

Abstract
Intraplaque hemorrhage accelerates atherosclerosis via oxidant stress and contributes to lesion development and destabilization. Normally, macrophages scavenge hemoglobin-haptoglobin (HbHp) complexes via CD163, and this process provokes the secretion of the anti-inflammatory atheroprotective cytokine interleukin (IL)-10. We therefore tested the hypothesis that HbHp complexes may drive monocyte differentiation to an atheroprotective phenotype. Examination of the macrophage phenotype in hemorrhaged atherosclerotic plaques revealed a novel hemorrhage-associated macrophage population (HA-mac), defined by high levels of CD163, but low levels of human leukocyte antigen-DR. HA-mac contained more iron, a pro-oxidant catalyst, but paradoxically had less oxidative injury, measured by 8-oxo-guanosine content. Differentiating monocytes with HbHp complexes reproduced the CD163high human leukocyte antigen-DRlow HA-mac phenotype in vitro. These in vitro HA-mac cells cleared Hb more quickly, and consistently showed less hydrogen peroxide release, highly reactive oxygen species and oxidant stress, and increased survival. Differentiation to HA-mac was prevented by neutralizing IL-10 antibodies, indicating that IL-10 mediates an autocrine feedback mechanism in this system. Nonlinear dynamic modeling showed that an IL-10/CD163-positive feedback loop drove a discrete HA-mac lineage. Simulations further indicated an all-or-none switch to HA-mac at threshold levels of HbHp, and this conversion was experimentally verified. These data demonstrate the creation of a novel atheroprotective (HA-mac) macrophage subpopulation in response to intraplaque hemorrhage and raise the possibility that therapeutically reproducing this macrophage phenotype may be cardio-protective in cases of atherosclerosis.
Atherosclerotic intraplaque hemorrhage is an important contributor to lesion development and destabilization.1,2,3,4,5 In the carotid artery, hemorrhage promotes progression and clinical symptoms.6 Furthermore, coronary intralesional hemorrhages are tightly associated with thrombosis.7 Mechanisms for atherogenicity of hemorrhage include the delivery of cholesterol-rich erythrocyte membranes and hemoglobin-derived iron, which can catalyze hydrogen peroxide conversion into highly reactive oxygen species (hROS).1,2 Thus a clearer understanding of macrophage responses to intraplaque hemorrhage is critical.
Hemoglobin (Hb) is principally cleared by complexing with haptoglobin (Hp), followed by uptake via CD163, a macrophage scavenger receptor.3,8 The association of Hb with Hp is normally crucial to its binding to CD163, although in diabetes there is evidence for Hp-independent CD163 binding of glycosylated Hb.3,9 The importance of Hp is strongly supported by the association of Hp genotypes with many forms of vascular disease.10,11 As a consequence of CD163 binding, HbHp induces interleukin 10 (IL10) and heme oxygenase-1 (HO-1), which are anti-inflammatory and atheroprotective.12,13,14 However, it is possible that CD163 functions primarily as an endocytic receptor, with down-stream signaling being induced by internalized heme.13 Taken together, experimental studies favor a protective role of CD163, and CD163 expression in atherosclerotic tissues may therefore represent an adaptation limiting the atherogenicity of plaque hemorrhage.
Hypercholesterolaemia-sensitive monocyte subsets (CD14highCD16null & CD14lowCD16pos) have been described in blood.15,16 However, macrophage differentiation to separate phenotypes occurs primarily in response to tissue micro-environmental influences, which in plaques include oxidatively modified low density lipoproteins (OxLDL). Indeed, Kruth has recently published evidence that macrophage subsets defined by the presence or absence of CD14 can be found in human atherosclerotic tissue.17
We tested the hypothesis that intraplaque hemorrhage results in monocyte differentiation to macrophages specialized for safe hemoglobin disposal. We show that culprit atherosclerotic plaques contain a novel anti-oxidant hemorrhage-associated macrophage subset (HA-mac) defined by high CD163 and low HLA-DR, which are distinct from pro-inflammatory lipid core macrophages. HA-mac could be generated in vitro by culturing monocytes with HbHp complexes through a mechanism centered on an IL-10 autocrine feedback loop. Our data indicate that monocytes entering atherosclerotic plaques may be adaptively modeled by plaque hemorrhage. Understanding this differentiation pathway may allow intervention to induce protective macrophage differentiation therapeutically.
Materials and Methods
Pathology, Immunohistochemistry, Confocal, Image Analysis
Human plaques were from a series of paraffin-embedded plaques that derive from consecutive West of Scotland hospital autopsies and have already been fully described.18 Tissues were studied with consent of next of kin for autopsy and for use of tissues for research. Local Research Ethics Committee and Central Office for Research Ethics Committees approved the research, and the tissues and site were registered under the UK Human Tissue Act. Exclusion criteria were severe sepsis or hematological malignancy.
To study the effects of hemorrhage on macrophages, we selected for study one lesion per patient with both evident hemorrhage and macrophage infiltrates on H&E sections. Consistent with the literature,7,19,20 these lesions were thrombosed due either to rupture (six cases) or intimal erosion (two cases). Crosschecking with clinical information indicated that the selected plaques corresponded to a tightly defined group of fatal myocardial infarction, occurring within 48 hours of event onset.18 As a consecutive hospital series, this cohort is very different to studies of selected autopsies for community sudden cardiac death.1,21
For immunostaining, sections were deparaffinized, microwaved for 20 minutes in citrate buffer (600W), blocked in serum, and peroxidase-blocked. Sections were incubated with anti-CD163 (clone 10D6, Novocastra, Newcastle on Tyne, UK), then with biotinylated anti-mouse secondary (1:200, Dako Ltd, Ely, UK) and streptavidin-alkaline phosphatase (1:100, Roche Diagnostics Ltd., Lewes, UK). Color was developed with Vector Blue (Vector Laboratories Ltd., Peterborough, UK). Sections were then incubated with anti-HLA-DR (clone CR3/43, Dako), which was detected with polymer-peroxidase kit (A. Menarini Diagnostics UK, Wokingham, UK) followed by color development with amino-ethyl-carbazole (Vector Laboratories). CD68 was probed with antibody clone PG-M1 (Dako) and detected with polymer-peroxidase kit (Menarini) and color was developed with 3′-5′-di-amino-benzidine (DAB, Sigma-Aldrich, Poole, UK) and hematoxylin counterstaining (Pioneer Research Chemicals, Colchester, UK). For CD163/iron dual staining, sections were probed for CD163 as above with polymer immunoperoxidase–amino-ethyl-carbazole and counterstained with Perl’s. Erythrocytes were detected with anti-glycophorin C (clone Ret40F, Dako), then detected with polymer immunoperoxidase/amino-ethyl-carbazole as above. Eight-oxo-guanosine was specifically probed with goat polyclonal IgG (1:1000 dilution in PBS, Calbiochem DR1001, Calbiochem, Beeston, Nottingham, UK), followed by detection with polymer-peroxidase/DAB/hematoxylin as above. Negative controls [pre-immune goat serum at equivalent concentration, and adsorption with 8-oxo-guanosine (8-oxo-G, Calbiochem)] were without brown staining.
Counting of CD68-positive macrophages (in total 9326) on CD163/HLA-DR dual-stained sections was performed manually. We measured the shortest straight line distance from the macrophages to the specified feature (lipid core, hemorrhage, or CD34-positive neovessels) with an eyepiece graticule. Neovessels were defined as CD34-bounded spaces of any size. Iron was assessed semiquantitatively on a standard hemochromatosis histology scale of 0 (no iron) to 3+ (maximum iron), where 3+ is equivalent to the images shown.
Quantification of HO-1 and 8-oxo-G intensity was by minor adaptation of validated image morphometry protocols,22,23 using Image J (a PC-compatible version of NIH-Image), rather than NIH-Image (Macintosh version). Digitally captured RGB images (Olympus Camedia C5050, Olympus, Watford, UK) were RGB split, after which the blue channel was subtracted from the red, producing a black and white image of brown density. This was measured by the Image J intensity histogram function to generate the arithmetic mean intensity. A region of interest was manually drawn to measure only parts of the image that included macrophages, and this was repeated for ≥5 representative images per area. Mean density units of intensity were then expressed as a continuous variable. Since normality testing by Kolmogorov-Smirnov indicated a non-Gaussian distribution, significance testing was by Mann-Whitney.
Confocal Microscopy
For confocal detection, we substituted fluorescent secondary antibodies for the enzyme-labeled second layers, with staining and storage in the dark. Primary antibodies, blocking and microwaving were as above. Anti-CD163 (clone 10D6) was detected with goat-anti-mouse AlexaFluor 647 (Molecular Probes, Invitrogen, Paisley, UK; excitation [Ex.] 633 nm, emission [Em.] 650 to 700 nm); anti-HLA-DR with goat-anti-mouse AlexaFluor 568 (Molecular Probes; Ex. 568 nm, Em. 580 to 610 nm); and anti-CD68 with fluorescein-isothiocyanate (FITC)-labeled KP1 (Dako; Ex. 488 nm, Em. 500 to 535 nm). Sections were counterstained for 5 minutes in nuclear dye 7-actinomycin D (Molecular Probes) and mounted in 80% glycerol/20% PBS (both Sigma-Aldrich). We used a Zeiss LSM 510 Meta inverted confocal microscope, with three laser lines. Pinhole and unable filter settings were at defaults. One scan was performed using differential interference contrast to define general topography. A further confocal scan was then performed at FITC excitation and Cy5 emission wavelengths (Ex. 488 nm, Em. 650 to 700 nm), yielding a selective image of 7-actinomycin D due to its characteristic wide Stoke’s shift. Scan and photomultiplier settings were set to optimize signal:noise ratio for each emission wavelength. Processing was with Zeiss LSM Image Browser, and comprised addition of scalebars, and adjustment of brightness and contrast, before import into Microsoft Powerpoint for assembly of montage Figures.
In Vitro Macrophage Cultures and Reagents
Human monocytes were isolated as described from venous blood of healthy consenting volunteers, following Local Research Ethics Committee approval.14,24 In brief, 50 ml of donor blood was collected aseptically, immediately citrated, and centrifuged to separate plasma from cells. Plasma was then recalcified to produce autologous serum. Mononuclear cells were purified by centrifugation through Histopaque 1077 (10771, Sigma-Aldrich) at 500 × g for 20 minutes.24 In some experiments, erythrocytes were removed by dextran sedimentation (Pharmacia, GE Health care, Amersham, UK) and Percoll (Pharmacia) density gradient centrifugation.14 Macrophages were purified on the culture plastic by adherence incubation for 1 hour followed by three washes of warm Iscove’s Modified Dulbecco’s Medium (IMDM, Cat. No. 21056, Invitrogen). These cells had >95% purity by flow cytometry for forward scatter, side scatter, and CD14 staining. Unless otherwise stated, macrophages were cultured for 7 days at approximately 105 cells per well in 24 well plates in 10% autologous donor serum, IMDM (10% AHS IMDM), with penicillin (100 IU/ml) and streptomycin (100 μg/ml). Hemoglobin (lyophilized stabilized purified Ao ferrous hemoglobin, H0267, Sigma-Aldrich) and haptoglobin phenotype 1-1 (lyophilized purified, H0138, Sigma-Aldrich) were reconstituted in PBS at 1 mg/ml. HbHp complexes were generated by dissolving equimolar amounts of Hb and Hp in growth medium. Hb and Hp were tested for endotoxin (Limulus amebocyte lysate, sensitivity <0.125 EU/ml, LAL Pyrogent Plus kit, Cat.No. N289, Cambrex, Walkersville, MD). Approximately 1 batch in 10 of each was endotoxin-positive and excluded from further use. Hb, Hp, or HbHp complexes were added at the indicated concentrations to monocyte cultures immediately following purification by adherence. The following antibodies, drug inhibitors, or vehicle were added at concentrations indicated 10 minutes before the HbHp complexes: antagonistic anti-CD163 monoclonal antibodies (RM3/1, Bachem AG, St Helens, UK; EdHu-1, Serotec, Oxford, UK); neutralizing anti-IL10 Mab 217 (R&D Systems); lipopolysaccharide (LPS, O111:B4), cytochalasin D, pepstatin-A, chloroquine (Sigma-Aldrich); and interferon gamma (IFNγ, Peprotech, London, UK).
Human LDL was collected from clinical apheresis supernatant and purified by ultracentrifugation and dialysis as before (gift of D. Patel).25 The LDL was oxidized in 20 μmol/L CuSO4 for 24 hours at 37°C, with oxidation validated by thiobarbituric acid and agarose gel electrophoresis as before.26
For flow cytometric staining, macrophages were harvested with a disposable sterile plastic policeman. Cells were centrifuged 13,000 rpm for 60 seconds in a microcentrifuge (Eppendorf, Fisher Scientific, Loughborough, UK), resuspended in 50 μl PBS (Sigma-Aldrich), and stained for 20 minutes at 4°C in 2 μg/ml FITC-labeled anti-CD163 (56C-FAT, Bachem). Isotype control was 2 μg/ml IgG1-FITC (Serotec). For double staining, the cells were co-incubated with anti-HLA-DR-PE (clone HK14, Sigma-Aldrich) or isotype control (mouse IgG1-PE, Sigma-Aldrich). The cells were then washed by centrifugation in 1 ml PBS, and resuspended in 400 μl PBS for flow cytometry. The cells were read on a Coulter EpicsXL flow cytometer, FL1–600V, FL2–600V, 1% compensation. Data were analyzed and presented using WinMDI software.
For cytokine assays, macrophages were cultured in 10% AHS-IMDM for 24 hours at approximately 103 cells per well in 96-well microplates (Nunc, VWR), in the presence or absence of 10 μg/ml HbHp complexes (approximately 10−7mol/L complexes), 10−8mol/L dexamethasone, or 10 ng/ml LPS. The supernatants were then stored at −80°C before analysis. Cytokines were measured by DuoSets according to manufacturer’s instructions (R&D Systems, Abingdon, UK).
For the DCF oxidant stress assay, macrophages were differentiated for 7 days in 96-well microplates, in either control medium or with added HbHp complexes. Macrophages were then loaded with 10 μmol/L 5′, 6′-di-chloromethyl, di-hydro, di-chloro-fluorescein diacetate (Molecular Probes). Cells were then stimulated with phorbol myristate acetate (Sigma-Aldrich; 10−7mol/L). Fluorescence (Ex. 485 ± 10 nm, Em. 535 ± 10 nm) was measured at the time points indicated in a 96 well plate reader (Synergy HT, Biotek, Fisher Scientific) at 37°C.
For peroxide assays, macrophages were differentiated and stimulated as before in 96-well plates, but in the presence of Amplex Red (1 μg/ml, Molecular Probes, Invitrogen) and horseradish peroxidase (1:1000, Sigma-Aldrich). Fluorescence was read after 24 hours (Ex. 535 nm, Em. 590 nm).
hROS exhibit electrophilic attack on the amino-moiety of aminophenylfluorescein (APF), which rearranges to form fluorescein.27 We validated the specificity of APF for HONOO·, OH·, OCl− rather than H2O2 in our own hands. To assess specific production of hROS, macrophages were differentiated as before in 96-well plates. We then added 5 μmol/L APF (Molecular Probes) and stimuli. After 24 hours, fluorescence was measured by microplate reader (Ex. 484 nm, Em. 515 to 550 nm) and expressed as arbitrary fluorescence units.
Supernatant Hb concentrations were measured spectrophometrically at the characteristic peak Hb absorbance (Soret peak, A412 nm, 100 μl volume, plate reader above). Calibration was performed using Hb at known concentrations of 10−8, 10−7, 10−6 mol/L, and full absorption spectrum (400 to 700 nm in 1-nm intervals).
Small Interfering RNA
Adherence-purified monocytes were transferred to 0.1% AHS in IMDM without antibiotics. Complexes were prepared from 5 fmol small interfering (si)RNA (Dharmacon, CD163; scramble). These were complexed with 10 μl Genlantis liposomes (AMS Biotechnology) for 30 minutes and complexes were then incubated with monocytes for 24 hours. Western blotting was done with Novex precast system (Invitrogen). Immunodetection was in PBS with 4% nonfat milk powder 0.1% Tween-20 (Sigma). We used a Western-blot validated monoclonal antibody GHI/61 (1:1000; Santa Cruz)28; anti-mouse peroxidase (1:10,000; Dako), ECL-Plus, and hyperfilm (both Amersham).
Hb was labeled with Alexa488 (Invitrogen) following manufacturer’s instructions, and as published.13 Hb-Alexa488 was complexed with Hp and added to macrophages at culture day 1 at 10−7mol/L. Cells were also assessed by RM3/1 flow cytometry and the effects of RM3/1 on Hb phagocytosis was assessed.
Mathematical Modeling Using Nonlinear Dynamics
Standard modeling approaches were used.29,30,31 The mathematical model was formed from a coupled pair of nonlinear ordinary differential equations describing the feedback loop (see supplemental Figure S1 at http://ajp.amjpathol.org) [equations (1) & (2)]. The model has two variables [CD163] and [IL10] that describe the time course of the concentrations of the two species. The rate of change of each species is determined by the production and degradation of each. We therefore obtain:
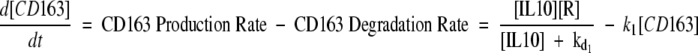 |
1 |
 |
2 |
where [HbHp] is a parameter indicating the HbHp concentration (in arbitrary units) and [R] is a parameter that describes the sensitivity of CD163 up-regulation to IL10 levels. One possible biological interpretation is that the first term of Equation 1 represents the concentration of IL10 receptors available for CD163 binding on the cell surface. Degradation is modeled using simple exponential decay with rates k1 and k2. The parameters kd1 and kd2 determine the saturation of the production rates of IL10 and CD-163 respectively.
These were solved numerically and graphed using Maple 11 (Maplesoft, Waterloo, Ontario, Canada). Since this was a proof-of-concept model, it was kept as simple as possible and simulations used arbitrary units, and illustrative values were assigned to parameters. An advantage to formulating a simplified model is that the steady state values can be derived analytically and it is possible to verify that these arbitrary choices do not affect conclusions. Equilibrium solutions and threshold values were obtained by algebraic rearrangement and graphed in Maple 11. To determine the robustness of the observed dichotomy, we ran simulations using a range of arbitrary parameters. These all predicted qualitatively similar (ie, dichotomous) behavior around a threshold. To validate the model, we then experimentally tested the behavior it predicted.
Statistical Analysis
The inverse relationship between CD163high and HLA-DRhigh macrophages in coronary plaques was examined by both χ2 analysis and by analysis of variance (ANOVA) with Bonferroni’s correction. Normality was tested by Kolmogorov-Smirnov and normally distributed data were tested parametrically. The data in Table 1 appeared approximately normally distributed, but normality testing was equivocal due to low n-values. They were therefore expressed parametrically as mean ± SE but tested using a rank method (Mann-Whitney), which is a more stringent assessment. Where indicated, statistical testing of parametric continuous data were performed using ANOVA with Bonferroni correction for multiple simultaneous comparisons, or a two-sample Student’s t-test. Statistical analysis was performed using Excel (Microsoft, Seattle, WA) or SigmaStat (Systat, San Jose, CA). Exact P values are presented to clarify the strength of the statistical differences.
Table 1.
Morphometry of Foam Cell and Hemorrhage-Associated Macrophage Subsets
| Measurement | Distance to lipid core (μm) M ± SE (Range) | Distance to neovessels (μm) M ± SE (Range) | Distance to hemorrhage (μm) M ± SE (Range) | Iron score (0–3 scale) M ± SE (Range) | HO-1 intensity (arbitrary density units) M ± SE | 8-oxo-G intensity (arbitrary density units) M ± SE | IL-10 intensity (arbitrary density units) M ± SE |
|---|---|---|---|---|---|---|---|
| Subset | |||||||
| HA-mac | 439 ± 14 | 15.7 ± 5.7 | 3.88 ± 1.8 | 2.25 ± 0.47 | 22.2 ± 3.9 | 14.0 ± 0.96 | 12.3 ± 1.5 |
| (CD163high HLADRlow) | (0 to 1000)** | (10 to 50)*** | (0 to10)** | (1+ to 3+)* | *** | ** | **** |
| FC-mac | 29 ± 15.5 | 785 ± 214 | 150 ± 86 | 0 ± 0 (0 to 0) | 7.95 ± 1.03 | 35.8 ± 4.7 | 4.2 ± 0.4 |
| (CD163low HLADRhigh) | (1 to 100) | (500 to 2000) | (50 to 750) |
P < 0.05, Mann-Whitney.
P < 0.01, Mann-Whitney.
P < 0.005, Mann-Whitney.
P < 0.001, Mann-Whitney.
Morphometric comparisons of the two macrophage subsets. Rows, data gathered in the same way (see Materials and Methods) for HA-mac (CD163highHLA-DRlow) and FC-mac (CD163lowHLA-DRhigh). Columns, measurement of respectively: nearest distance to lipid core, nearest distance to microvessels, nearest distance to hemorrhage, cellular iron (where Figure 2D = 3+), HO-1 staining intensity, 8-oxo-G staining intensity.
Results
Culprit Human Lesions Contain a Novel Hemorrhage-Associated Macrophage Subpopulation Identified by CD163
We examined the effect of atherosclerotic plaque hemorrhage on macrophage phenotype, assessing 351 plaques from 83 consecutive autopsies.18,19 Eight had confluent intraplaque hemorrhage and macrophage infiltrates. Each of these was a thrombosed atherosclerotic lesion with a large hemorrhage and numerous macrophages (Figure 1A, and (see supplemental Figure S2 at http://ajp.amjpathol.org). All eight corresponded to fatal myocardial infarction (5 males, 3 females, ages 60 to 80 years, death within 48 hours of the onset of clinical event).
Figure 1.

Human culprit plaques contain a hemorrhage-associated macrophage subset. Results are representative of eight ruptured plaques. Scale bars = 500 μm. A: H&E stain, low power view of a culprit lesion with plaque rupture. T = thrombus. Arrow = plaque fissure. B: CD68 immunolabeling (immunoperoxidase-DAB, brown), using clone KP1. Clone PG-M1 staining was equivalent. Arrow = plaque fissure. C: CD163/HLA-DR double immunolabeling, low power. Blue = CD163, red = HLA-DR. Box = area shown at higher magnification in (D), which includes a fibrous cap infiltrated by macrophages and an area of hemorrhage. D: CD163/HLA-DR double immunolabeling, high power. Blue = CD163, red = HLA-DR. FC = foam cell macrophages. HA = hemorrhage associated macrophages. Serial sections of these are also shown in zones HA & FC in Supplemental Figure S4 at http://ajp.amjpathol.org. E: CD163/erythrocyte (RBC) double immunolabeling. Blue = CD163, red = RBC marker glycophorin-c. T = thrombus. F: α-smooth muscle actin immunolabeling (immunoperoxidase-DAB, brown). Arrow = plaque fissure. G: Quantification of subsets. Macrophages of each subset were counted and expressed as a proportion of macrophages in each lesion and then summarized as mean and SE for all eight available ruptured plaques. *Statistically significant distribution, analysis of variance, P < 10−8.
CD68 immunostaining identified fibrous cap and lipid core macrophages (Figure 1B). Two-color immunostaining with CD163 and HLA-DR revealed that these were either predominantly CD163highHLA-DRlow or CD163lowHLA-DRhigh (Figure 1, C and D). Double immunostaining for CD163 and the erythrocyte marker glycophorin-c indicated that the CD163high macrophages colocalized with plaque hemorrhage (Figure 1E). CD163lowHLA-DRhigh cells were α-smooth muscle actin negative (Figure 1F). Findings were remarkably consistent in all eight thrombosed hemorrhaged plaques: 75 ± 2.4% macrophages were CD163lowHLA-DRhigh and 25 ± 2.9*% were CD163highHLA-DRlow (Figure 1G). We identified minimal numbers of CD68posCD163lowHLA-DRlow cells (Figure 1G). While CD163highHLA-DRlow macrophages were consistently found in culprit lesions (see supplemental Figure S3 at http://ajp.amjpathol.org) twenty consecutive stable plaques from the same study contained only pro-inflammatory CD163lowHLA-DRhigh macrophages (not shown).
We then assessed whether the CD163highHLA-DRlow and CD163lowHLA-DRhigh cells were definitively CD68pos macrophages. We used confocal immunofluorescence to analyze all three markers simultaneously and relate them to lesion topography (see supplemental Figure S4 at http://ajp.amjpathol.org). Two collections of CD68-positive macrophages are shown. One subset is mixed with hemorrhage, ie, extravasated erythrocytes, and is characterized by high CD163 and a low HLA-DR (ie, CD68posCD163highHLA-DRlow). In contrast, macrophages more distant to hemorrhage were conventional foam cells (FC), with an abundant foamy cytoplasm and high HLA-DR, but low CD163 (CD68posCD163lowHLA-DRhigh. These observations therefore define a novel subset of CD68posCD163highHLA-DRlow HA-mac.
HA-mac Are Protected from Oxidant Stress Despite Iron Loading
We next characterized the possible pathophysiological role of HA-mac by assessing oxidant stress with antibodies specific for oxygen-adducted guanosine (8oxoG).32 While FC macrophages contained 8oxoG, as reported,32 HA-mac in the same sections were 8oxoG-negative, indicating reduced oxidative stress (Figure 2A). Reduced HA-mac oxidative stress was in the face of increased loading with iron (a pro-oxidant catalyst), evidenced by CD163/Perl’s double-staining (Figure 2A).33 In serial sections, HA- macrophages reciprocally expressed HO-1 strongly and myeloperoxidase weakly (Figure 2A). FC macrophages had the reverse pattern (Figure 2A). CD163high macrophages were found adjacent to extravasated erythrocytes and intraplaque microvessels and co-expressed IL10 (Figure 2, B–E).
Figure 2.

A: Reciprocal oxidative stress and iron content in CD163high (HA-mac) and CD163low (foam cell) macrophages Left column: photomicrographs of macrophages in area identified as hemorrhage-associated (HA). Right column, photomicrographs of macrophages in area identified as foam cell rich (FC). Scale bars = 40 μm. Rows, immunolabeling as indicated, respectively for 8-oxo-guanosine (8-oxo-G); heme-oxygenase-1 (HO-1), and myeloperoxidase (MPO). The bottom row are double-labeled for CD163 (red)/iron (Perl’s stain; blue). Note Perl’s negative golden particles in the FC region are well described as OxLDL-derived ceroid.33 Brown is immunoperoxidase-DAB, and blue is hematoxylin counterstain. B–E: Ruptured plaque CD163+ macrophages, hemorrhage, iron, and IL10. Images correspond to area HA of (A), in serial sections, and are representative of the eight culprit plaques. Scale bars = 100 μm. B: − CD163 (blue immuno-alkaline phosphatase)/glycophorin C (erythrocytes, red immunoperoxidase) C: CD163/CD34. CD163 (red immunoperoxidase)/CD34 (endothelial cells, blue immuno-alkaline phosphatase); D: CD163/Perl’s iron histochemistry. CD163 (red immunoperoxidase)/iron (blue, histochemistry with Perl’s stain; ie, Prussian blue ferricyanide reaction). E: CD163/IL10 CD163 (blue immuno-alkaline phosphatase)/IL10 (red immunoperoxidase).
These associations were tested statistically. We measured distances of the two macrophage populations, HA-mac (CD163highHLA-DRlow) and FC-mac (CD163lowHLA-DRhigh) to key plaque features (Table 1). FC-mac were closer to the lipid core (29.0 ± 15.5 μm [mean ± SE]) than were HA-mac (439 ± 14 μm; P < 0.01, Wilcoxon). In contrast, HA-mac preferentially localized to CD34+ neovessels (P < 0.005, Wilcoxon) and hemorrhage (P < 0.01, Wilcoxon). Perl’s stain revealed intracellular iron sequestration (ie, from Hb) in HA-mac but not FC-mac (P < 0.05, Wilcoxon). HO-1 expression, quantified by image analysis, was higher in HA-mac than in FC-mac (P < 0.01, Wilcoxon). In contrast, 8oxoG was significantly reduced in the HA-mac versus FC-mac (P < 0.01, Wilcoxon). IL10 expression, quantified by image analysis, was higher in CD163highHLA-DRlow (HA) than in CD163lowHLA-DRhigh (FC) macrophages (P < 0.001, Wilcoxon).
Differentiation of Human Monocytes with HbHp Complexes Reproduces HA-mac
Using flow cytometric analysis of peripheral blood monocytes, we demonstrated that approximately 90% were CD14highCD16null and 10% were CD14lowCD16pos in keeping with previous reports.24 CD163 expression was restricted to monocytes, and there was no detectable difference in CD163 expression between the CD14lowCD16pos and CD14highCD16null subsets (see supplemental Figure S5 at http://ajp.amjpathol.org).
Whether HbHp complexes induce the CD163highHLA-DRlow phenotype was tested by culturing human monocytes for 8 days in the presence of Hb, Hp or HbHp complexes (Figure 3). HbHp complexes (10−7mol/L), but neither constituent protein alone induced a CD163highHLA-DRlow phenotype (Figure 3, A–D). This effect was blocked separately by two antagonistic anti-CD163 antibodies (clones RM3/1, EdHu-1),14 or by neutralizing anti-IL10 antibodies, but not by isotype-matched neutralizing anti-tumor necrosis factor (TNF)-α antibodies (Figure 3, E–H). These observations indicated dependence on CD163 and autocrine IL10 for CD163highHLA-DRlow polarization. This polarization was also blocked by specific inhibitors of phagocytosis or lysosomal processing (Figure 3, I–L): respectively cytochalasin-D (phagocytosis inhibitor, 10−6 mol/L), chloroquine (lysosomal inhibitor, 10−6 mol/L), or pepstatin-A (lysosomal protease inhibitor, 10−9 to 10−6 mol/L).
Figure 3.

Mechanisms causally linking Hb to HA-mac phenotype. Human peripheral blood monocytes were incubated in the presence of Hb, Hp, HbHp complexes, with vehicle, cytokines, or antagonists at the indicated concentrations. After 8 days of culture, macrophages were detached and dual stained for CD163 (FITC) and HLA-DR (PE). Representative flow cytometric dot plots are presented for at least five experiments from separate donors, with greater than twenty experiments examining the effects of HbHp complexes versus control. For each, the x axis is CD163 and the y axis is HLA-DR (isotype controls were in the first quadrant, not shown). The vehicle control for cytochalasin-D and chloroquine was DMSO. Dex = dexamethasone. Anti-TNF and anti-IL10 = functionally antagonistic antibodies to TNF-α and IL10 respectively. LPS = E. Coli lipopolysaccharide. Hp:Hp complexes were used at 100 nmol/L (10−7mol/L). Key: A: Unstimulated culture of day 8 macrophages. B: Incubated with HbHp complexes at 10−7mol/L, 100 nmol/L. C: Incubated with Hb at 10−7mol/L, 100 nmol/L. D: Incubated with Hp at 10−7mol/L, 100 nmol/L. E: As (B), with addition of anti-CD163 clone RM3/1, 70 nmol/L. F: As (B), with addition of anti-CD163 clone EdHu-1, 70 nmol/L. G: As (B), with addition of anti-IL10 Mab217, 70 nmol/L. H: As (B), with addition of anti-TNF-α Mab225, 70 nmol/L. I: As (B), with addition of DMSO, 1:1000. J: As (B), with addition of 10−6 M/L cytochalasin-D in DMSO, 1:1000. K: As (B), with addition of 10−6 M/L chloroquine in DMSO, 1:1000. L: As (B), with addition of 10−7 M/L, pepstatin-A in DMSO, 1:1000. M: As (B), with addition of LPS, 1 ng/ml. N: Control (unstimulated) culture of macrophages at day 8. O: Incubated with 5 × 10−11mol/L (50 pM) IL10 from outset of culture. P: As (O), but with 5 × 10−10mol/L (500 pM) IL10. Q: As (O), but with 5 × 10−9mol/L (5 nmol/L) IL10. R: As (O), but with 5 × 10−8mol/L (50 nmol/L) IL10. S: Unstimulated culture of macrophages at day 8. T: Incubated with 10−9mol/L (1 nmol/L) dexamethasone from outset of culture. U: As (T), but with 10−8mol/L (10 nmol/L) dexamethasone. V: As (T), but with 10−7mol/L (100 nmol/L) dexamethasone. W: As (T), but with 10−6mol/L (1 μmol/L) dexamethasone.
The prototypic pro-inflammatory stimuli IFN-γ (5 × 10−8 mol/L, not shown) and LPS (1 to 10 ng/ml) prevented the HbHp-induced phenotype, producing CD163lowHLA-DRhigh cells even in the presence of HbHp (Figure 3M). Adenovirus (multiplicity of infection = 200) also had this effect (not shown). In contrast, exogenous recombinant IL10 (5 × 10−11 mol/L to 5 × 10−8 mol/L) evoked differentiation to the full CD163highHLA-DRlow phenotype (Figure 3, N–R). Dexamethasone (10−9 mol/L to 10−7mol/L; 1 to 100 nmol/L), a prototypic anti-inflammatory agent, increased CD163 and reduced HLA-DR, but not to the extent seen with HbHp complexes (Figure 3, S–W). Hb freshly prepared by hypotonic lysis of autologous erythrocytes had the same effect as commercial Hb (see supplemental Figure S6 at http://ajp.amjpathol.org). Although HbHp complexes maximally induced CD163 at day 4, they needed to be added at culture outset for full induction, suggesting the programming of macrophage differentiation (see supplemental Figure 7 at http://ajp.amjpathol.org).
HA-mac Are Anti-Inflammatory and Antioxidant, and Promote HbHp Clearance in Vitro
We then investigated the anti-inflammatory and antioxidant phenotype of the in vitro differentiated HA-mac (Figure 4, A–C). We examined pro- or anti-inflammatory cytokines, for reference comparing with dexamethasone (10−8 mol/L) and LPS (10 ng/ml) (Figure 4). HbHp complexes (10−7 mol/L) or dexamethasone increased IL10 (Figure 4A) and increased the IL10/TNF ratio (Figure 4C), consistent with net anti-inflammatory activity. LPS increased TNF more than IL10, decreasing the IL10/TNF ratio, consistent with net pro-inflammatory activation. Moreover, HA-mac had increased survival compared with control cultures (Figure 4D), and were more capable of clearing HbHp complexes relative to untreated controls (Figure 4E). For concentration-effect curves for HbHp on IL10 secretion see supplemental Figure S8 at http://ajp.amjpathol.org.
Figure 4.

Homeostatic phenotype of HA-mac A–C: x-axes, respective stimuli, control medium (10% AHS IMDM); LPS = E. Coli lipopolysaccharide (10 ng/ml); HbHp = HbHp complexes (100 nmol/L; 10−7mol/L); Dx = dexamethasone (10 nmol/L; 10−8mol/L). A: y axis, Absolute IL10 levels in supernatants in monocyte-macrophage cultures 24 hours after addition of stimuli at the start of culture. (*P < 0.001 analysis of variance overall, with P < 0.05 relative to control, Bonferroni adjusted post-test, values are mean + SEM of five donors). B: y axis, Absolute TNF-α levels in supernatants in monocyte-macrophage cultures 24 hours after addition of stimuli at the start of culture. (*P = 0.006 analysis of variance overall with P < 0.05 relative to control, Bonferroni adjusted post-test, values are mean + SEM of five donors). C: y axis, Ratio of IL10/TNF in panels A and B. (*P = 0.0001 analysis of variance overall with P < 0.05 relative to control with Bonferroni adjusted post-test, n = 5 donors.). Concentration-effect curves (not shown) indicated that the direction of IL10/TNF ratio was maintained at all concentrations of LPS (1 to 1000 ng/ml) and HbHp (10−8 to 10−6 mol/L) D: y axis, macrophage survival as measured by reduction of the colorimetric formazan dye MTS. Viability was measured relative to control cultures (100%). Control = unmodified 10% AHS IMDM; HbHp = 10−7mol/L HbHp complexes added at the start of culture. Student’s t-test *P = 0.000143, values are mean + SEM of five donors). E: y axis, Hb concentration in medium supernatant at 4 minutes after addition. x axis, time after addition of HbHp complexes (10−7mol/L), measuring supernatant [Hb] spectrophometrically at 412 nm. Open circles, macrophages were incubated in control medium (10% AHS IMDM) for 8 days. Filled triangles, macrophages were differentiated with HbHp complexes (10−7mol/L) for 8 days. Student’s t-test *P = 0.000283. F: y axis, specific H2O2 production as measured by Amplex Red/peroxidase (see Materials and Methods), calibrated by a H2O2 standard curve. Open bars, Control – differentiation in 10% AHS IMDM for 8 days. Filled bars, HbHp – differentiation in added HbHp complexes (10−7mol/L). x axis, addition of opsonised zymosan, a prototypical macrophage oxidative burst stimulant, in two doses to control or HbHp differentiated macrophages. The dose of OpZ, is expressed as particles/well. Data are representative of five independent experiments using separate donors. Student’s t-test *P = 0.0017. G: y axis, H2O2 production as measured by Amplex Red/peroxidase, calibrated by a H2O2 standard curve. x axis, addition of OxLDL (30 μg/ml) to control or HbHp differentiated (10−7mol/L) macrophages. Open bars, Control – differentiation in 10% AHS IMDM for 8 days. Filled bars, HbHp – differentiation in added HbHp complexes (10−7mol/L). Data are representative of five independent experiments using separate donors. Student’s t-test *P = 0.034. H: y axis, oxidative stress as measured by DCFDA fluorescence in macrophages at 8 days cultured in the presence or absence of HbHp (see Materials and Methods). Data are representative of five independent experiments using separate donors. Student’s t-test *P < 0.005. I: y axis, hROS measured as fluorescence of the specific reporter amino-phenyl-fluorescein (APF) (see Materials and Methods). Open bars, Control – differentiation in 10% AHS IMDM for 8 days. Filled bars, HbHp – differentiation in added HbHp complexes (10−7mol/L). x axis, Cu-Ox-LDL, human LDL oxidized in 10μmol/L CuSO4 at 37°C for 18 hours, added at 30 μg/ml. Serum deprivation – medium changed to matched IMDM without AHS. Student’s t-test *P = 9 × 10−5 (OxLDL) and *P = 3 × 10−6 (SFM). Data are representative of five independent experiments using separate donors.
Since iron-loading may promote oxidant stress, we used multiple complementary readouts to assess whether HA-mac differentiation modulates production of reactive oxygen species (ROS). Compared with control day 8 macrophages, H2O2 production was suppressed in HA-mac, either in response to opsonised zymosan or to copper-oxidized low density lipoprotein (OxLDL, Figure 4, F and G). Hb-associated peroxidase activity was excluded as a confounder for H2O2 measurements (see supplemental Figure S9 at http://ajp.amjpathol.org). Likewise, HA-mac were protected from phorbol-ester induced oxidant stress as measured by 6′-di-chloromethyl, di-hydro, di-chloro-fluorescein diacetate fluorescence in the absence of serum (P < 0.005, Figure 4H). H2O2 is converted to hROS (specifically HONOO·, OH·, OCl−), which are selectively detected with APF (see Materials and Methods27). APF fluorescence indicated that HA-mac produced less hROS in response to OxLDL or serum deprivation (Figure 4I) or opsonised zymosan (not shown). Time-course and concentration-effect curves for these stimuli are given in supplemental Figure S9 at http://ajp.amjpathol.org. All of the protective effects of HbHp were concentration-related and optimal at 10−7 mol/L (see supplemental Figure S10 at http://ajp.amjpathol.org). The protective response was reversed at the highest HbHp concentration, a feature we attribute to overload (the cultures were opaque red).
CD163-siRNA Specifically Reduces Hb Uptake, RM3/1 Binding
CD163-specific siRNA duplexes suppressed CD163, evidenced by Western blotting using antibody GHI/61 (see supplemental Figure S11A at http://ajp.amjpathol.org). This corresponded to reduced RM3/1 surface binding—even on a log scale (see supplemental Figure S11B at http://ajp.amjpathol.org). We measured uptake of AlexaFluor488-labeled HbHp complexes over 60 minutes after addition to macrophage cultures (Supplemental Figure S11C at http://ajp.amjpathol.org). CD163-siRNA suppressed uptake of Alexa488-labeled HbHp complexes (see supplemental Figure S11D at http://ajp.amjpathol.org). RM3/1 also reduced Hb uptake, proving functional antagonistic activity (see supplemental Figure S11E http://ajp.amjpathol.org). Finally incubating the cells for 4 days with unlabeled HbHp complexes in the presence or absence of scramble or CD163-siRNA, showed that macrophage survival was reduced by CD163-knockdown; see supplemental Figure S11F at http://ajp.amjpathol.org).
Nonlinear Dynamics and Experimental Validation Show that HbHp/IL10/CD163 Positive Feedback Evokes All-or-None Commitment to CD163high versus CD163low Phenotypes at a Threshold Concentration of HbHp Complexes
We observed that HbHp binding to CD163 leads to IL10 secretion, and that IL10 up-regulates CD163. Since their output promotes their input, positive feedback loops are associated with exponential phenomena, as in the action potential or in pathophysiological vicious cycles. In contrast, positive feedback loops have been recently implicated in stable commitment to divergent states in T-helper cells.34 Here, the positive feedback loop enhances subset distinctiveness by abolishing intermediate states. More generally, similar regulatory architectures are being observed in a variety of systems biology contexts.29
We tested whether, in principle, a positive feedback loop between IL10 and CD163 in response to HbHp would generate discrete subsets governed by HbHp levels. We therefore constructed a proof of concept model. This is shown diagrammatically (see supplemental Figure S1 at http://ajp.amjpathol.org). We converted the diagram to a mathematical model to test whether a threshold level of HbHp switched the system between two lineages, deliberately keeping the model as simple as possible rather than providing a complete quantitative description of the regulatory system. The model uses arbitrary variables and arbitrary parameters to probe a qualitative pattern of cell behavior.
In multiple simulations of equations (1) and (2) (see Materials and Methods), CD163 fell to negligible levels at low concentrations of HbHp, irrespective of starting concentrations (Figure 5A, blue curves). On the other hand, at HbHp concentrations above threshold, CD163 rose to a stable plateau with the level of final CD163 independent of initial CD163 (Figure 5A, red curves). Thus the model predicts that CD163-IL10 positive feedback leads to binary CD163high or CD163low phenotypes depending on HbHp input.
Figure 5.

Nonlinear dynamics demonstrate that Hb-gated CD163-IL10 feedback drives switching to HA-mac phenotype A: Representative simulated time course of CD163 levels in a simple mathematical model describing the positive feedback loop between CD163 and IL10 given by Equations 1 and 2, see Materials and Methods. Both axes are in arbitrary units. Blue curves show the behavior of CD163 for a low HbHp input ([HbHp] = 0.01 in arbitrary units) for a number of starting values ([CD163] = 0, 2, …, 8). Red curves show behavior for a high HbHp input ([HbHp] = 1) with the same starting levels. Other parameter values are = 10, k1 = 1, k2 = 1, kd1 = 1, kd2 = 1 (arbitrary units) and the initial level of IL10 is 0.5 (arbitrary units) in all cases. The qualitative behavior is independent of this choice, and similar threshold behavior can be observed for all choices of k1, k2, kd1 and kd2 as long as the sensitivity of CD-163 to IL10 is sufficiently high ([R] > k1 k2 kd1, SOM). B: Variation of final steady state values of CD163 and IL10 with HbHp input (all given in arbitrary units). Other parameter values are = 10, k1 = 1, k2 = 12, kd1 = 0.75, kd2 = 10−5 (arbitrary units). The steady states were obtained from Equations 3 and 4, SOM and clearly illustrate the threshold, which for these parameter values occurs at H* = 9 × 10−5. Similar qualitative behavior is seen for all other parameter values as long as > k1 k2 kd1. C: Observed concentration-effect curves for HbHp complexes on CD163 levels by flow cytometry. x axis, fluorescence intensity (CD163 staining (blue or red colors) or isotype control (gray). y axis, cell number. HbHp concentration in culture (expressed in mol/L) is given by the histogram color code as indicated. To match (A), red colors are above threshold and the blue colors below threshold. The experiment is representative of n = 5 donors.
The model was then used to predict behavior of the final steady state levels of CD163 and IL10 with varying [HbHp] (Equations 3 and 4). Steady states occur when the concentrations of CD163 and IL10 do not change with time. These can be obtained by setting the left hand sides of the above equations to 0 and solving for [CD163] and [IL10]. This can be done algebraically. One set of solutions is simply [CD163]* = 0, [IL10]* = 0, corresponding to the low CD163 state. It is perhaps unrealistic that this is exactly at 0. However, if the state was a small non-zero level it would not affect any of the subsequent qualitative conclusions (eg, by adding a small basal production rate to the above equations). We have chosen not to do this as it complicates the model, and in particular, makes it impossible to obtain analytic expressions for the other steady state and the threshold in HbHp. With the equations as given, straightforward manipulation gives the other steady state as
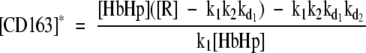 |
3 |
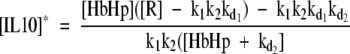 |
4 |
Only non-negative values of protein levels are of biological relevance. For the above expressions to satisfy this constraint we require [R] > k1 k2 kd1 and [HbHp] ≥ H* where H* is the threshold given by
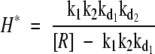 |
5 |
At [HbHp] = H* the two steady states intersect in the well known transcritical bifurcation, exchanging stability.35 Further analysis shows that for [HbHp] < H*, the 0 steady state is stable and all solutions converge to it. At [HbHp] = H* the 0 steady state loses stability. For [HbHp] > H* it is the steady state given by Equations 3 and 4.
Therefore, H* is the threshold level of HbHp. H* forms the boundary between the two CD163 phenotypes. With [HbHp] below this value, CD163 levels drop to a steady state of negligible levels, irrespective of starting conditions (eg, blue curves in Figure 5A). This is interpreted as the CD163lowHLA-DRhigh subset. In contrast, for [HbHp] above H* above the H* threshold, CD163 rises to a stable plateau, again irrespective of initial value. This corresponds to the red curves in Figure 5A and the CD163highHLA-DRlow subset. This threshold is shown in Figure 5B. The final steady state level of CD163 rises rapidly as HbHp concentration reaches the threshold, before saturating at a fixed level. The simulations were run using a wide range of arbitrary parameters, and consistently gave these qualitative effects.
Finally, we tested this prediction. We showed by flow cytometry that CD163 increased in an all-or-none manner at a threshold concentration of 10−9mol/L (Figure 5C). It should be noted that the conceptual model uses arbitrary parameters, and does not predict the precise threshold. Moreover, the model was not designed to explore the effects of frank HbHp overload. Evidence from plaques showing expression of IL-10 by HA-mac was also consistent with the predictions of the model (Table 1, Figure 2).
Discussion
Intraplaque hemorrhage1,2 and plaque macrophages36,37 play key roles in atherosclerotic lesion progression and vulnerability. We do not challenge this view, but instead define a novel macrophage subset HA-mac. These cells are identified within hemorrhaged inflamed lesions, and have several atheroprotective and compensatory properties. Our data provide a logical extension of previous work on CD163, and add to conceptual understanding of macrophage subsets in relation to human vascular disease.10,13,38
Our in vitro data support that plaque hemorrhage adaptively modifies macrophage differentiation. Here, hemoglobin directs a discrete subset with increased hemoglobin uptake, reduced oxidant stress and generation of the anti-inflammatory cytokine IL10. This interpretation is biologically plausible, as it is an everyday observation that bruising is not itself inflammatory. However, since our starting material was postmortem specimens, the phenomenon is self-evidently not fully atheroprotective. The HA-mac were a minority (approximately 25%), and conventional foamy macrophages predominated even in the plaques with large hemorrhages. Moreover, the known up-regulation of HLA-DR by modified lipoproteins may accentuate the difference between the two subsets (FC versus HA).39 Consistent with this, we have found that CD163high macrophages are absent in lesions with lipid core but no hemorrhage.
By defining a distinct macrophage subset, our data extend earlier descriptions that HO-1 and CD163 are found in atherosclerosis.13 Similarly, a substantial body of work, mainly by Levy and colleagues, indicates that haptoglobin genotype Hp2-2 is less protective than Hp1-1 in a wide range of vascular diseases.10,40 We now give a mechanism contributing to these observations by defining how plaque hemorrhage drives a specific antioxidant hemorrhage-associated macrophage subset.
The concept of functionally distinct leukocyte subsets is well established for lymphocytes in vitro and in vivo (eg, CD4 vs CD8, Th1 vs Th2).36 Macrophage subsets have been defined by in vitro differentiation with cytokines.38 Thus, classically activated macrophages are generated by pro-atherogenic stimuli eg, IFN-γ, TNF-α, and LPS, and alternatively-activated macrophages are induced by IL-4.38,41,42 Loss of the key Th1 transcription factor T-bet suppresses macrophage HLA-DR in atherosclerosis in vivo, via reduction of CD4 T-helper derived IFN-γ and increased IL-4.43 This is consistent with a skew from classical to alternative activation, suggesting that classically activated plaque macrophages are pro-atherogenic. However, HA-mac were characterized in the present study as more closely corresponding to the deactivated macrophage subset induced by IL10 or glucocorticoids.38 Interestingly, although dexamethasone up-regulated CD163 and suppressed HLA-DR, we found that its effect was weak relative to that of HbHp.14,44 This may be in part related to different signaling pathways, since dexamethasone signals primarily via the glucocorticoid receptor, while IL10 signals via silencer of cytokine signaling proteins, themselves therapeutic targets.44,45 Understanding different effects on macrophage function of glucocorticoids and HbHp therefore requires further study.
Well-established roles of macrophages in atherosclerosis include foam cell formation, lipid necrotic core formation, matrix metalloproteinase production, oxidant production, and inflammatory cytokine secretion.36,37,46 However recent evidence describes active cholesterol export from the plaque via macrophage lymphatic emigration,47 or via the adenosine triphosphate binding cassette pathway to high-density lipoprotein.48 This necessitates a more complex paradigm encompassing homeostatic macrophage roles. Our observations add to this by describing a non-lipid-driven macrophage homeostatic mechanism.
We are not aware of a previous mathematical description of a bistable system driving macrophage differentiation, either in vascular disease or other situations. The nonlinear dynamic modeling defined a role for the IL10 autocrine positive feedback loop. The key feature of the model is the stability produced by the loop’s saturability. At its simplest, this may equate to saturation of promoter regions of specific gene regulatory elements with transcription factors. The molecular nature of these effects is beyond the scope of the present paper but under active study. Critically, this feedback accurately predicted the existence of a threshold HbHp concentration for CD163 induction. Thus we experimentally validated the model at the qualitative level of all-or-none commitment to respective subsets. The system features positive feedback, threshold, and an all-or-none response, which promote rapid decisive commitment. This permits the vascular wall to respond rapidly and consistently to sudden pro-oxidant threats such as plaque hemorrhage. We found experimentally that 6 to 8 days differentiation in 1 μmol/L HbHp concentrations reversed the protective phenotype, a feature consistent with toxicity from Hb overload. At this concentration, the cultures are macroscopically red and the cells brown. Thus this protective system can be overwhelmed, leading to maladaptive function.
In conclusion, we have characterized a novel atheroprotective macrophage subset (HA-mac) associated with intralesional hemorrhage. This subset contrasts strongly with pro-atherogenic conventional (classical) lipid core macrophages. HA-mac are adapted to swiftly clear Hb and contain oxidative stress. We predict that HA-mac would suppress the impact of hemorrhage on atherosclerosis progression. In culprit lesions, the effects of HA-mac are by definition too little and too late, but therapeutic modification of HA-mac pathways may prevent plaque destabilisation.
Supplementary Material
Acknowledgments
We thank Viola Leung, Sandra Cantilena, and Donna Horncastle for technical support, and Dr. Justin Mason for critical review of the manuscript.
Footnotes
Address reprint requests to J.J. Boyle, Vascular Sciences Section, National Heart and Lung Institute, Faculty of Medicine, Imperial College London, Hammersmith Hospital, Du Cane Road, London W12 0NN, U.K., E-mail: joseph.boyle@imperial.ac.uk.
Supported by grants from the British Heart Foundation (FS/07/010 BHF Gerry Turner Intermediate Clinical Research Fellowship to J.J.B., and professorial awards to K.T. and D.O.H.) and Biomedical Research Centre funding. J.S. is supported by the UK Biotechnology and Biological Sciences Research Council via the Centre for Integrative Systems Biology at Imperial College, BB/C519670/1. H.A.H. is supported by an Imperial College Deputy Rector’s Award, the IC Department of Mathematics, and a National Science Foundation Graduate Research Fellowship.
R.C.L. and D.O.H. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2325. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, Tulenko TN, Wrenn SP, Narula J. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- Levy AP, Moreno PR. Intraplaque hemorrhage. Curr Mol Med. 2006;6:479–488. doi: 10.2174/156652406778018626. [DOI] [PubMed] [Google Scholar]
- Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. doi: 10.1161/01.cir.92.3.657. [DOI] [PubMed] [Google Scholar]
- Lin HL, Xu XS, Lu HX, Zhang L, Li CJ, Tang MX, Sun HW, Liu Y, Zhang Y. Pathological mechanisms and dose dependency of erythrocyte-induced vulnerability of atherosclerotic plaques. J Mol Cell Cardiol. 2007;43:272–280. doi: 10.1016/j.yjmcc.2007.05.023. [DOI] [PubMed] [Google Scholar]
- Takaya N, Yuan C, Chu B, Saam T, Underhill H, Cai J, Tran N, Polissar NL, Isaac C, Ferguson MS, Garden GA, Cramer SC, Maravilla KR, Hashimoto B, Hatsukami TS. Association between carotid plaque characteristics and subsequent ischemic cerebrovascular events: a prospective assessment with MRI–initial results. Stroke. 2006;37:818–823. doi: 10.1161/01.STR.0000204638.91099.91. [DOI] [PubMed] [Google Scholar]
- Davies MJ, Thomas AC. Plaque fissuring–the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina. Br Heart J. 1985;53:363–373. doi: 10.1136/hrt.53.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabriek BO, Dijkstra CD, van den Berg TK. The macrophage scavenger receptor CD163. Immunobiology. 2005;210:153–160. doi: 10.1016/j.imbio.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, Schaffner A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood. 2006;107:373–380. doi: 10.1182/blood-2005-03-1014. [DOI] [PubMed] [Google Scholar]
- Levy AP, Purushothaman KR, Levy NS, Purushothaman M, Strauss M, Asleh R, Marsh S, Cohen O, Moestrup SK, Moller HJ, Zias EA, Benhayon D, Fuster V, Moreno PR. Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2-2 genotype: implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ Res. 2007;101:106–110. doi: 10.1161/CIRCRESAHA.107.149435. [DOI] [PubMed] [Google Scholar]
- Asleh R, Guetta J, Kalet-Litman S, Miller-Lotan R, Levy AP. Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ Res. 2005;96:435–441. doi: 10.1161/01.RES.0000156653.05853.b9. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611–616. doi: 10.1161/01.atv.19.3.611. [DOI] [PubMed] [Google Scholar]
- Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res. 2006;99:943–950. doi: 10.1161/01.RES.0000247067.34173.1b. [DOI] [PubMed] [Google Scholar]
- Philippidis P, Mason JC, Evans BJ, Nadra I, Taylor KM, Haskard DO, Landis RC. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ Res. 2004;94:119–126. doi: 10.1161/01.RES.0000109414.78907.F9. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van RN, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2. CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–1126. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle JJ. Association of coronary plaque rupture and atherosclerotic inflammation. J Pathol. 1997;181:93–99. doi: 10.1002/(SICI)1096-9896(199701)181:1<93::AID-PATH696>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- van der Wal AC, Becker AE, van der Loos CM, Das PK. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994;89:36–44. doi: 10.1161/01.cir.89.1.36. [DOI] [PubMed] [Google Scholar]
- Falk E. Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br Heart J. 1983;50:127–134. doi: 10.1136/hrt.50.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani R, Roberts WC. Extravasated erythrocytes, iron, and fibrin in atherosclerotic plaques of coronary arteries in fatal coronary heart disease and their relation to luminal thrombus: frequency and significance in 57 necropsy patients and in 2958 five mm segments of 224 major epicardial coronary arteries. Am Heart J. 1983;105:788–797. doi: 10.1016/0002-8703(83)90242-9. [DOI] [PubMed] [Google Scholar]
- Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol. 2001;23:291–299. [PubMed] [Google Scholar]
- Ruifrok AC. Quantification of immunohistochemical staining by color translation and automated thresholding. Anal Quant Cytol Histol. 1997;19:107–113. [PubMed] [Google Scholar]
- Boyle JJ, Bowyer DE, Weissberg PL, Bennett MR. Human blood-derived macrophages induce apoptosis in human plaque-derived vascular smooth muscle cells by Fas-ligand/Fas interactions. Arterioscler Thromb Vasc Biol. 2001;21:1402–1407. doi: 10.1161/hq0901.094279. [DOI] [PubMed] [Google Scholar]
- Eden ER, Patel DD, Sun XM, Burden JJ, Themis M, Edwards M, Lee P, Neuwirth C, Naoumova RP, Soutar AK. Restoration of LDL receptor function in cells from patients with autosomal recessive hypercholesterolemia by retroviral expression of ARH1. J Clin Invest. 2002;110:1695–1702. doi: 10.1172/JCI16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant CE, Law NS, Van DV, Hardwick SJ, Carpenter KL, Mitchinson MJ. Oxidized low-density lipoprotein is cytotoxic to human monocyte-macrophages: protection with lipophilic antioxidants. FEBS Lett. 1995;358:175–178. doi: 10.1016/0014-5793(94)01393-f. [DOI] [PubMed] [Google Scholar]
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem. 2003;278:3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- Hogger P, Dreier J, Droste A, Buck F, Sorg C. Identification of the integral membrane protein RM3/1 on human monocytes as a glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163) 152. J Immunol. 1998;161:1883–1890. [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Di VB, Lemerle C, Michalodimitrakis K, Serrano L. From in vivo to in silico biology and back. Nature. 2006;443:527–533. doi: 10.1038/nature05127. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN. Cell-signalling dynamics in time and space. Nat Rev Mol Cell Biol. 2006;7:165–176. doi: 10.1038/nrm1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, De Meyer GR, Herman AG, Kockx MM. Reactive oxygen species induce RNA damage in human atherosclerosis. Eur J Clin Invest. 2004;34:323–327. doi: 10.1111/j.1365-2362.2004.01343.x. [DOI] [PubMed] [Google Scholar]
- Carpenter KL, Van DV, Taylor SE, Hardwick SJ, Clare K, Hegyi L, Mitchinson MJ. Macrophages, lipid oxidation, ceroid accumulation and alpha-tocopherol depletion in human atherosclerotic lesions. Gerontology. 1995;41 Suppl 2:53–67. doi: 10.1159/000213725. [DOI] [PubMed] [Google Scholar]
- Yates A, Bergmann C, Van Hemmen JL, Stark J, Callard R. Cytokine-modulated regulation of helper T cell populations. J Theor Biol. 2000;206:539–560. doi: 10.1006/jtbi.2000.2147. [DOI] [PubMed] [Google Scholar]
- Fall C, Marland E, Wagner J, Tyson J. Computational Cell Biology. New York: Springer,; Interdisciplinary Applied Mathematics. 2002:pp 378–408. [Google Scholar]
- Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–291. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Shashkin P, Gleissner CA, Dunson D, Jain N, Lee JK, Miller Y, Ley K. Induction of dendritic cell-like phenotype in macrophages during foam cell formation. Physiol Genomics. 2007;29:149–160. doi: 10.1152/physiolgenomics.00051.2006. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy JE, Kalet-Litman S, Miller-Lotan R, Levy NS, Asaf R, Guetta J, Yang C, Purushothaman KR, Fuster V, Moreno PR. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2007;27:134–140. doi: 10.1161/01.ATV.0000251020.24399.a2. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA. 2005;102:1596–1601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaer DJ, Boretti FS, Schoedon G, Schaffner A. Induction of the CD163-dependent haemoglobin uptake by macrophages as a novel anti-inflammatory action of glucocorticoids. Br J Haematol. 2002;119:239–243. doi: 10.1046/j.1365-2141.2002.03790.x. [DOI] [PubMed] [Google Scholar]
- Tang J, Raines EW. Are suppressors of cytokine signaling proteins recently identified in atherosclerosis possible therapeutic targets? Trends Cardiovasc Med. 2005;15:243–249. doi: 10.1016/j.tcm.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–11784. doi: 10.1073/pnas.0403259101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.