

Abstract
Inter-individual and regional variability in recombination rates cannot be fully explained by the DNA sequence itself. Epigenetic mechanisms might be one additional factor affecting recombination. A biochemical approach to studying human germline methylation is difficult. We used the density of the 434,198 nonredundant methylation-associated SNPs (mSNPs) in the derived allele HapMap data set as a surrogate marker for germline DNA methylation. We validated our methodology by demonstrating that the mSNP density confirmed known patterns of genomic methylation, including the hypermutability of methylated cytosine and hypomethylation of CpG islands. Using this approach, we found a genome-wide positive correlation between germline methylation and regional recombination rate (500-kb windows: r = 0.622, P < 10−15). This remained significant with multiple correlations correcting for sequence features known to affect recombination, such as GC content and CpG dinucleotides (500-kb windows: r = 0.172, P < 10−15). Using the ENCODE data set for increased resolution, we found a positive correlation between germline DNA methylation and recombination rate (50-kb windows: r = 0.301, P = 0.002). This correlation was further strengthened when corrected for sequence features affecting recombination (50-kb windows: r = 0.445, P < 0.0001). In the Human Epigenome Project data set there was increased DNA methylation in regions within recombination hot spots in male germ cells (0.632 vs. 0.557, P = 0.007). The relationship we observed between germline DNA methylation and recombination could be explained in two ways that are not mutually exclusive: DNA methylation could indicate preferred sites for recombination, or methylation following recombination could inhibit further recombination, perhaps by being part of the enigmatic molecular pathway mediating crossover interference.
Modern genome-scale analysis of genetic linkage has increased the resolution of the human recombination map and provided an opportunity to study this fundamental evolutionary mechanism. The Marshfield recombination map provided sex-averaged and sex-specific recombination rates with a resolution of ∼3 centimorgans (cM) and demonstrated significant individual and sex-specific variations in the recombination rate (Broman et al. 1998). Further progress was made with the creation of the deCODE recombination map, which increased the resolution over prior genetic maps approximately fivefold and replicated observations of inter-individual and sex-specific recombination variability (Kong et al. 2002). Analysis suggested that the CpG fraction, GC content fraction, and poly(A)n/poly(T)n,n ≥ 4 fraction explained a significant amount of the variation in the recombination rate when the genome was analyzed in 3-Mb windows. Further analysis of the data using 5-Mb and 10-Mb windows gave similar correlations and suggested that several additional sequence features, i.e., Wn ≥ 10 (W = A or T), Rn ≥ 10 (R = A or G), distance from the centromere, and chromosome length, contributed to the variability of human recombination (Jensen-Seaman et al. 2004).
Finer-scale recombination maps have more recently been produced, utilizing linkage disequilibrium, a measure of nonrandom association between adjacent single nucleotide polymorphisms (SNPs), to predict recombination rates (Myers et al. 2005). These maps have sufficient power to detect recombination hot spots, i.e., regions of ∼1–2 kb interspersed throughout the genome that account for the majority of genomic recombination activity (Jeffreys et al. 2001). The latest high-resolution recombination map of the genome is based on the second generation of the human haplotype map (www.hapmap.org) (Frazer et al. 2007). Analysis of sequence features revealed that only the GC content correlated significantly with recombination rate over a wide range of window sizes (8–512 kb). In addition, several DNA motifs correlated with recombination at window sizes <8 kb, and exons and repeat content had a negative correlation with recombination at larger window sizes (16, 128, 256, and 512 kb) (Myers et al. 2006).
Several observations suggest that the DNA sequence itself does not provide a full explanation for the different recombination rates of individual genomic regions (Winckler et al. 2005; Neumann and Jeffreys 2006). Almost no recombination hot spots that are known in humans were found at orthologous locations in the chimpanzee genome despite a 98.6% similarity in the regions studied (Winckler et al. 2005). Furthermore, inter-individual variation in recombination hot spot activity without differences in adjacent DNA sequence has been demonstrated (Neumann and Jeffreys 2006). One explanation would be that the recombination rate is not determined by regional DNA sequence, but rather is affected by variant distant DNA sequences. However, an alternative mechanism for the different recombination rates of individual genomic regions could be that these regions are marked by epigenetic modifications (Winckler et al. 2005; Neumann and Jeffreys 2006; Sandovici et al. 2006), either locally or by distal elements brought into close proximity (Ling et al. 2006). This notion is supported indirectly by the increased recombination rate of genome regions containing clusters of imprinted genes (Lercher and Hurst 2003; Sandovici et al. 2006).
Epigenetics is the study of modifications of DNA-associated information that can be transmitted through either meiosis or mitosis, but does not involve the DNA sequence itself. Methylation of cytosine is one of these modifications, affecting 3% of cytosines (Weisenberger et al. 2005) but 70% of all CpG dinucleotides (Robertson and Wolffe 2000) within the human genome. It serves as a critical regulator within the genome, controlling tissue-specific gene expression (Song et al. 2005; Weber et al. 2007) and mediating X chromosome inactivation (Mohandas et al. 1981; Venolia et al. 1982; Hellman and Chess 2007). Methylation has also been suggested as a part of the defense mechanism against potentially harmful transposon activity (Yoder et al. 1997). Patterns of DNA methylation differ between healthy and cancerous tissue (Feinberg and Vogelstein 1983; Badal et al. 2003). Recently, both global and regional DNA methylation levels have been demonstrated to change significantly during the lifetime of individuals (Bjornsson et al. 2008), supporting a possible role in the pathogenesis of common age-related disorders (Bjornsson et al. 2004).
We hypothesized that germline DNA methylation might be the prime epigenetic mechanism affecting meiotic recombination. Methylation is the only epigenetic modification proven to be established at prophase I in meiosis when recombination occurs. This has been demonstrated genome-wide (Oakes et al. 2007) as well as for retrotransposons (Bourc'his and Bestor 2004) and imprinted genome sites (Davis et al. 1999). It is possible, however, that other epigenetic modifications (such as histone modifications) participate in the process.
We chose to focus our efforts on germline DNA methylation and took a novel approach to investigate this relationship. Deamination of methylated cytosine results in a C → T transition (and a G → A transition on the opposite strand) likely to be missed by the DNA repair system (Coulondre et al. 1978; Cooper and Youssoufian 1988). The resulting hypermutability of methylated CpG has resulted in a great under-representation of CpGs in the human genome (Bird 1980). According to the neutral theory of molecular evolution, most substitutions in the DNA sequence are caused by genetic drift rather than selection and have little effect on fitness (Kimura 1991). As SNPs are generally considered functionally neutral, they can be considered to reflect the local mutation rate (Kimura 1989). Their frequency of fixation and elimination is dependent on the population size but is independent of the type of SNP (base change). A small subset is undergoing positive selection (The International HapMap Consortium 2005). It is rare that SNPs result in a positive attribute to an organism, and such mutations are quickly fixed. We assume that the probability of a SNP being selected for is essentially independent of the type of SNP in this small subset. Single nucleotide mutations with deleterious consequences in the SNP data set should be rare because they are quickly eliminated by purifying selection. Current estimates of hypermutability of methylated cytosines are fivefold based on the frequency of disease-causing single nucleotide mutations in human genes (Krawczak et al. 1998) and sixfold based on the frequency of CpG sequences in SNPs in the human genome (Zhao and Zhang 2006). Both the absolute and relative density of methyl-associated SNPs (mSNPs) can therefore be used as a surrogate marker to reflect the degree of methylation in the germline. This has previously been used to predict germline DNA methylation on a smaller scale (Bjornsson et al. 2006). Using the recently released HapMap data set of 3.1 million SNPs, we created a model of human germline methylation. We then used this model to test if DNA methylation of the human germline was directly and independently correlated with meiotic recombination. The results suggest that regional DNA methylation in the germline affects the local recombination rate.
Results
Identification of methylation-associated SNPs within the HapMap database, including the ENCODE regions
The second-phase HapMap database has 2,252,113 nonredundant C/T or G/A SNPs in the autosomal chromosomes. Of those, 763,035 (33.9%) are located within a CpG dinucleotide. A derived allele data set was created by mapping each SNP to the corresponding base in the chimpanzee or macaque genomes, thus determining which SNP allele was the ancestral one (Thomas et al. 2007). We used this data set to search for C/T or G/A SNPs where either C or G was the ancestral allele (thus excluding noninformative T → C or A → G SNPs). A total of 1,239,485 C/T or G/A nonredundant polymorphisms fulfilled this criterion for the autosomal chromosomes. Of these, 434,198 (35.0%, 79 ± 36 per 500-kb window) were within a CpG dinucleotide, thus meeting the criteria of a mSNPgenome. There was no appreciable difference between mSNPs and non-mSNPs using the integrated haplotype score (iHS) parameter for selection (Supplemental Table S1) (Voight et al. 2006).
A search within the ENCODE HapMap data set found 9809 C/T or G/A nonredundant polymorphisms. Of those, 2987 (30.5%, 299 ± 123 per 500-kb window) were within a CpG dinucleotide, thus meeting the criteria of a mSNPENCODE. Due to the limited derived allele data for this subset, the additional criteria used in the genome-wide data set was not applied to the ENCODE data set.
We examined the SNP allele counts for evidence that mSNPgenome counts are representative of hypermutable methylated sequences. Out of 548,370,281 sequenced cytosine bases in the autosomal chromosomes of the hg17 release of the human genome, 26,635,559 (4.9%) were within a CpG dinucleotide. If no specific dinucleotide is hypermutable, we would expect the distribution of C/T polymorphisms within dinucleotides to be the same as the distribution of cytosine bases within dinucleotides, so 4.9% of these polymorphisms (60,205 and 476 in the genome-wide-derived allele data set and the ENCODE data set, respectively) should be within a CpG dinucleotide. The observed number of polymorphisms was significantly greater than the expected number for both data sets (ratio of 7.2 and 6.3 for the genome-wide-derived allele data set and the ENCODE data set, respectively; P < 10−15 for both values), suggesting that these data sets represent the known hypermutability of methylated cytosine. Furthermore, if we expect that methylation explains the difference between the expected and observed number of polymorphisms within CpGs, ∼373,993 (86%) of our mSNPgenome are due to methylation. This is consistent with the observed fivefold increased frequency of single nucleotide mutations identified in the coding sequences of human genes (Krawczak et al. 1998), and a sixfold increased frequency of CpG sites in human SNPs (Zhao and Zhang 2006).
Analysis of the derived allele data set revealed significantly more mSNPs with either C or G as the ancestral allele than with T or A as the ancestral allele (443,657 vs. 333,606, ratio 1.32, P < 10−15). The directionality observed in these numbers is consistent with the fact that deamination of methylated cytosine results in C/T or G/A mutations that likely remain unrepaired, and therefore become polymorphisms. Additionally, counts of C/T SNPs were compared with that of G/T SNPs, which have been categorized as to whether the 3′ adjacent base is G or another base (Table 1). For SNPs with C as the ancestral allele, their proportion is significantly increased with G as the adjacent allele relative to the control G/T counts. In contrast, very similar proportions were observed for T/C and T/G SNPs when T was the ancestral allele (Table 1). The increased relative proportion of C/T SNPs with C as the ancestral allele and an adjacent G further suggests that mSNPs are representative of hypermutable methylated sequences. These results together suggest that the subset of mSNPs in the derived allele data set is not a random subset of the genome. The observed overrepresentation of mSNPs in all data sets suggests that the mSNPs variable is indicative of the previously known hypermutability of methylated cytosines, thus supporting the methodology of our approach.
Table 1.
Absolute counts of SNPs in the derived allele data set within dinucleotides containing G and dinucleotides not containing G (H = A,C,T) for SNPs linked to methylation and control SNPs

The ancestral allele is marked with (*). Our model is based on the premise that the (C*/T)pG count is informative of cytosine methylation.
For all genome-wide analyses, we used mSNPs determined by the derived allele data set (mSNPgenome) to minimize inclusion of noninformative mSNPs.
We calculated a methylation index (MI) as explained in Methods. We found that the MI at a given window (Xi) correlated positively with the MI at the adjacent window (Xi + 1) (500-kb windows: r = 0.362, P < 10−15) (data not shown). This result suggests that the MI reflects a genome characteristic extending over a length at least comparable to the 500-kb window studied.
To support the theory that the MI was indicative of DNA methylation, we correlated the CpG island base count with the MI for each window. CpG islands are long (>200 bp) stretches of sequence with an unusually high frequency of CpGs. They are commonly found near the transcriptional start sites of genes (Antequera and Bird 1999). In general, CpG islands are known to be hypomethylated compared with other CpGs in the genome (Bird 1986). We found a strong negative correlation between the MI and number of CpG island bases per window (500-kb windows: r = −0.483, P < 10−15; Supplemental Fig. S1). This result is consistent with lower mutation rates of CpG dinucleotides in CpG islands, suggesting that MI is representative of genome methylation. Since the correlation between CpG islands and 1/NSNP is positive (500-kb windows: r = 0.461, P < 10−15) and the correlation between CpG islands and 1/NCpG is mildly negative (500-kb windows: r = −0.120, P < 10−15), it is unlikely that the strong negative correlation is due to the CpG count (data not shown).
Genome-wide map of germline methylation
We constructed a genome-wide map of the methylation index (MI) of the human genome in 500-kb windows (Fig. 1). The map suggested both inter- and intra-chromosomal variability in germline methylation (Fig. 1; Supplemental Fig. S2). Chromosome 19 had the lowest average germline methylation, and one of the lowest intra-chromosomal variabilities. While this could in part be explained by a higher proportion of CpG islands (Grimwood et al. 2004), it is notable that this chromosome has a lower recombination hot spot density than any other chromosome (Myers et al. 2005).
Figure 1.

A genome-wide map of human germline methylation. A methylation index (MI) was calculated for each 500 kb of genomic sequence with satisfactory data (no gaps in sequencing and at least 20 known SNPs). The y-axis value and bar color (see color index) demonstrate methylation as the ratio of average genome MI (MIav).
Genome-wide analysis of the correlation between germline methylation and recombination
Two parameters can be used to describe the recombinational activity of a window within the genome: the recombination rate of the window and the number of bases within recombination hot spots. The correlation between these parameters was high (r = 0.725, 0.747, 0.782, 0.822 in window sizes 125 kb, 250 kb, 500 kb, and 1000 kb, respectively, P < 10−15 for all correlations).
The quantitative relationship between germline methylation and recombination was tested by several methods. The MI index does not allow for simultaneous correction of multiple confounding factors (see Methods). For this analysis, we therefore used the absolute mSNP count per window while correcting for the number of CpGs and SNP density as well as other relevant sequence factors, rather than the methylation index.
We observed a significant positive correlation between the absolute number of mSNPs per window and both the recombination rate (500-kb windows: r = 0.622, P < 10−15; Fig. 2A; Table 2) and the number of bases within recombination hot spots (500-kb windows: r = 0.508, P < 10−15; Fig. 2B). The correlation increased with increasing window sizes and appears to be higher for recombination rates than recombination hot spots (Supplemental Table S2A), suggesting that methylation might affect the recombination rate beyond recombination hot spots alone. Several other variables might be influencing both variables, explaining this correlation. Therefore, we did a partial correlation correcting for previously known sequence factors influencing recombination at window sizes of 125–1000 kb (GC content, repeats, and exons) (Smith et al. 2005; Myers et al. 2006) as well as factors specific for our methylation model (CpG density and SNP density). The density of recently described DNA motifs associated with recombination (Myers et al. 2005) is not significantly correlated with recombination at window sizes >8 kb, and therefore was not specifically modeled in our analysis (Myers et al. 2006).
Figure 2.

A significant positive correlation was found between number of mSNPs per 500-kb window and both the recombination rate (r = 0.622, P < 10−15) (A) and number of bases within recombination hot spots per window (r = 0.508, P < 10−15) (B).
Table 2.
Correlation of several sequence and model features with recombination rate in the high-resolution ENCODE regions and genome-wide

aStatistically significant values (P < 0.05/6) are in bold.
After correcting for these factors, we still found a significant positive correlation between the number of mSNPs in windows and either the recombination rate (500-kb windows: r = 0.172, P < 10−15) or bases within recombination hot spots (500-kb windows: r = 0.152, P < 10−15) in all window sizes (Supplemental Table S2A).
We created a linear model of either recombination rate or bases per recombination hot spot as a response variable, and several sequence and model features as the predictor variable. When looking at the known predictors of recombination rate in addition to mSNPs in a linear model, mSNPs contributed significantly to the model in all window sizes (Supplemental Table S2B). We found that for recombination rate, mSNPs were the third and fourth strongest predictor in window sizes of 250 kb and 500 kb, respectively (Table 3; Supplemental Table S2B). For recombination hot spots, mSNPs were the second strongest and strongest predictor in window sizes of 250 kb and 500 kb, respectively (Supplemental Table S2B). The proportion of recombination rate variability explained by the linear model was 0.339–0.518, and the proportion of recombination hot spot variability explained by the linear model was 0.193–0.369 depending on window sizes (Supplemental Table S2B). This proportion is within a similar range or higher than previous models have been able to explain (Kong et al. 2002; Smith et al. 2005).
Table 3.
Multiple linear regression of recombination rate as a response to sequence and model feature predictors in the high-resolution ENCODE regions and genome-wide

aStandardized beta values are shown. β is the number of standard deviations that the outcome variable will change as a result of one standard deviation change in the predictor variable.
bStatistically significant values (P < 0.05/6) are in bold.
c(RM) Value not included in linear model.
We repeated the analysis using non-log-transformed data and obtained similar results (data not shown). However, the amount of variability explained by the model was decreased, presumably due to more outliers in the nontransformed data set.
Analysis of the correlation between germline methylation and recombination at a higher resolution
Given the size of the human genome and the latest observed number of recombination hot spots in the genome (Frazer et al. 2007), the average distance between recombination hot spots is 80 kb. Our absolute maximal resolution using the genome-wide HapMap data set is 125 kb, and in that resolution finer-scale effects might be obscured from the relatively coarse measurements. Therefore, an alternative approach was needed to study the relationship between germline methylation and recombination hot spots in greater detail.
The ENCODE HapMap data set contains more detailed haplotype analysis of 5 Mb of human genome sequence. It has roughly three times more known SNPs per sequenced base than the genome-wide data set. We used the ENCODE HapMap data set to create windows of 25 and 50 kb for the 5 Mb of available sequence. In this increased resolution subset, we found a significant positive correlation between mSNP and recombination rate in both window sizes (50-kb windows: r = 0.301, P = 0.002; 25-kb windows: r = 0.319, P < 0.0001; Table 2; Supplemental Table S3A). Furthermore, when we performed correlation correcting for factors known to affect recombination rate at the given window sizes (GC ratio, repeats, exons) as well as factors affecting the methylation model (CpG count, SNP density), the correlation was further strengthened (50-kb windows: r = 0.445, P < 0.0001; 25-kb windows: r = 0.335, P < 0.0001; Supplemental Table S3A). In the linear model of recombination rate, mSNP was the strongest predictor of recombination rate for both window sizes (Table 3; Supplemental Table S3B). The ENCODE data set had insufficient power to create a linear model of recombination hot spots (Supplemental Table S3B), since 153 out of 200 25-kb windows and 60 out of 100 50-kb windows did not contain any recombination hot spot.
Correlation of recombination and Human Epigenome Project data
Given the results obtained using the model of germline methylation, we investigated the correlation between recombination and methylation in an independent set of data. The aim of the Human Epigenome Project (HEP) is to identify, catalog, and interpret genome-wide DNA methylation profiles of all human genes in all major tissues utilizing bisulfite sequencing, the gold standard of methylation profiling (Rakyan et al. 2004). The latest data release from the HEP project contains the methylation status of 2524 amplicons within three human chromosomes in 12 different tissues. The average length of each amplicon is 411 bp, and they contain on average 16 CpGs (Eckhardt et al. 2006). The data set contains information about the percentage of methylation of each cytosine base within a CpG dinucleotide. The short amplicon length made it impossible to calculate the recombination rate of each amplicon, so location within recombination hot spots was used instead.
We screened the whole data set for results from sperm tissue, the final product of the male germ line. Then we calculated the average methylation of each amplicon by averaging the methylation percentage of each sequenced CpG within the amplicons. Since recombination hot spots are on average three to four times larger than the amplicons from the HEP data set, the amplicons were divided into two groups. One group contained amplicons located within recombination hot spots (n = 219) and the other contained amplicons not located within recombination hot spots (n = 1745). For both groups, the distribution of methylation was still bimodal after averaging the CpG methylation values within each amplicon (Fig. 3). The average amplicon methylation was significantly higher for the group located within recombination hot spots (0.632 vs. 0.557, P = 0.007; Fig. 3). We assigned each amplicon a methylation status of one of three categories: unmethylated (<20% methylation), heterogeneously methylated (20%–80% methylation), and hypermethylated (>80% methylation), as described in the HEP data release (Eckhardt et al. 2006). For amplicons not within a recombination hot spot, the distribution of amplicons into the three categories (no methylation, heterogeneous methylation, and hypermethylation) was 36%, 13%, and 50%, respectively. For amplicons within recombination hot spots, the distribution into the three categories was 28%, 16%, and 56%, respectively. These differences in distribution were significantly different between the two groups (P = 0.03). Comparable results were obtained when methylation was split into groups of even ranges (<33%, 33%–66%, and >66% methylation) or into two groups (0%–50% and 51%–100%) (data not shown).
Figure 3.

Density map of the distribution of the average methylation of amplicons located inside recombination hot spots (black, n = 219) or outside of recombination hot spots (gray, n = 1745). Average methylation was significantly more in regions located within hot spots (0.632 vs. 0.557, P = 0.007, t-test).
Discussion
Several recent observations suggest that the DNA sequence itself is not the sole determinant of region-specific and inter-individual variation in recombination activity. Therefore, other alternatives have been proposed, including epigenetic mechanisms. DNA methylation is a prime candidate, either by itself or by interacting closely with another unknown epigenetic mechanism (i.e., histone modifications) conserved through meiosis. To study this mechanism in the germline, we have proposed a novel approach using methylation-associated SNPs (mSNPs) as a surrogate marker for DNA methylation.
Our methodology was supported by the observed sevenfold overrepresentation of mSNPs in the HapMap data set compared with control SNPs. This supports that the mSNPs reflect the previously demonstrated hypermutability of methylated CpGs. Furthermore, the mSNP marker detected the previously known hypomethylation of CpG islands. A genome-wide map of the human germline methylation detected both inter- and intrachromosomal differences. Chromosome 19, known for the lowest recombination hotspot intensity, had the lowest average germline methylation.
We found a statistically significant positive correlation between the number of mSNPs and both recombination rate and recombination hot spots in a genome-wide resolution of 125–1000 kb. This remained significant after correcting for known sequence properties influencing recombination (such as GC content and CpG dinucleotides) and factors influencing the methylation model.
We found an even stronger correlation between recombination rate and mSNPs in the ENCODE regions. These regions comprise a 5-Mb subset of the human genome with an increased density of known SNPs and sequence information, thus allowing analysis with greater resolution. In this data set, germline methylation was found to be the strongest predictor of recombination rate in our linear model of recombination rate in 25- and 50-kb window sizes. This suggests that epigenetic modification might affect recombination more strongly at a smaller (kilobase) rather than on a larger (megabase) scale.
Finally, we used a biological data set from the Human Epigenome Project (HEP) to provide independent results supporting the correlation between methylation and recombination. The project has released a limited but high-quality data set including the bisulfite sequencing results of ∼2000 stretches of DNA in sperm. This is, to date, the largest data release of direct DNA methylation measurements. We found that the average methylation was significantly higher in amplicons located within recombination hot spots compared with amplicons that are not. Furthermore, the distribution of amplicons into groups containing different amounts of methylation was significantly different between amplicons located within recombination hot spots and amplicons not located within recombination hot spots.
Our approach of using mSNPs as a surrogate marker for methylation has several limitations. Not all SNPs fulfilling the criteria of mSNPs are representative of methylation; and, conversely, not all methylation is represented by mSNPs. The difference in recombination rates between males and females poses an additional problem when a sex-averaged data set is used to estimate recombination. This phenomenon has been demonstrated when recombination patterns are examined at large scales (megabases) (Broman et al. 1998; Kong et al. 2002), but the difference seems to decrease when recombination is studied on a finer scale (Myers et al. 2006; Coop et al. 2008). Finally, a cause-and-effect relationship can never be determined based on a correlation between two variables. Both variables might reflect a third variable more proximal to the cause.
Given the limitation of our methylation marker, it is remarkable how consistent and strong our correlations are. Although linear models are commonly used to model recombination, the contribution from nonlinear effects cannot be excluded. Nonlinear effects are, however, more likely to affect the exact magnitude of effects rather than their directionality. While methylation appears to influence recombination, other factors certainly play a role. For instance, recently described motifs determining a significant amount of hot spot-dependent recombination do not contain CpG dinucleotides (Myers et al. 2005).
A relationship between germline DNA methylation and recombination could be interpreted in two ways that are not mutually exclusive. Areas of the genome undergoing recombination could be methylated secondarily, perhaps inhibiting further recombination. A second recombination event (re-recombination) close to a previous one in the same meiosis would negate the potentially beneficial effects of recombination. An interesting consequence of this possibility is that methylation might be a part of the enigmatic signaling pathway mediating crossover interference. At the molecular level, it would be of interest to study if methylation is induced by repair of double-stranded breaks by homologous recombination either in the context of chromosome recombination at meiosis or as a DNA damage response (O'Hagan et al. 2008). This also applies to the relationship between methylation and gene conversion with associated meiotic drive, two important processes closely linked to homologous recombination (Webb et al. 2008).
Alternatively, it is possible that methylation is a potentiator of recombination. Perhaps areas marked by methylation are preferred sites for recombination. Such areas might be those where potentially mutagenic recombination is unlikely to cause harmful effects. This hypothesis is supported by the fact that DNA methylation patterns are already established in prophase I when recombination occurs (Davis et al. 1999; Bourc'his and Bestor 2004; Oakes et al. 2007).
The finding of a positive correlation between methylation and homologous recombination contrasts with the observation of chromosomal instability caused by mutations in DNMT3B, a de novo methylation enzyme of the human genome (Xu et al. 1999). This suggests that methylation might have a role in promoting homologous recombination, but suppresses nonhomologous recombination. Another possibility, as previously explained, is that methylation is used to mark regions that have already undergone recombination. A lack of methylation could then result in frequent re-recombination, eventually leading to chromosomal instability. This would be an interesting avenue to pursue in further studies.
Our results suggest that differences in methylation provide an explanation for a previously unexplained phenomenon of inter-individual differences in recombinational activity despite identical DNA sequence, as well as different locations of recombination hot spots between species with high sequence homology (Winckler et al. 2005; Neumann and Jeffreys 2006). Previously, CpG density has been correlated with recombination (Kong et al. 2002; Myers et al. 2006). Also, an epigenetic mechanism has been proposed as the explanation for recombination (Winckler et al. 2005; Neumann and Jeffreys 2006; Sandovici et al. 2006). Our results support and extend these results by showing a positive correlation between germline DNA methylation and human recombination.
Methods
Definitions
In the following paragraphs, we define methylation-associated SNP (mSNP) as any C/T or G/A (corresponding to a C/T polymorphism on the opposite strand) polymorphism with an adjacent 3′ guanine (for C/T polymorphism) or adjacent 5′ cytosine (for G/A polymorphism). Therefore, the subset of mSNPs includes all possible methylation-associated mutations occurring in the CpG dinucleotide within a given database of SNPs. For the genome-wide associations, an additional criterion for a mSNP to become a mSNPgenome was that the ancestral allele was either C or G. The mSNPENCODE were defined as mSNPs in the ENCODE regions.
We took two different approaches to correct for possible confounding factors of our methylation model. For graphical representation and mapping purposes we calculated the MI for the genome in various window sizes. The index should be considered as a ratio of observed mSNP (NmSNP) to the expected (NCpG · NSNP) times a constant. The index is defined as:
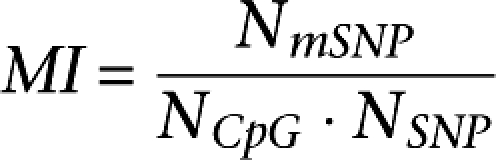 |
where NmSNP stands for the number of mSNPgenome within a given window. NCpG stands for the number of CpG dinucleotides within the window and is directly proportional to the probability of a SNP occurring in a CpG. NSNP stands for the number of all SNPs except mSNPs detected within the window. The density reflects both functional constraints in the region and the sensitivity of the method used for SNP detection. The product NCpG · NSNP is therefore directly proportional to the expected number of mSNPs.
The MI only provides correction for two confounding factors. When several factors can affect a variable, and the factors can be intercorrelated, a calculation of an index such as the MI is statistically insufficient. The MI index was therefore used only for imaging purposes. For exploration of the relationship between recombination rate and germline methylation, the absolute number of mSNPs per window was used while correcting for the effects of other confounding factors using partial correlation or multiple linear regression (see below), since these methods allow for the correction of multiple variables simultaneously.
HapMap data sets
For genome-wide searches for mSNPs, we used release 21 (July 2006) of the genotype data from the International HapMap Consortium. We downloaded the nonredundant genotype set for all 22 autosomal chromosomes for all populations from the HapMap website (http://www.hapmap.org). We also downloaded a derived alleles data set (http://hgwdev.cse.ucsc.edu/∼daryl/HapMap_rel21_derived_alleles/). This data set includes information about the chimpanzee and macaque states for each HapMap allele set when available, thus determining which allele was the ancestral one (Thomas et al. 2007). We analyzed data for all 22 autosomal chromosomes. We wrote programs to extract the appropriate iHS value for mSNPs and non-mSNPs from the entire database of calculated iHS values for HapMap phase II, downloaded from http://haplotter.uchicago.edu/selection/ on 01/01/2009 (Voight et al. 2006).
The ENCODE (ENCyclopedia of DNA Elements) project aims to identify all functional elements in the human genome (The ENCODE Project Consortium 2004). We downloaded the full HapMap genotype data set for the currently available 10 500-kb human genome regions from the HapMap ENCODE website (http://www.hapmap.org/downloads/encode1.html, accessed 7/11/2008).
For both the genome-wide data set and the ENCODE data set, we searched within all four populations for the SNPs of interest. We then pooled the populations and erased redundant polymorphisms so our final data set used in further analysis contained a single copy of each SNP.
Sequence features
For information on GC content, location of gaps, and CpG dinucleotide count, we created programs to search within the human genome sequence (NCBI Build 35, UCSC hg 17), downloaded from the UCSC Genome Browser (http://genome.ucsc.edu) (Kent et al. 2002). Information on recombination rate and recombination hot spots based on phase II of the International HapMap Consortium project was downloaded from the HapMap website. The creation of this data set is described in detail elsewhere (The International HapMap Consortium 2005). For all other sequence features, both for genome-wide analysis and the ENCODE region analysis, we used tables derived from the UCSC Table Browser (Karolchik et al. 2004). For genome-wide analysis, all data tables were based on NCBI Build 35 of the human genome (UCSC hg 17); for the ENCODE regions, the tables were based on NCBI Build 34 (UCSC hg 16). Specifically, we used the cpgIslandExt table for the location and properties of CpG islands (Gardiner-Garden and Frommer 1987), the knownGene table for the location of exons of known genes (Hsu et al. 2006), and the rmsk table for the location and properties of repeated elements created using the RepeatMasker (http://www.repeatmasker.org) based on the Repbase database of repeated elements (Jurka et al. 2005). Due to the multiple data sets underlying the known gene table, redundant data were removed from the table, and only one copy of each exon was included in the analysis.
HEP data set
Data from the Human Epigenome Project (HEP) were downloaded from the project website (http://www.epigenome.org). We used the most recent data release containing the results from bisulfite sequencing of 2524 amplicons chosen from chromosomes 6, 20, and 22 in 12 different tissue types (Eckhardt et al. 2006). We then wrote programs selecting out data from sperm as well as programs to determine if an amplicon was located within a recombination hot spot or not.
Programs
We developed several programs in the JAVA programming language using the Textpad editor (Helios Software Solutions). For statistical and figure creations, scripts were also developed in the R statistical language. The source code of all programs used in the paper is available at www.hi.is/∼mis.
Sliding windows correlation and statistical analysis
Our genome-wide analyses were done using four different window sizes (125 kb, 250 kb, 500 kb, and 1000 kb). For each window size, we divided the genome into nonoverlapping windows. Each window was then assigned values according to its genetic properties (recombination rate, number of bases within CpG dinucleotides and repeats, GC content, mSNP amount, SNP density, and calculated MI). Windows containing any sequencing gaps were removed prior to analysis. The genome-wide map of the MI only displays windows where more than 20 SNPs were available for analysis. The ENCODE regions were analyzed in a similar fashion using nonoverlapping windows of two different sizes (25 kb and 50 kb). Each window was assigned values according to their genetic properties in the same way as for the genome-wide analysis.
For simple correlation, we first explored the distribution of all variables by applying the Kolmogorov–Smirnov normality test. If any of the variables were not normally distributed, a Spearman ranked correlation coefficient was calculated; otherwise a Pearson correlation coefficient was calculated. Partial correlations were also done using either ranked data or unranked data based on tests of normality.
Prior to multiple linear regression analysis, we first transformed the data to provide a better fit to normal distribution using Box–Cox transformation, a form of lognormal transformation. Linear regression was then done using either recombination rate or number of bases within recombination hot spots as the response variable using a stepwise backward method.
When comparing the summary statistics of two different groups, either χ2 or t-tests were done. For the genome-wide and ENCODE regions analysis, multiple correlation (six correlations) was performed between sequence features and recombination. A P-value < 0.05/6 was therefore considered statistically significant. We note, however, that certain features tested were substantially intercorrelated (such as GC content and amount of CpG dinucleotides), possibly increasing the likelihood of type II errors. For other analysis where multiple testing was not done, a P-value < 0.05 was considered statistically significant. Linear regression was done using SPSS version 15 (SPSS, Inc.). All other statistical analysis as well as figure preparation was done using R package version 2.5.1.
Acknowledgments
This work was supported by a grant to M.I.S. by the Icelandic Student Innovation fund and by grants from the Icelandic Science Fund, University of Iceland Research Fund, and Landspitali Research Fund.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are http://www.genome.org/cgi/doi/10.1101/gr.086181.108.
References
- Antequera F., Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol. 1999;9:R661–R667. doi: 10.1016/s0960-9822(99)80418-7. [DOI] [PubMed] [Google Scholar]
- Badal V., Chuang L.S., Tan E.H., Badal S., Villa L.L., Wheeler C.M., Li B.F., Bernard H.U. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: Genomic hypomethylation correlates with carcinogenic progression. J. Virol. 2003;77:6227–6234. doi: 10.1128/JVI.77.11.6227-6234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A.P. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Bjornsson H.T., Fallin M.D., Feinberg A.P. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Bjornsson H.T., Ellingsen L.M., Jonsson J.J. Transposon-derived repeats in the human genome and 5-methylcytosine-associated mutations in adjacent genes. Gene. 2006;370:43–50. doi: 10.1016/j.gene.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Bjornsson H.T., Sigurdsson M.I., Fallin M.D., Irizarry R.A., Aspelund T., Cui H., Yu W., Rongione M.A., Ekstrom T.J., Harris T.B., et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc'his D., Bestor T.H. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431:96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- Broman K.W., Murray J.C., Sheffield V.C., White R.L., Weber J.L. Comprehensive human genetic maps: Individual and sex-specific variation in recombination. Am. J. Hum. Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G., Wen X., Ober C., Pritchard J.K., Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395–1398. doi: 10.1126/science.1151851. [DOI] [PubMed] [Google Scholar]
- Cooper D.N., Youssoufian H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Coulondre C., Miller J.H., Farabaugh P.J., Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Davis T.L., Trasler J.M., Moss S.B., Yang G.J., Bartolomei M.S. Acquisition of the H19 methylation imprint occurs differentially on the parental alleles during spermatogenesis. Genomics. 1999;58:18–28. doi: 10.1006/geno.1999.5813. [DOI] [PubMed] [Google Scholar]
- Eckhardt F., Lewin J., Cortese R., Rakyan V.K., Attwood J., Burger M., Burton J., Cox T.V., Davies R., Down T.A., et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Feinberg A.P., Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M., Frommer M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Grimwood J., Gordon L.A., Olsen A., Terry A., Schmutz J., Lamerdin J., Hellsten U., Goodstein D., Couronne O., Tran-Gyamfi M., et al. The DNA sequence and biology of human chromosome 19. Nature. 2004;428:529–535. doi: 10.1038/nature02399. [DOI] [PubMed] [Google Scholar]
- Hellman A., Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Hsu F., Kent W.J., Clawson H., Kuhn R.M., Diekhans M., Haussler D. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys A.J., Kauppi L., Neumann R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 2001;29:217–222. doi: 10.1038/ng1001-217. [DOI] [PubMed] [Google Scholar]
- Jensen-Seaman M.I., Furey T.S., Payseur B.A., Lu Y., Roskin K.M., Chen C.F., Thomas M.A., Haussler D., Jacob H.J. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004;14:528–538. doi: 10.1101/gr.1970304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. The neutral theory of molecular evolution and the world view of the neutralists. Genome. 1989;31:24–31. doi: 10.1139/g89-009. [DOI] [PubMed] [Google Scholar]
- Kimura M. Recent development of the neutral theory viewed from the Wrightian tradition of theoretical population genetics. Proc. Natl. Acad. Sci. 1991;88:5969–5973. doi: 10.1073/pnas.88.14.5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A., Gudbjartsson D.F., Sainz J., Jonsdottir G.M., Gudjonsson S.A., Richardsson B., Sigurdardottir S., Barnard J., Hallbeck B., Masson G., et al. A high-resolution recombination map of the human genome. Nat. Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Krawczak M., Ball E.V., Cooper D.N. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am. J. Hum. Genet. 1998;63:474–488. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher M.J., Hurst L.D. Imprinted chromosomal regions of the human genome have unusually high recombination rates. Genetics. 2003;165:1629–1632. doi: 10.1093/genetics/165.3.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J.Q., Li T., Hu J.F., Vu T.H., Chen H.L., Qiu X.W., Cherry A.M., Hoffman A.R. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science. 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- Mohandas T., Sparkes R.S., Shapiro L.J. Reactivation of an inactive human X chromosome: Evidence for X inactivation by DNA methylation. Science. 1981;211:393–396. doi: 10.1126/science.6164095. [DOI] [PubMed] [Google Scholar]
- Myers S., Bottolo L., Freeman C., McVean G., Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- Myers S., Spencer C.C., Auton A., Bottolo L., Freeman C., Donnelly P., McVean G. The distribution and causes of meiotic recombination in the human genome. Biochem. Soc. Trans. 2006;34:526–530. doi: 10.1042/BST0340526. [DOI] [PubMed] [Google Scholar]
- Neumann R., Jeffreys A.J. Polymorphism in the activity of human crossover hotspots independent of local DNA sequence variation. Hum. Mol. Genet. 2006;15:1401–1411. doi: 10.1093/hmg/ddl063. [DOI] [PubMed] [Google Scholar]
- Oakes C.C., La Salle S., Smiraglia D.J., Robaire B., Trasler J.M. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev. Biol. 2007;307:368–379. doi: 10.1016/j.ydbio.2007.05.002. [DOI] [PubMed] [Google Scholar]
- O'Hagan H.M., Mohammad H.P., Baylin S.B. Double-strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008;4:e1000155. doi: 10.1371/journal.pgen.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan V.K., Hildmann T., Novik K.L., Lewin J., Tost J., Cox A.V., Andrews T.D., Howe K.L., Otto T., Olek A., et al. DNA methylation profiling of the human major histocompatibility complex: A pilot study for the Human Epigenome Project. PLoS Biol. 2004;2:e405. doi: 10.1371/journal.pbio.0020405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K.D., Wolffe A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000;1:11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- Sandovici I., Kassovska-Bratinova S., Vaughan J.E., Stewart R., Leppert M., Sapienza C. Human imprinted chromosomal regions are historical hot-spots of recombination. PLoS Genet. 2006;2:e101. doi: 10.1371/journal.pgen.0020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.V., Thomas D.J., Munro H.M., Abecasis G.R. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res. 2005;15:1519–1534. doi: 10.1101/gr.4421405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F., Smith J.F., Kimura M.T., Morrow A.D., Matsuyama T., Nagase H., Held W.A. Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc. Natl. Acad. Sci. 2005;102:3336–3341. doi: 10.1073/pnas.0408436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D.J., Trumbower H., Kern A.D., Rhead B.L., Kuhn R.M., Haussler D., Kent W.J. Variation resources at UC Santa Cruz. Nucleic Acids Res. 2007;35:D716–D720. doi: 10.1093/nar/gkl953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venolia L., Gartler S.M., Wassman E.R., Yen P., Mohandas T., Shapiro L.J. Transformation with DNA from 5-azacytidine-reactivated X chromosomes. Proc. Natl. Acad. Sci. 1982;79:2352–2354. doi: 10.1073/pnas.79.7.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight B.F., Kudaravalli S., Wen X., Pritchard J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb A.J., Berg I.L., Jeffreys A. Sperm cross-over activity in regions of the human genome showing extreme breakdown of marker association. Proc. Natl. Acad. Sci. 2008;105:10471–10476. doi: 10.1073/pnas.0804933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M., Hellmann I., Stadler M.B., Ramos L., Paabo S., Rebhan M., Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- Weisenberger D.J., Campan M., Long T.I., Kim M., Woods C., Fiala E., Ehrlich M., Laird P.W. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33:6823–6836. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler W., Myers S.R., Richter D.J., Onofrio R.C., McDonald G.J., Bontrop R.E., McVean G.A., Gabriel S.B., Reich D., Donnelly P., et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- Xu G.L., Bestor T.H., Bourc'his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Yoder J.A., Walsh C.P., Bestor T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335–340. doi: 10.1016/s0168-9525(97)01181-5. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Zhang F. Sequence context analysis of 8.2 million single nucleotide polymorphisms in the human genome. Gene. 2006;366:316–324. doi: 10.1016/j.gene.2005.08.024. [DOI] [PubMed] [Google Scholar]