cells may be a potential source of visceral podocytes. We then evaluated the number of parietal podocytes along the Bowman’s capsule in Wistar and in MWF rats. As shown in Figure 2E, PEC number over Bowman’s capsule length ratio did not differ between Wistar and MWF rats, independently of age and treatment. The percentage of parietal podocytes over the total PECs was comparable between Wistar and MWF rats at 40 weeks, averaging about 4.6% of PECs (Figure 2F). With age this percentage numerically decreased in MWF rats (averaging 2.1 ± 0.9%), but the difference was not statistically significant. At variance, in lisinopril-treated rats, the percentage of parietal podocytes was significantly higher than in untreated MWF at the same age (P < 0.01) and in Wistar rats (P < 0.05), averaging 7.5 ± 3.2% (Figure 2F).
The results of GDNF expression at the glomerular level are reported in Figure 4, A–D. In Wistar and MWF rats, selective GDNF immunostaining was visualized in smooth muscle cells of renal arteries and arterioles, and at the glomerular vascular pole. Rarely glomeruli of Wistar rats showed focal and weak staining that was confined to single cells in mesangial area. This GDNF staining was slightly increased in untreated MWF rats, affecting about 30% of glomeruli at 60 weeks, but remaining confined to small regions of the glomerular capillary. GDNF expression was absent in the Bowman’s capsule of Wistar animals (see Figure 4A). At variance, strong GDNF expression was observed in untreated MWF rats as shown in Figure 4, B–C, affecting 43 ± 9% and 53 ± 19% of glomeruli at 40 and 60 weeks, respectively. Lisinopril treatment was associated with a marked decrease in GDNF expression in Bowman’s capsule, as compared with untreated animals (see Figure 4D) being partially present in only 15 ± 3% of glomeruli.
Figure 4.

Immunohistochemistry of GDNF and RECA-1. A–D: Representative confocal images of GDNF expression in glomeruli from Wistar rats (A), untreated MWF rats at 40 (B) and at 60 weeks of age (C), and in lisinopril-treated MWF rats (D). Staining for GDNF is in red, WGA in green, and DAPI in blue. GDNF is strongly expressed in the Bowman’s capsule of glomeruli from untreated MWF rats. E–L: Representative confocal images of glomeruli labeled with RECA-1 (in red) showing EC immunostaining in Wistar rats (E, F), in untreated MWF rats at 40 (G, H) and at 60 weeks of age (I, J), and in lisinopril-treated MWF rats (K, L). Reduction in glomerular EC volume is observed in untreated MWF rats. The area of glomerular tuft positive for RECA-1 is increased by ACE inhibition (merged images of WGA, in green, and RECA-1). Final magnification = original ×400, (A–D); ×630, (E–L).
Effect of ACE Inhibition on Glomerular Endothelial and Mesangial Cells
Beside podocytes, we estimated the number of ECs per glomerulus and their volume density in the glomerular tuft, as well as the number of MCs per glomerulus. As shown in Figure 5A, independently of treatment and age, no significant change in the mean number of ECs per glomerulus was observed between MWF and control Wistar rats. By contrast, as shown in representative images of Figure 4, E–L, the density of glomerular tuft volume occupied by glomerular ECs was importantly reduced in untreated MWF rats at 40 and 60 weeks of age, as compared with control Wistar rats at 40 weeks (see also Figure 5B). In lisinopril treated rats Vv of glomerular ECs was significantly higher (P < 0.01) than in untreated MWF rats at both 40 and 60 weeks of age (Figure 5B). As shown in Figure 5C, linear regression analysis showed an important and significant positive correlation (r = 0.7, P < 0.01) between the number of podocytes per glomerulus and the mean value of glomerular EC Vv.
Figure 5.

Effect of ACE inhibition on glomerular ECs and MCs. Estimation of (A) the mean number of ECs per glomerulus, and (B) glomerular EC volume density (Vv) in Wistar rats and in untreated or lisinopril-treated MWF rats. C: Statistically significant correlation, by linear regression analysis, between Vv of glomerular EC and the number of podocytes per glomerulus in untreated MWF rats at 40 and 60 weeks of age and in lisinopril-treated MWF rats. D: Mean number of MCs per glomerulus in Wistar rats and in untreated or lisinopril-treated MWF rats. Results are mean ± SD. *P < 0.05 and **P < 0.01 vs. untreated MWF rats at 40 weeks; °P < 0.05 and °°P < 0.01 vs. untreated MWF rats at 60 weeks; §P < 0.01 vs. all MWF groups.
The mean number of MCs per glomerulus is reported in Figure 5D. The number of MCs was significantly higher in glomeruli from untreated MWF rats as compared with controls and tended to increase with age. Treatment with lisinopril halted the progressive increase in MC number.
Discussion
In MWF rats, compensatory glomerular tuft enlargement developed with age, due to nephron inborn deficit,29 with proteinuria, progressive podocyte loss, and glomerulosclerosis.16 Recently, we have reported that ACE inhibition treatment, even when started in the advanced stage of glomerular injury, induced glomerular capillary restructuring with increase in volume of normal glomerular capillary.14 To elucidate the mechanisms involved in this process of capillary regeneration, here we investigated whether ACE inhibition affected changes in cellular components of the glomerular capillary tuft and Bowman’s capsule. Our present data demonstrate that glomerular cell number was significantly higher in MWF rats than in normal Wistar rats of same age, likely as a result of compensatory growth. Of interest, among resident cells of the glomerular tuft, while podocytes were lower in number in MWF rats than in normal controls, the mean glomerular EC number was comparable between the two strains. On the contrary MCs significantly increased in MWF rats. Infiltrating/inflammatory cells, although present in these animals, were only quantitatively marginal, making their contribution to glomerular tuft hypercellularity negligible.
Previous estimation of podocyte number in Wistar and MWF rats16 is in line with our present results, although in our earlier study podocyte count in Wistar rats was numerically higher. The difference between the two studies may depend on the variability between individual animal data and on slightly different experimental conditions. We are confident that, on the basis of the validation of our measurements (see Materials and Methods), our present results are reliable.
At variance to the progressive loss observed with age, when animals were treated with the ACE inhibitor, podocyte number per glomerulus selectively increased, and this may have contributed to maintain glomerular podocyte architecture, reducing the filtration surface area covered by each podocyte, and likely preventing cell stress and dysfunction.16,30,31 The reversal of glomerulosclerosis associated with reversal of podocyte loss in this model is different from what reported for the subtotal nephrectomy model. In the latter, high dose enalapril induced no changes in podocyte number.8 However, different mechanisms may underlie glomerular injury between these two models. Five/sixth nephrectomy is characterized by sudden hemodynamic changes, while the MWF rat model is related to a slow adaptation to a reduced number of nephrons.29 In addition, glomerular damage induced by 5/6 nephrectomy may be more extensive than in MWF rats because of massive reduction in functioning nephrons (>80%) and very large increase in glomerular volume (>250%). In MWF rats VG increased by 90%, as compared with Wistar rats and was slightly although significantly reduced by the ACE inhibition (<20%). This reduction in VG was not observed previously in these strain of rats treated with comparable dose of lisinopril,14 likely because of a shorter treatment period (10 vs. 20 weeks) and a later start (50 vs. 40 weeks of age).
So far the beneficial effect of ACE inhibitors on podocyte loss has been documented only in experimental diabetes, starting the treatment in the early phase of the disease.32 Our present data show that in the MWF rat, delayed treatment with an ACE inhibitor is indeed beneficial on glomerular podocytes, not only by halting their loss, but also by increasing their number per capillary tuft. This key finding of the present investigation poses new questions on its mechanism. First, how does podocyte number increase in response to ACE inhibition. It has been shown that normal differentiated podocytes have limited capability to proliferate,33 consistent with our data in normal Wistar rats, and this may indeed be the cause of podocyte depletion we observed in aged MWF rats. However, in untreated MWF rats, a number of WT1-positive cells also expressed Ki-67 suggesting entering of these cells in the cell cycle as a response to injury of the capillary structure. The fact that WT1-positive cells still decreased with age might be explained by the increasing sclerosis volume with age. Of interest, on lisinopril treatment, WT1+ Ki-67+ cells were increased, and at least two possibilities can be taken into account to explain this finding. One is that in MWF rats, some podocytes may re-engage the cell cycle as adaptive response to injury in the attempt to replace podocyte loss. Conversely to untreated MWF rats, podocytes in lisinopril-treated rats may progress throughout the cell cycle. This is supported by our present finding that ACE inhibition induced a loss in p27 expression in podocytes. Along this line, studies in podocytes in culture have documented that angiotensin II increases both p27 mRNA and protein expression, and these effects are inhibited by an AT1R antagonist.34 In addition, reduced p27 expression has been shown to favor podocyte proliferation, whereas an increase prevents podocyte proliferation by inducing cell cycle arrest.35 The second possibility is that WT1-positive cells expressing Ki-67, we have observed in lisinopril-treated MWF rats, may derive from podocyte precursors. The results of the present investigation provide the first demonstration of a strong induction of GDNF in the Bowman’s capsule in this model of spontaneous glomerular injury. GDNF, a member of transforming growth factor family, is up-regulated together with its receptor tyrosine kinase Ret in injured podocyte, likely acting in an autocrine way in favoring podocyte survival.36 Notably, GDNF also improves survival of kidney-derived mesenchymal stem cells.37 It is tempting to speculate that in case of progressive glomerular dysfunction associated with podocyte loss, an adaptive response develops, supporting cell survival in the Bowman’s capsule through GDNF induction. This process seems to be regressed by ACE inhibition, since reduced GDNF expression was observed in the Bowman’s capsule of lisinopril-treated MWF rats.
A subset of PECs of Bowman’s capsule of adult human kidney has been identified as a renal stem cell niche with self-renewal potential and high cloning efficiency.38 A recent study documented that parietal podocytes express epitopes of mature visceral podocytes, and some of them retain the ability to divide.28 Our data that parietal podocytes are increased with ACE inhibition would support a possible role for parietal cells as potential progenitors of visceral podocytes. We speculate that parietal podocytes might serve to repopulate visceral podocytes, by migration into the capillary tuft through the vascular pole or through cellular bridges between capillary tuft and Bowman’s capsule.28,36 To this regard, that some PECs have ultrastructural features of podocytes, as shown by the representative image in supplemental Figure S2 (see http://ajp.amjpathol.org), have been reported in the literature.27 The fact that there is continuity of the inner surface of the Bowman’s capsule and the outer glomerular capillary membrane, as shown by transmission electron microscopy in supplemental Figure S3 (see http://ajp.amjpathol.org), opens the possibility for parietal cells to migrate from the Bowman’s capsule to the capillary tuft, even in normal physiological condition. Finally, that PECs can migrate to the capillary tuft also in different areas than the vascular pole is suggested by occasional findings, such as those reported in supplemental Figure S4 (see http://ajp.amjpathol.org), that we obtained in normal Wistar rats, that are in line with similar findings reported in literature by others.39
The second question that arises from our present data is whether extra-renal progenitor/stem cells could contribute to podocyte repopulation. The presence of recipient-derived podocytes in biopsies from female kidney, transplanted into male recipients supports the concept that immigrating progenitor cells can replace podocytes in human.40 Bone marrow-derived stem cells have been recently documented to be a potential reservoir of glomerular podocytes in vivo in the absence of tissue injury41 and also to contribute to podocyte repopulation in the damaged kidney.42 That this process can take place also in MWF rats on ACE inhibition cannot be ruled out by our present data, and needs further and more complex experimental approach that merits, however, further investigation.
Besides the effect on WT1-positive cells, our present data show that in lisinopril-treated MWF rats, glomerular EC number was not affected by treatment, but rather the glomerular tuft occupied by ECs increased in volume implying restructuring, rather than regeneration of endothelial component in response to ACE inhibition. In vitro studies have recently documented43 that AT1R blocker can restore podocyte potential to promote glomerular EC sprouting, proliferation, and migration through the induction of podocyte-derived angiogenic factors, such as vascular endothelial growth factor-A and angiopoietin 1. Altogether these evidences would indicate that the increase in podocyte density we have observed might be the driving event for EC remodeling in treated MWF rats. Finally, consistent with its effect on glomerular hypercellularity, lisinopril was beneficial in halting MC hyperplasia. Along this line, delayed treatment with the ACE inhibitor enalapril was also shown to reduce the number of cells within the mesangium in subtotally nephrectomized rats.8
Our present results indicate that restoration of podocyte number per glomerulus by ACE inhibition is a key determinant for glomerular restructuring and repair. The reduction of the tuft volume occupied by each podocyte and the increase in glomerular EC volume density are likely responsible for remission of glomerular injury and may contribute to its regression. In a recent editorial,44 focused on the role of podocytes in focal sclerosis, Kretzler asked whether it is possible to establish a critical threshold of podocyte depletion that define the point of no return. Our study does not directly estimate this threshold, but does documents that in MWF rats, the important and progressive reduction in podocyte number per glomerulus (less than 30% of Wistar rats) was reversed by ACE inhibitor treatment with beneficial effects on structural lesions. This would indicate, in line with recent clinical observations,1,4,5 that ACE inhibition may be effective also in advanced stages of nephropathy.
In conclusion, our study demonstrates that ACE inhibition induces podocyte repopulation and glomerular EC remodeling, even when glomerular injury already is advanced. These selective effects on glomerular cellular components seem to be the basis of regression of glomerular structural injury. Thus, experimental demonstration of drug-induced podocyte repopulation supports the evidence that in patients affected by chronic nephropathies renal disease regression can be effectively induced by ACE inhibitors, and opens new insights on the mechanisms responsible for tissue regeneration.
Supplementary Material
Footnotes
Address reprint requests to Daniela Macconi, Biol.Sci.D., “Mario Negri” Institute for Pharmacological Research, Department of Biomedical Engineering, Laboratory of Renal Biophysics, Via Gavazzeni, 11, 24125 Bergamo, Italy. E-mail: macconi@marionegri.it.
Supported in part by the European Commission within the EuReGene project (LSHG-CT-2004-005085). S.C. is recipient of Fondazione Aiuti per la Ricerca sulle Malattie Rare (ARMR; Bergamo, Italy).
Supplemental material for this article can be found on http://ajp.amjpathol.org.
Part of this work was presented at the 39th Annual Meeting of the American Society of Nephrology, November 15-19, 2006, San Diego, CA.
References
- Ruggenenti P, Schieppati A, Remuzzi G. Progression, remission, regression of chronic renal diseases. Lancet. 2001;357:1601–1608. doi: 10.1016/S0140-6736(00)04728-0. [DOI] [PubMed] [Google Scholar]
- Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, Mustonen J. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351:1952–1961. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- Wilmer WA, Hebert LA, Lewis EJ, Rohde RD, Whittier F, Cattran D, Levey AS, Lewis JB, Spitalewitz S, Blumenthal S, Bain RP. Remission of nephrotic syndrome in type 1 diabetes: long-term follow-up of patients in the Captopril Study. Am J Kidney Dis. 1999;34:308–314. doi: 10.1016/s0272-6386(99)70360-4. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P, Perna A, Benini R, Bertani T, Zoccali C, Maggiore Q, Salvadori M, Remuzzi G, Investigators of the GISEN Group Gruppo Italiano Studi Epidemiologici in Nefrologia In chronic nephropathies prolonged ACE inhibition can induce remission: dynamics of time-dependent changes in GFR. J Am Soc Nephrol. 1999;10:997–1006. doi: 10.1681/ASN.V105997. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P, Brenner BM, Remuzzi G. Remission achieved in chronic nephropathy by a multidrug approach targeted at urinary protein excretion. Nephron. 2001;88:254–259. doi: 10.1159/000045998. [DOI] [PubMed] [Google Scholar]
- Marinides GN, Groggel GC, Cohen AH, Border WA. Enalapril and low protein reverse chronic puromycin aminonucleoside nephropathy. Kidney Int. 1990;37:749–757. doi: 10.1038/ki.1990.42. [DOI] [PubMed] [Google Scholar]
- Boffa JJ, Lu Y, Placier S, Stefanski A, Dussaule JC, Chatziantoniou C. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol. 2003;14:1132–1144. doi: 10.1097/01.asn.0000060574.38107.3b. [DOI] [PubMed] [Google Scholar]
- Adamczak M, Gross ML, Krtil J, Koch A, Tyralla K, Amann K, Ritz E. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol. 2003;14:2833–2842. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- Fujihara CK, Velho M, Malheiros DM, Zatz R. An extremely high dose of losartan affords superior renoprotection in the remnant model. Kidney Int. 2005;67:1913–1924. doi: 10.1111/j.1523-1755.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- Ma LJ, Nakamura S, Aldigier JC, Rossini M, Yang H, Liang X, Nakamura I, Marcantoni C, Fogo AB. Regression of glomerulosclerosis with high-dose angiotensin inhibition is linked to decreased plasminogen activator inhibitor-1. J Am Soc Nephrol. 2005;16:966–976. doi: 10.1681/ASN.2004060492. [DOI] [PubMed] [Google Scholar]
- Ma LJ, Nakamura S, Whitsitt JS, Marcantoni C, Davidson JM, Fogo AB. Regression of sclerosis in aging by an angiotensin inhibition-induced decrease in PAI-1. Kidney Int. 2000;58:2425–2436. doi: 10.1046/j.1523-1755.2000.00426.x. [DOI] [PubMed] [Google Scholar]
- Remuzzi A, Fassi A, Bertani T, Perico N, Remuzzi G. ACE inhibition induces regression of proteinuria and halts progression of renal damage in a genetic model of progressive nephropathy. Am J Kidney Dis. 1999;34:626–632. doi: 10.1016/S0272-6386(99)70385-9. [DOI] [PubMed] [Google Scholar]
- Remuzzi A, Gagliardini E, Donadoni C, Fassi A, Sangalli F, Lepre MS, Remuzzi G, Benigni A. Effect of angiotensin II antagonism on the regression of kidney disease in the rat. Kidney Int. 2002;62:885–894. doi: 10.1046/j.1523-1755.2002.00526.x. [DOI] [PubMed] [Google Scholar]
- Remuzzi A, Gagliardini E, Sangalli F, Bonomelli M, Piccinelli M, Benigni A, Remuzzi G. ACE inhibition reduces glomerulosclerosis and regenerates glomerular tissue in a model of progressive renal disease. Kidney Int. 2006;69:1124–1130. doi: 10.1038/sj.ki.5000060. [DOI] [PubMed] [Google Scholar]
- Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- Macconi D, Bonomelli M, Benigni A, Plati T, Sangalli F, Longaretti L, Conti S, Kawachi H, Hill P, Remuzzi G, Remuzzi A. Pathophysiologic implications of reduced podocyte number in a rat model of progressive glomerular injury. Am J Pathol. 2006;168:42–54. doi: 10.2353/ajpath.2006.050398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remuzzi A, Puntorieri S, Alfano M, Macconi D, Abbate M, Bertani T, Remuzzi G. Pathophysiologic implications of proteinuria in a rat model of progressive glomerular injury. Lab Invest. 1992;67:572–579. [PubMed] [Google Scholar]
- Mundlos S, Pelletier J, Darveau A, Bachmann M, Winterpacht A, Zabel B. Nuclear localization of the protein encoded by the Wilms’ tumor gene WT1 in embryonic and adult tissues. Development. 1993;119:1329–1341. doi: 10.1242/dev.119.4.1329. [DOI] [PubMed] [Google Scholar]
- Duijvestijn AM, van Goor H, Klatter F, Majoor GD, van Bussel E, van Breda Vriesman PJ. Antibodies defining rat endothelial cells: rECA-1, a pan-endothelial cell-specific monoclonal antibody. Lab Invest. 1992;66:459–466. [PubMed] [Google Scholar]
- Ricono JM, Xu YC, Arar M, Jin DC, Barnes JL, Abboud HE. Morphological insights into the origin of glomerular endothelial and mesangial cells and their precursors. J Histochem Cytochem. 2003;51:141–150. doi: 10.1177/002215540305100202. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shirato I, Asanuma K, Takeda Y, Hayashi K, Tomino Y. Protein gene product 9.5 is selectively localized in parietal epithelial cells of Bowman’s capsule in the rat kidney. J Am Soc Nephrol. 2000;11:2381–2386. doi: 10.1681/ASN.V11122381. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Stereological methods. London: Academic Press Inc.,; Practical methods for biological morphometry. 1979:40–116. [Google Scholar]
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Shankland SJ, Wolf G. Cell cycle regulatory proteins in renal disease: role in hypertrophy, proliferation, and apoptosis. Am J Physiol Renal Physiol. 2000;278:F515–F529. doi: 10.1152/ajprenal.2000.278.4.F515. [DOI] [PubMed] [Google Scholar]
- Marshall CB, Shankland SJ. Cell cycle regulatory proteins in podocyte health and disease. Nephron Exp Nephrol. 2007;106:e51–59. doi: 10.1159/000101793. [DOI] [PubMed] [Google Scholar]
- Gibson IW, Downie I, Downie TT, Han SW, More IA, Lindop GB. The parietal podocyte: a study of the vascular pole of the human glomerulus. Kidney Int. 1992;41:211–214. doi: 10.1038/ki.1992.29. [DOI] [PubMed] [Google Scholar]
- Bariety J, Mandet C, Hill GS, Bruneval P. Parietal podocytes in normal human glomeruli. J Am Soc Nephrol. 2006;17:2770–2780. doi: 10.1681/ASN.2006040325. [DOI] [PubMed] [Google Scholar]
- Fassi A, Sangalli F, Maffi R, Colombi F, Mohamed EI, Brenner BM, Remuzzi G, Remuzzi A. Progressive glomerular injury in the MWF rat is predicted by inborn nephron deficit. J Am Soc Nephrol. 1998;9:1399–1406. doi: 10.1681/ASN.V981399. [DOI] [PubMed] [Google Scholar]
- Wiggins JE, Goyal M, Sanden SK, Wharram BL, Shedden KA, Misek DE, Kuick RD, Wiggins RC. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol. 2005;16:2953–2966. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- Petermann AT, Pippin J, Durvasula R, Pichler R, Hiromura K, Monkawa T, Couser WG, Shankland SJ. Mechanical stretch induces podocyte hypertrophy in vitro. Kidney Int. 2005;67:157–166. doi: 10.1111/j.1523-1755.2005.00066.x. [DOI] [PubMed] [Google Scholar]
- Gross ML, El-Shakmak A, Szabo A, Koch A, Kuhlmann A, Munter K, Ritz E, Amann K. ACE-inhibitors but not endothelin receptor blockers prevent podocyte loss in early diabetic nephropathy. Diabetologia. 2003;46:856–868. doi: 10.1007/s00125-003-1106-8. [DOI] [PubMed] [Google Scholar]
- Kriz W. Progressive renal failure–inability of podocytes to replicate and the consequences for development of glomerulosclerosis. Nephrol Dial Transplant. 1996;11:1738–1742. [PubMed] [Google Scholar]
- Xu ZG, Yoo TH, Ryu DR, Cheon Park H, Ha SK, Han DS, Adler SG, Natarajan R, Kang SW. Angiotensin II receptor blocker inhibits p27Kip1 expression in glucose-stimulated podocytes and in diabetic glomeruli. Kidney Int. 2005;67:944–952. doi: 10.1111/j.1523-1755.2005.00158.x. [DOI] [PubMed] [Google Scholar]
- Griffin SV, Petermann AT, Durvasula RV, Shankland SJ. Podocyte proliferation and differentiation in glomerular disease: role of cell-cycle regulatory proteins, Nephrol Dial Transplant. 2003;18 Suppl 6:vi8–13. doi: 10.1093/ndt/gfg1069. [DOI] [PubMed] [Google Scholar]
- Tsui CC, Shankland SJ, Pierchala BA. Glial cell line-derived neurotrophic factor and its receptor ret is a novel ligand-receptor complex critical for survival response during podocyte injury. J Am Soc Nephrol. 2006;17:1543–1552. doi: 10.1681/ASN.2005080835. [DOI] [PubMed] [Google Scholar]
- Shi H, Patschan D, Dietz GP, Bahr M, Plotkin M, Goligorsky MS. Glial cell line-derived neurotrophic growth factor increases motility and survival of cultured mesenchymal stem cells and ameliorates acute kidney injury. Am J Physiol Renal Physiol. 2008;294:F229–F235. doi: 10.1152/ajprenal.00386.2007. [DOI] [PubMed] [Google Scholar]
- Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol. 2006;17:2443–2456. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- Gibson IW, Downie TT, More IA, Lindop GB. Tuft-to-capsule adhesions and their precursors: differences between the vascular and tubular poles of the human glomerulus. J Pathol. 1998;184:430–435. doi: 10.1002/(SICI)1096-9896(199804)184:4<430::AID-PATH1226>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Becker JU, Hoerning A, Schmid KW, Hoyer PF. Immigrating progenitor cells contribute to human podocyte turnover. Kidney Int. 2007;72:1468–1473. doi: 10.1038/sj.ki.5002524. [DOI] [PubMed] [Google Scholar]
- Poulsom R, Forbes SJ, Hodivala-Dilke K, Ryan E, Wyles S, Navaratnarasah S, Jeffery R, Hunt T, Alison M, Cook T, Pusey C, Wright NA. Bone marrow contributes to renal parenchymal turnover and regeneration. J Pathol. 2001;195:229–235. doi: 10.1002/path.976. [DOI] [PubMed] [Google Scholar]
- Prodromidi EI, Poulsom R, Jeffery R, Roufosse CA, Pollard PJ, Pusey CD, Cook HT. Bone marrow-derived cells contribute to podocyte regeneration and amelioration of renal disease in a mouse model of Alport syndrome. Stem Cells. 2006;24:2448–2455. doi: 10.1634/stemcells.2006-0201. [DOI] [PubMed] [Google Scholar]
- Liang XB, Ma LJ, Naito T, Wang Y, Madaio M, Zent R, Pozzi A, Fogo AB. Angiotensin type 1 receptor blocker restores podocyte potential to promote glomerular endothelial cell growth. J Am Soc Nephrol. 2006;17:1886–1895. doi: 10.1681/ASN.2005020205. [DOI] [PubMed] [Google Scholar]
- Kretzler M. Role of podocytes in focal sclerosis: defining the point of no return. J Am Soc Nephrol. 2005;16:2830–2832. doi: 10.1681/ASN.2005080841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.