

Abstract
The high mutation rate of HIV is linked to the generation of viruses expressing proteins with altered function whose impact on disease progression is unknown. We investigated how HIV-1 viruses lacking Env, Vpr, and Nef affect CD4+ T cell survival. We found that in the absence of these proteins, HIV-1-infected CD4+ primary T cells progress to the G0 phase of the cell cycle and to cell death, indicating that viruses expressing inactive forms of these proteins can contribute to the CD4+ T cell decline as the wild-type virus, suggesting that other HIV proteins are responsible for inducing apoptosis. Apoptosis in these cells is triggered by the alteration of the Egr1-PTEN-Akt (early growth response-1/phosphate and tensin homolog deleted on chromosome 10/Akt) and p53 pathways, which converge on the FOXO3a (Forkhead box transcription factor O class 3a) transcriptional activator. The FOXO3a target genes Fas ligand and TRAIL, involved in the extrinsic apoptotic pathway, and PUMA, Noxa, and Bim, which are part of the intrinsic apoptotic pathway, were also up-regulated, indicating that HIV infection leads to apoptosis by the engagement of multiple apoptotic pathways. RNAi-mediated knockdown of Egr1 and FOXO3a resulted in reduced apoptosis in HIV-infected HeLa and CD4+ T cells, providing further evidence for their critical role in HIV-induced apoptosis and G0 arrest. We tested the possibility that Tat is responsible for the T cell apoptosis observed with these mutant viruses. The induction of Egr1 and FOXO3a and its target genes was observed in Jurkat cells transduced by Tat alone. Tat-dependent activation of the Egr1-PTEN-FOXO3a pathway provides a mechanism for HIV-1-associated CD4+ T cell death.
Human exposure to HIV results in an acute systemic infection that becomes chronic once virus-specific immune responses are established, as these responses are not sufficient to clear the infection. Initially, there is a relatively balanced coexistence of host and pathogen and this chronic infected state is not associated with disease. However, with time, the balance between HIV-1 replication and host immune control breaks down and a progressive loss of CD4+ T cells occurs. This leads to clinical AIDS and, in the absence of therapy, to the death of the infected host, primarily due to opportunistic infections that cannot be controlled by the damaged immune system (reviewed in Refs. 1, 2).
We know much about the direct cytopathic effect that the virus has on CD4+ T cells, but this effect is insufficient to explain the dramatic losses of CD4+ T cells in the infected hosts that progress to AIDS. Studies have shown a correlation between disease progression, immunoactivation, and T cell apoptosis, suggesting that HIV-1 can eliminate both infected and uninfected CD4 cells via activation of programmed cell death (3–9).
Apoptosis involves multiple biochemical events leading to cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation. The process is controlled by a variety of cell signals, which may originate either extracellularly or intracellularly. Two separate pathways lead to caspase activation: the extrinsic pathway and the intrinsic pathway. The extrinsic pathway is activated by the binding of ligands such as Fas ligand (FasL),3 TNF, and TRAIL/Apo-2 ligand to their death receptors FAS/CD95, TNFR1, DR4, and DR5. This binding leads to the activation of membrane-proximal caspases (caspase-8 and caspase-10), which activate effector caspase-3 and caspase-7. The intrinsic pathway is initiated by the disruption of the mitochondrial membrane and the release of mitochondrial proteins, such as cytochrome c into the cytoplasm. Cytochrome c is crucial for the assembly of a caspase-activating complex, which activates caspase-9, leading to the initiation of the apoptotic caspase cascade (10 –13). The Bcl-2 family proteins regulate the release of cytochrome c (11, 14–16). Based on their function in apoptosis, the Bcl-2 family can be divided into proapoptotic and antiapoptotic members that can physically interact. The interaction of proteins with BH-3 (Bcl-2 homology region-3) motifs (e.g., Noxa, p53 up-regulated modulator of apoptosis (PUMA), Bad, and Bim) with antiapoptotic Bcl-2 proteins (e.g., Bcl-2 and Bcl-xL) promotes the activation of Bax and Bak (15, 16). Additionally, there is a substantial cross-talk between the extrinsic and intrinsic pathways. Caspase-8 can proteolytically activate Bid, which can then facilitate the release of cytochrome c and amplify the apoptotic signal that follows the engagement of death receptors (10 –13).
The molecular basis of how this phenomenon is triggered in HIV-infected primary cells and possibly in bystander noninfected CD4+ T cells is not completely understood and it is likely to depend on the interplay of viral and cellular proteins. A better understanding of how viral proteins interact with and alter host cell metabolism might provide insights important for improved understanding of AIDS pathogenesis.
HIV-1 expresses a number of proteins that can have significant effects on host cell functions. Several different HIV-1 proteins have been shown to interfere with cellular proteins implicated in the control of cell death (reviewed in Ref. 17). Among these proteins, the role of Env in apoptosis is probably the best studied, particularly for bystander cell death (18–22). Env can vary significantly among different viral isolates and during the course of the infection within the same individual, leading to viruses with different cytopathic characteristics (23–25). The contribution to the disease course by the different mutants that can occur in vivo is not known.
Other HIV-1 proteins that have been implicated in aspects of apoptosis include Tat, the protease, Nef, and Vpr. Exposure to Tat can induce cell death by apoptosis in cultured PBMC (5). Tat has also been shown to play an important role in neuronal apoptosis (26). As Tat can be secreted by infected cells and taken up by uninfected cells, Tat-induced apoptosis could affect both infected and uninfected cells (4, 27, 28). HIV-1 Nef can induce chemokines in primary macrophages that may facilitate lymphocyte recruitment and activation (29). Nef can also facilitate infection of resting T cells by secreting paracrine factors such as ICAM and soluble CD23 (30) and can induce a T cell activation program similar to that induced by stimulation of the CD3 receptor (31). Vpr can affect cell death by affecting cell cycle progression and inducing the dissipation of the mitochondrial transmembrane potential (32, 33). Vpr mutants observed in long-term nonprogressors have been shown to have reduced or absent apoptotic potential. As a consequnce, it was suggested that mutations within the Vpr gene may be associated with prolonged survival (34, 35). The HIV-1 transcriptional transactivator Tat has also been reported to affect host cell functions (26, 36–39).
TRAIL, a member of the TNF family, and FasL have been shown to trigger apoptosis in T cells from HIV-1-infected individuals while T cells from uninfected controls are completely resistant to TRAIL-induced apoptosis (40–43). There are at least four receptors for this molecule, some of which mediate TRAIL signaling (DR4 and DR5) while others block TRAIL signal transduction (decoy receptor (DcR)1 and DcR2) (44). Differential expression of these receptors, which might be modulated directly or indirectly by HIV-1, may play a role in the sensitivity to TRAIL of cells from HIV-1+ individuals (45). TRAIL was found to be elevated in plasma of HIV-1-infected patients compared with uninfected individuals; DR5 expression was also elevated and associated with the apoptotic marker annexin V (45, 46). TRAIL was found to be induced in HIV-1- and SIV-infected macrophages and dendritic cells derived from species susceptible to AIDS, but not from species resistant to AIDS (36). FAS/FasL interaction is associated with the level of apoptosis observed in the lymphoid tissues of HIV-infected individuals, with their viral loads, and with CD4+ T cell number (40). The increased level of immunoactivation observed during HIV infection may explain the increased susceptibility to FasL-mediated apoptosis, since FAS+ resting T cells are resistant to apoptosis (47–49).
In this study we investigated how HIV-1 viruses that lack Env and Nef or Env, Vpr, and Nef affected CD4+ T cell survival. We found that in the absence of these proteins most HIV-1-infected CD4+ primary T cells progressed to the G0 phase of cell cycle followed by cell death, suggesting that these null mutants of these proteins can have an impact on the CD4+ T cell destruction observed during the infection. Apoptosis of these cells was triggered by the modulation of genes critical for the intrinsic and extrinsic apoptotic pathways that are regulated via the Forkhead box transcription factor O class 3a (FOXO3a) transcriptional activator, which was found induced by both HIV mutants and wild-type strains with different tropism.
Materials and Methods
Plasmids
Vectors for production of retroviruses (pCMMP-eGFP, pMMP-Tat1b-IGFP, pHDM-G/VSV-G envelope, and pMD.MLVgp) were generously provided by Dr. Jeng-Shin Lee from the Harvard Gene Therapy Initiative. Vectors for HXB2 and Bal envelopes were obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health: HXB2-env from Dr. Kathleen Page and Dr. Dan Littman and HIV-1 clone Bal.01 (catalog no. 11445) from Dr. Mascola. To construct pNL4-3.eGFP.R+E− (HIVΔ2GFP) and pNL4-3.eGFP.R−E− (HIVΔ3GFP), the GFP coding sequence was amplified by PCR using primers containing a 5′ NotI and a 3′ XhoI site. The PCR product was then cleaved with NotI and XhoI and ligated to similarly cleaved pNL4-3.HSA.R+E− or pNL4-3.HSA.R−E− vectors obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health: pNL4-3.HSA.R+E− and pNL4-3.HSA.R−E− from Dr. Nathaniel Landau (Germantown, MD). These vectors contain the gene for murine heat-stable Ag (HSA), CD24, fused in frame to the nef initiator methionine codon. This resulted in a replacement of the HSA gene with GFP. The HIVΔ2GFP and HIVΔ3GFP are competent for a single round of replication due to a frame-shift near the 5′ end of env to block production of gp160, and requires cotransfection with Env expression vector to produce infectious virus. Recombinant adenoviruses Ad-tet transactivator (tTA) and Ad-TatSF2 were constructed according to established protocols (50) in HEK 293 T (293T) cells. The Tat coding region is under a tetracycline inducible promoter. Tat is expressed in cells coinfected with Ad-tTA, which expresses the tetracycline-responsive transactivator.
Cell lines, primary cells, treatments, Abs, and reagents
The human cervical cancer cell line HeLa, human kidney cell line 293T, or Jurkat T cell lines were maintained in DMEM or in RPMI 1640 (Invitrogen), respectively, supplemented with 10% (v/v) FBS (Gemini Bio-Products) and 100 μg of penicillin and streptomycin/ml and grown at 37°C and 5% CO2. Human PBMCs were obtained from healthy donors from the Children's Hospital Boston blood bank by Ficoll-Hypaque density gradient centrifugation. Human CD4+ T cells were isolated using a CD4+ T cell negative selection kit from Miltenyi Biotec according to the manufacturer's instructions. The purity of CD4+ lymphocytes was between 97% and 99%. Isolated human CD4+ T cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated human AB serum (Valley Biomedical), 600 U/ml recombinant human (rh) IL-2, 5 μg/ml anti-CD3, and 0.3 μg/ml anti-CD28 (R&D Systems) for a period of 2–3 days, followed by a 3-day incubation in the presence of rhIL-2 alone, before transduction. Following transduction, primary lymphocytes were cultured in RPMI 1640 supplemented with 10% human AB serum and 600 U/ml rhIL-2. Human CD4+ cells were stained with Abs against CD3, CD4, CD95/Fas, CD95L/FasL, p53, membrane TRAIL (mTRAIL, BD Biosciences), DR4, and DR5 (eBioscience), CD8 (BD Biosciences), CD14, CD16, CD19 (Beckman Coulter), and CD123 (Miltenyi Biotec). Other Abs used in the studies were against HIV-1 p24 (KC57-FITC) (Beckman Coulter), human Ki67 (BD Biosciences), human Egr1 (early growth response protein-1), PTEN (phosphate and tensin homolog deleted on chromosome 10), p-Akt (Ser473), FOXO3a, ATM (mutated in ataxia-telangiectasia) (Cell Singaling Technology), human activating transcription factor 3 (ATF3), cyclin E, p130 (Santa Cruz Biotechnology), and human cyclin A-PE (BD Pharmingen). For blocking experiment the following mAbs were used: anti-human DR4 (HS101), DR5 (HS201) (Alexis Biochemicals), TRAIL (RIK-2), CD95L/FasL (NOK-1) (eBioscience), and CD95/Fas (ZB4) (Gene Tex). As an isotype control, a mouse IgG (catalog no. 16-4714, R&D Systems) was used.
Transfection
293T cells were transfected with plasmid DNA of appropriate expression vectors by calcium phosphate precipitation. Supernatants containing appropriate viruses were harvested 72 h after transfection, filtered through a 0.45-μm-pore-size membrane, concentrated using a 100,000 MWCO column (Centricon Plus-20, Millipore) according to the manufacturer's instructions, and titrated by infection.
Transduction of primary T lymphocytes and infection of cell lines
For primary cells, a day before infection, the media was replaced with fresh IL-2-supplemented media to ensure cell cycling. T cells were transduced with vector-containing supernatants of different vesicular stomatitis virus-G protein (VSV-G)-, HXB2-, or Bal-pseudotyped vectors (enhanced GFP (eGFP), Tat-GFP, HIVΔ2GFP or HIVΔ3GFP) at multiplicity of infection (MOI) of 1, and supplemented with 4 μg/ml protamine sulfate (Sigma-Aldrich). Cells were exposed to the virus-containing supernatants for 6 h at 37°C and 5% CO2 and then washed in plain RPMI 1640 medium. Forty-eight hours after transduction T cells were harvested, sorted, and GFP-positive cells collected for PCR and microarrays or functional assays.4 HeLa cells were transduced in 24-well plates with viruses diluted into cell culture media with 8 μg/ml polybrene. After 6 h, virus was replaced with fresh culture media. Jurkat T cells were exposed to adenoviruses expressing tTA or Tat protein (HIVSF2) at MOI of 20 for 2 h. After 2 h, cells were washed and fresh media was replaced.
RNA isolation, labeling, and array hybridization
Ten hours postsorting, CD4+ cells were harvested and total RNA was isolated using TRIzol reagent (Invitrogen). Total RNA (0.1 μg) was prepared for hybridization to HG U133 2.0 oligonucleotide array (Affymetrix) according to the small sample expression protocol (Affymetrix). Hybridization was conducted overnight with 1.5 μg of labeled cRNA product, and arrays were scanned on Affymetrix scanners.
DNA array data analysis
GeneChip arrays were scanned using a GeneArray scanner (Affymetrix). The CEL files were generated from DAT files using GeneChip operating software (GCOS, Affymetrix). Expression ratios were calculated for each probe set relative to the intensity in the eGFP control. Changes in gene expression were considered significant if each of the following criteria was met: 1) expression changed at least 1.6-fold in two of the three donors compared with eGFP control, and 2) increased gene expression included at least one “present” call (Affymetrix algorithm), or both eGFP control samples were present when gene expression decreased.
Quantitative real-time RT-PCR
One hundred nanograms of RNA was reverse transcribed using an iScript cDNA kit (Bio-Rad) followed by amplification of HIV-1 Tat or human genes for GAPDH and other genes selected from the microarray analysis using a SYBR Green SuperMix with ROX kit (Bio-Rad) and primers specific for the genes of interest and human GAPDH as a control.
Immunofluorescent staining and flow cytometry
For surface staining, isolated CD4+ T lymphocytes (1 × 106 cells) were washed in PBS and incubated with Abs against CD4, mTRAIL, DR4, DR5, and Fas for 20 min at room temperature, washed in PBS, and fixed in 1% paraformaldehyde (Sigma-Aldrich). For intracellular staining, cells were stained for surface markers as described above, fixed, and permeabilized with saponin-containing BD Cytofix/Cytoperm solution (BD Biosciences) for 20 min at room temperature according to the manufacturer's instructions. Cells were washed in BD Perm/Wash buffer and incubated with Abs against FasL, or p53 for 30 min at room temperature. Alternatively, cells were blocked with 1% donkey serum (Santa Cruz Biotechnology) for 30 min, stained for 1 h with Abs raised in rabbit against human ATF3, ATM, cyclin A, cyclin E, Egr1, PTEN, p-Akt, FOXO3a, or p130, followed by a 30-min incubation with a secondary donkey anti-rabbit IgG-PE (Santa Cruz Biotechnology) Ab. For intracellular staining with Ab against FasL, cells were pretreated for 4–6 h with protein transport inhibitor GolgiStop (BD Biosciences) containing monensin. For cell cycle analysis, CD3+CD4+ cells were fixed and permeabilized, stained for 30 min at room temperature with mAbs against HIV-1 p24 and Ki67, washed, and fixed in 0.6–1% paraformaldehyde for 5 min. After fixation, cells were washed in PBS and resuspended in 7-aminoactinomycin D (7AAD) dye (BD Biosciences) to visualize DNA content. Isotype-matched IgG for each fluorochrome was used as a negative control. For cell cycle staining of annexin V+ cells, cells were stained with Ab to CD4 followed by a 15-min staining with annexin V (BD Biosciences) according to the manufacturer's instructions. Cells were fixed and permeabilized as previously described, except that all fixation, permeabilization, and wash buffers were supplemented with 2.5 mM calcium chloride to maintain binding of annexin V to the cell surface. Flow cytometric acquisition was performed on MoFlow (Dako). Cell analysis was performed on the gated live-cell population using Summit software (Dako).
ELISA
ELISA for HIV-1 p24 from PerkinElmer was performed according to the manufacturer's instructions.
Apoptosis staining
CD4+ T lymphocytes were stained with Ab against CD4 as described above, followed by staining with annexin V-PE and 7AAD (BD Biosciences) according to the manufacturer's instructions, and analyzed by flow cytometry as described above. Level of HIV-1-induced apoptosis was expressed as the percentage of CD4+GFP+ T cells that stained for annexin V relative to the mock (eGFP) sample. To measure the effect of short interfering RNA (siRNA) against FOXO3a on HIV-1-induced apoptosis, siRNA-treated and HIV-1-transduced HeLa cells were harvested 48 h postinfection and stained with annexin V-PE and 7AAD. For blocking studies, activated CD4+ T lymphocytes were electroporated with siRNAs, treated for 4 h with mAbs against human TRAIL, DR4, DR5, CD95/Fas, or CD95L/FasL at a final concentration of 5 μg/ml, infected with HIV-1 virus pseudotyped with HXB2 envelope, and harvested 48 h postinfection. Apoptosis level was expressed as the percentage of GFP+ (infected) HeLa or CD4+ T cells that stained for annexin V relative to the control.
siRNA treatments
All siRNA treatments were performed with Dharmacon SMARTpool siRNA duplexes: ATF3, ATM, Egr1, FOXO3a, and nontargeting siRNA. SMARTpool siRNAs were transfected at a final concentration of 150–200 nM into exponentially growing HeLa cells with INTERFERin transfection reagent (Bridge Bioscience) or electroporated at a final concentration of 3 μg into stimulated CD4+ T lymphocytes using a Nucleofector kit (Amaxa), all according to the manufacturers' protocols.
Western blots
For Western blotting, 20–50 μg of total proteins was separated by SDS-PAGE, transferred onto the nitrocellulose membrane, and incubated with rabbit Abs raised against FOXO3a, phospho-FOXO3a (Ser318), PTEN, Akt, phospho-Akt (Ser473) (Cell Signaling Technology), ATF3, cyclin E, p130, phospho-p130 (Thr986) (Santa Cruz Biotechnology), and cyclin A (BD Pharmingen). The secondary Ab was detected by ECL (Pierce) according to the manufacturer's instructions.
Statistical analysis
A two-tailed, two-sample Student t test was used to calculate p-values for differences in means between groups. The data are expressed as means ± SEM. For statistical inference, a p-value of <0.05 was considered significant.
Results
HIV-1 can induce apoptosis in human primary CD4+ T cells independently of the envelope, Nef and Vpr proteins
The hallmark of HIV-1 infection is the progressive depletion of CD4+ T cells. The contribution of different HIV-1 proteins to this process and the mechanisms by which they can induce apoptosis in primary T cells is not fully understood. To investigate the role of HIV-1 in primary CD4+ T cell death we investigated the induction of apoptosis by single-cycle Nef-deleted viruses that carried a functional or a deleted vpr gene and were pseudotyped with HIV-related envelopes or VSV-G envelope. Pseudotyped viruses were utilized to take advantage of the single-cycle infection established by these viruses that permits the analysis of a homogeneous population after infection.
Purified populations of primary CD4+ lymphocytes, which had been activated in a TCR-dependent manner using Abs to CD3 and CD28 as well as recombinant IL-2 protein, were infected with two mutated HIV-1 viruses, HIVΔ2GFP (with env and nef deletions) and HIVΔ3GFP (vpr, env, and nef deletions). These mutants are competent for a single round of infection after pseudotyping and express the GFP protein that provides a marker for infectivity. As a control, we used a retroviral vector expressing only GFP (eGFP). CD3+CD4+ T lymphocytes were purified by negative selection to a purity of 97–99% (data not shown). The contamination of CD3+CD4+GFP+ T lymphocytes with CD14+GFP+ monocytes or CD123+GFP+ plasmacytoid dendritic cells was found negligible (Fig. 1A). Forty-eight hours after infection, GFP+ cells were sorted and placed in culture for further analysis. The purity of GFP+ cells after sorting was between 95% and 99% (Fig. 1, A and B, day 2). CXCR4 Env (HXB2) and CCR5 Env (HIV-1 Bal) were tested in these experiments and compared with the infection induced by a VSV-G-pseudotyped HIV-1. CD4+ T cells infected with pseudotyped HIV-1 viruses were sorted using GFP as a marker and monitored for cell proliferation, viability, and apoptosis progression. A significant decrease in the percentage of GFP+ cells in the HIVΔ2GFP- or HIVΔ3GFP-infected populations relative to the control (eGFP/VSV-G-infected cells) was observed regardless of the Env used in the pseudotyping (Fig. 1B). All cultures infected with HIV-1 viruses failed to proliferate (Fig. 1C), their viability showed a steady decline (Fig. 1D), and similar levels of apoptosis were observed when compared with cultures infected with VSV-G-pseudotyped HIV-1 (Fig. 1E–H). VSV-G-pseudotyped viruses infected naive and memory CD4+ T cells at rates comparable to those of dual-tropic HIV-1 (data not shown).
FIGURE 1.
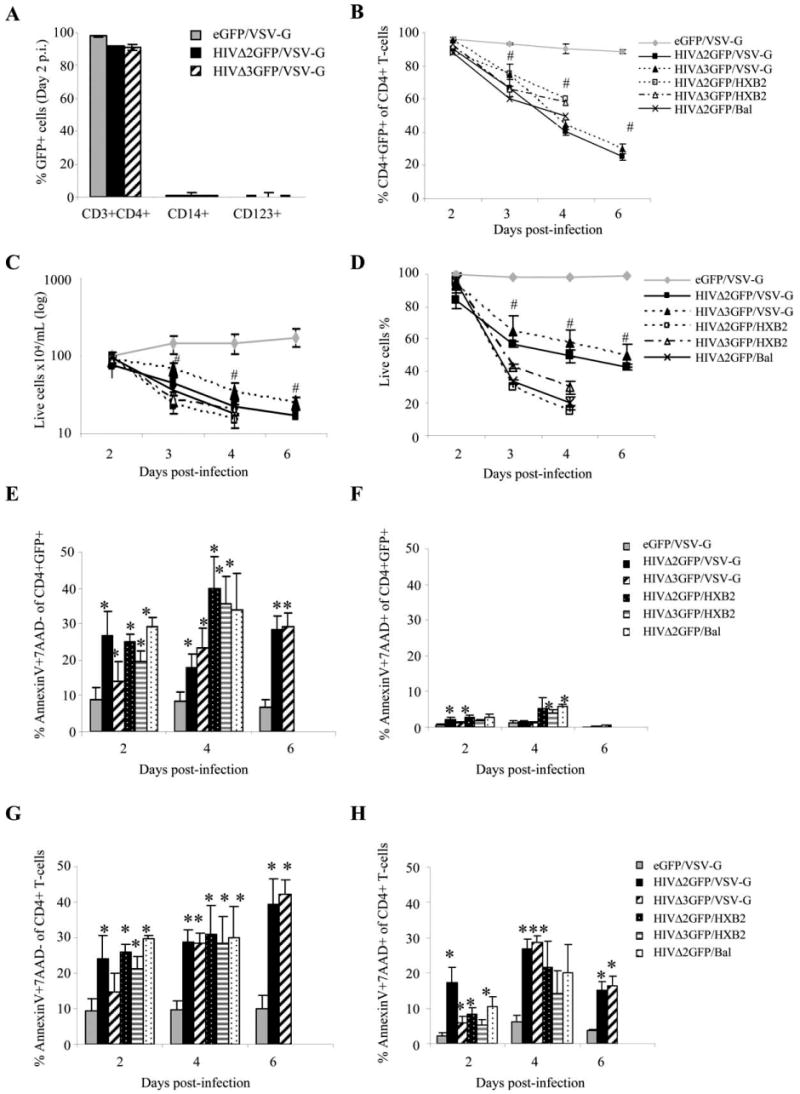
HIV-1 wild-type and mutants induce apoptosis in human primary CD4+ T cells at similar rates. A, Phenotype of sorted HIV-1-infected cells assessed by flow cytometry 2 days postinfection (day of sorting). The results are expressed as the means ± SE of three independent donors. B, Infected cell percentage measured by flow cytometric evaluation of GFP+ live cells. C, Total live cell count and (D) viability percentage of infected cultures monitored by trypan blue exclusion. E–H, Apoptosis assessment over time in GFP+CD4+ T cells (E–F) and in total CD4+ T lymphocytes (G–H). Level of early apoptosis in CD4+ T cells infected with HIVΔ2GFP or HIVΔ3GFP virus is expressed as percentage of CD4+GFP+ (E) or CD4+ (G) T cells that are annexin V and 7AAD-negative relative to mock sample (eGFP). Late apoptosis is expressed as percentage of CD4+GFP+ (F) or CD4+ (H) T cells that are positive for both annexin V and 7AAD relative to mock sample (eGFP). The data represent the means ± SE of three experiments. * and #, p < 0.05 relate to increase or decrease, respectively, relative to control (eGFP).
We measured the rate of early and late apoptosis by evaluating the number of annexin V+ cells and annexin V+7AAD+ cells and observed a significant (p < 0.05) increase in apoptosis levels in CD4+ cells infected with pseudotyped HIVs (Fig. 1E–H). The rate of early apoptosis was similar when HIVΔ2GFP and HIVΔ3GFP were compared, whether evaluated in cells gated on CD4+GFP+ (Fig. 1E) or in total CD4+ populations (Fig. 1G). At later time points the CD4+ T cell population includes cells that are GFP+ and GFP−, and the latter could be contaminating uninfected cells or infected cells in which GFP degraded as a consequence of apoptosis. Very moderate levels of late apoptosis (annexin V+7AAD+ cells) could be detected in CD4+GFP+ cells, whether infected with HIVΔ2GFP or HIVΔ3GFP (Fig. 1F), and these rates did not reflect the progressive increase of early apoptosis observed during the time course. However, late apoptosis was significantly higher when evaluated in CD4+ cells of HIV-1-infected cultures, regardless of GFP status (Fig. 1H), compared with the late apoptosis observed in CD4+ T cells of eGFP-infected cultures. These data indicate that there was a decrease in GFP expression in HIV-1-infected cells in late stages of apoptosis. Therefore, the evaluation of late apoptosis is more precise if evaluated independently of GFP as an infectivity marker. Taken together, these data indicate that HIV-induced apoptosis can occur independently of the nef, env, and vpr gene products. These three genes can be very diverse in different HIV-1 isolates, as in the case of the env gene that varies among subtypes and clades, or deleted, as in the case of nef. In the case of the vpr gene, mutations and loss of function have been detected in infected individuals and were associated with reduced pathology (35, 51). Our data indicate that the ultimate fate of most HIV-infected primary CD4+ T cells is cell death independent of the genotype of env, nef, and vpr.
CD4+ T cells infected with pseudotyped HIV-1 arrest in the G0 phase of the cell cycle
To investigate how HIV affects the progression of the cell cycle in cells infected with pseudotyped HIV-1 viruses, we examined the DNA content of infected (CD4+GFP+) cells during a 5-day time-course analysis by staining the cells with 7AAD dye followed by a flow cytometric analysis. Based on the DNA content, cells were defined as G1/G0 when the DNA amount is equal to 1, S phase when the DNA amount is between 1 and 2, and G2/M when the DNA amount is equal to 2. Fig. 2A (upper panel) shows representative cell cycle profiles of CD4+GFP+ cells 48 h after infection with different viruses. Because staining with 7AAD does not permit distinguishing between G0 and G1 phases, we also analyzed the Ki-67 Ag expression in CD3+CD4+GFP+ cells (Fig. 2A, lower panel). The Ki-67 mAb recognizes a nuclear Ag that is expressed exclusively in cells in late G1, S, and G2/M phases of the cell cycle (52). Quiescent or resting cells in the G0 phase of the cell cycle do not express the Ki-67 Ag (53). Nonactivated CD4+ T cells were used as a negative control for Ki-67 expression (first of lower panels in Fig. 2A). A similar analysis was performed in three independent donors, and the results are reported in Fig. 2B for the early time points after infection. Most uninfected CD4+ T cells (data not shown) or those infected with the eGFP virus were in the G0/G1 phase of the cell cycle (Fig. 2A–C). As expected, cells infected with pseudotyped HIVΔ2GFP virus were predominantly in the G2/M phase, indicating cell cycle arrest or delay (Fig. 2, A and B) and this increase was statistically significant over the 5-day analysis period when compared with the number of cells found in G2/M phase in the eGFP-infected cells. Cells infected by the HIVΔ2GFP virus accumulated predominantly in the G0 phase of the cell cycle at later time points (day 5, Fig. 2C) (p < 0.05), suggesting that these cells may have been delayed in G2/M but were not completely arrested. We also found that cells infected by HIVΔ3GFP showed an increased number of cells in the G2/M phase of the cell cycle, and this increase was statistically significant in early time points after infection, compared with eGFP control (Fig. 2B). However, the observed number of cells in the G2/M phase was not as high as in the case of HIVΔ2GFP virus. This phenomenon has been linked to the Vif protein, as the G2/M delay was abolished when Vif was eliminated from the Vpr− virus (54). During the 5-day cell cycle analysis, HIV-1-infected cells accumulated predominantly in the G0 phase of the cell cycle (Ki-67−), and this G0 arrest was statistically significant 5 days after infection (Fig. 2C). To further confirm this finding, we analyzed the expression of the p130 protein, a G0 marker, by Western blot (Fig. 2D) 3 days after infections. We found a 2-fold increase of both the total and the phosphorylated forms of p130 in HIVΔ2GFP-infected primary CD4+ T cells and HeLa. This increase was slightly lower in the HIVΔ3GFP-infected cells.
FIGURE 2.
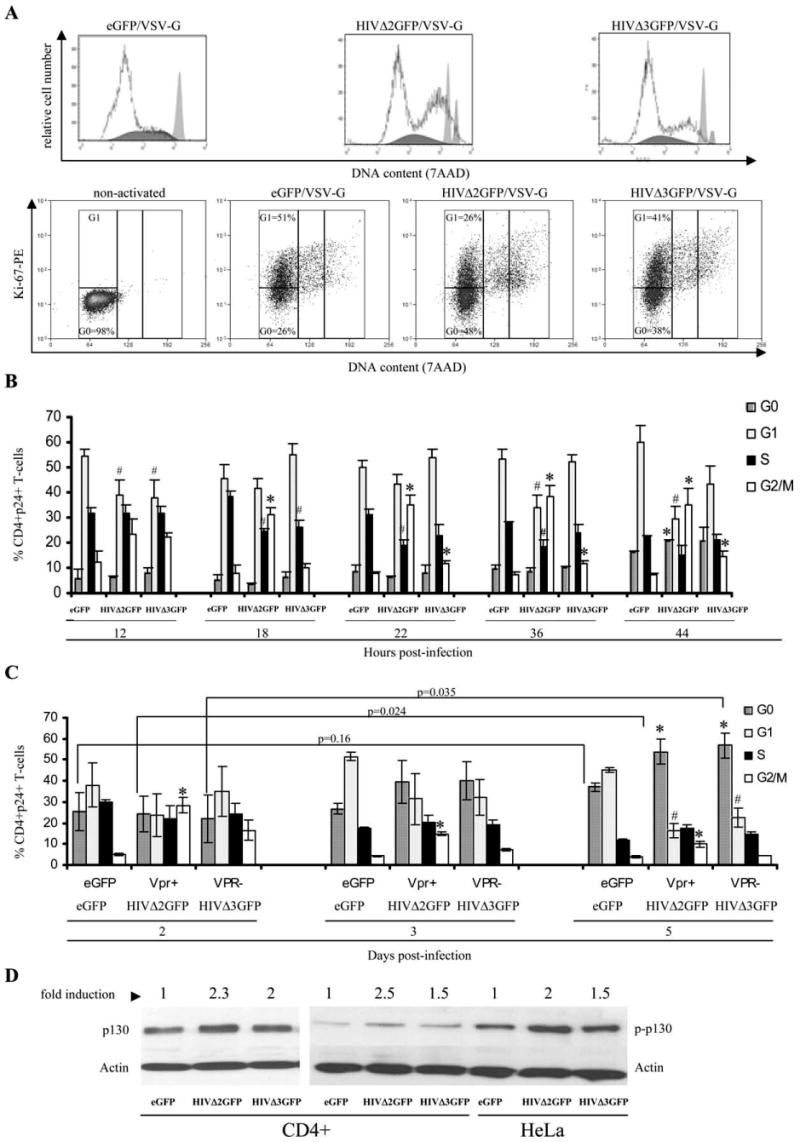
Cell cycle profiles of human CD4+ T cells infected with VSV-G-pseudotyped HIVΔ2GFP and HIVΔ3GFP (A). The results obtained with infected, sorted cells from three independent donors are reported in B and C. The results are expressed as the means ± SE of three independent experiments. *, p < 0.05 relative to eGFP control. D, Expression of p130 and phosphorylated p130 proteins measured by Western blot in infected and sorted CD4+GFP+ T cells or HeLa cells. Results are expressed as fold increase relative to eGFP control.
We were also interested in establishing the phase of the cell cycle during which HIV-1-infected cells become committed to death and whether some infected cells survive the HIV-1 infection. To address this issue we stained infected, CD4+GFP +-sorted T cells with Abs against Ki-67 as well as annexin V and 7AAD over a 10-day time course. CD3+CD4+ gated T cells were evaluated for annexin V and GFP expression and reported as GFP+ annexin V− cells (Fig. 3, A and B, left panels, live cells), GFP + annexin V+ cells (Fig. 3, A and B, middle panels, early apoptosis), and GFP− annexin V+ cells (Fig. 3, A and B, right panels, late apoptosis). In these different populations the cell cycle phase distribution was analyzed 3, 4, 6, and 10 days postinfection. We found that most GFP+ annexin V+ (early apoptosis stage) cells present in the HIVΔ2GFP-infected cells were found in the G2/M phase of the cell cycle, but GFP− annexin V+ cells of the same cell population accumulated predominantly in G0 at later time points. The bulk of HIVΔ3GFP-infected cells that were annexin V+ accumulated predominantly in the G0 phase of the cell cycle independently of GFP expression (Fig. 3B, middle and right panels). These data indicate that the apoptotic process initiates in the G2/M stage of the cell cycle but that cells do not arrest in this phase and instead move to G0, independently of Vpr expression. A more prolonged G2/M delay is Vpr-dependent, possibly linked to overexpression of proteins aimed at DNA repair, such as GADD45 (growth arrest and DNA-damage-inducible 45), that may slow down the apoptotic process. This delay may be critical to achieve the previously observed higher production of virus described for Vpr+ HIV compared to a syngeneic ΔVpr strain. We have confirmed this difference for our viruses HIVΔ2GFP and HIVΔ3GFP (Fig. 3C). Based on three independent donors there was a significantly higher amount of p24 in the supernatants of cells infected with HIVΔ2GFP compared with those infected with HIVΔ3GFP virus at earlier time points. As the cells became apoptotic over time this difference was no longer significant (p > 0.05). Interestingly, GFP+ annexin V− cells could be detected 10 days after HIV infection and they accumulated predominantly in G0 (Fig. 3, A and B, left panel). These cells may include a subpopulation of infected cells that survives the infection and has become resting. Additional studies are planned to further investigate this cell population.
FIGURE 3.
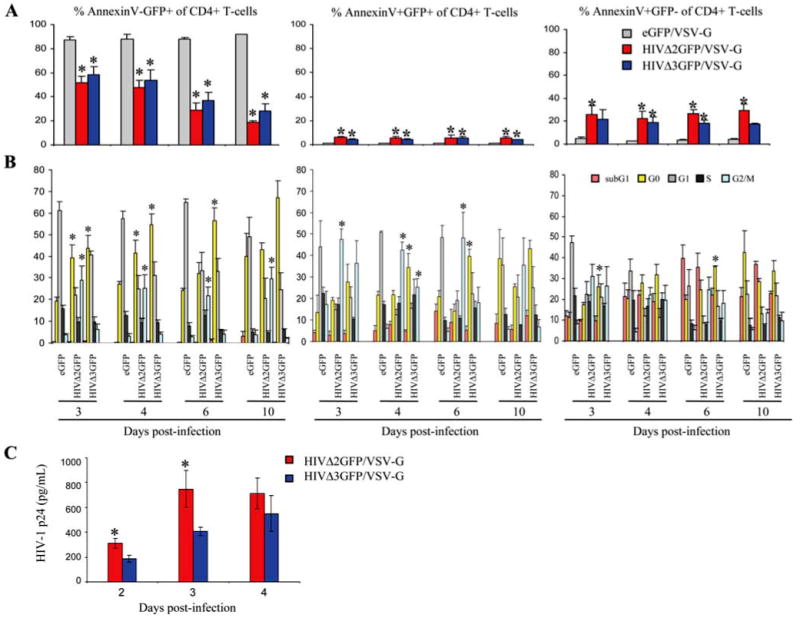
Cell cycle profiles of apoptotic human CD4+ T cells infected with VSV-G-pseudotyped HIVΔ2GFP and HIVΔ3GFP and sorted for GFP positivity. A, Percentage of annexin V−GFP+ (left panel), annexin V+GFP+ (middle panel), and annexin V+GFP− (right panel) cells. B, Flow cytometric analysis of cell cycle profiles in the cell populations reported in A determined by staining with an Ab against Ki-67 and 7AAD. C, Production of p24 in human CD4+ T cells infected with HIVΔ2GFP or HIVΔ3GFP. Production of HIV-1 p24 was measured in supernatants from infected, sorted cultures on days 2, 3, and 4 postinfection using a p24 ELISA. The results are expressed as the means ± SE of three independent donors. *, p < 0.05 related to decrease or increase.
Gene modulation by HIV-1 in human primary CD4+ T cells: activation of the Egr1-PTEN-FOXO3a pathway
To gain more insight into the molecular pathways leading to cell death in pseudotyped HIVΔ2GFP- and HIVΔ3GFP-infected cells, we conducted gene expression profiling using DNA array analysis. To identify genes whose expression was affected by both viruses, we isolated CD4+ T cells from three independent human donors and infected them with viruses pseudotyped with VSV-G envelope: HIVΔ2GFP, HIVΔ3GFP, and eGFP as a control, followed by cell sorting for CD4+GFP+ cells. The VSV-G envelope was given preference since the infection rates achieved with this Env were consistently higher than with the other envelope genes, and these experiments required sorting of a substantial number of cells to carry out additional analyses. The purity of sorting was evaluated in the sorted population by flow cytometry and was found to be between 95% and 99% (Fig. 4A). Additionally, the infection rate was evaluated by reverse transcription and real-ime PCR for HIV-1 Tat (Fig. 4B). Substantially lower Tat Ct values found in the HIV-infected cells supported significantly (p < 0.05) higher levels of Tat RNA in these cells compared with the negative eGFP cells. Ten hours after sorting, cells were harvested and total RNA was extracted and used for microarray analysis. We used an algorithm for identifying differentially expressed genes in human CD4+ T cells in response to HIV-1 infection (Fig. 4, C and D). Changes in gene expression were considered significant if expression changed at least 1.6-fold in two independent human donors compared with the expression in negative control (eGFP-infected cells). Of the 313 genes induced in HIVΔ2GFP, only 163 were also induced in HIVΔ3GFP (Fig. 4, C and D, red). Only 31 genes were found to be similarly down-regulated by two viruses (Fig. 4, C and D, green). Fig. 4D shows all the genes similarly regulated in human primary CD4+ T lymphocytes from three independent donors infected with HIVΔ2GFP and HIVΔ3GFP. The genes are annotated according to their known function. A brief name description as well as the fold expression relative to control (eGFP-expressing cells) are given for all the genes in the three independent samples in supplemental Table I.5
FIGURE 4.
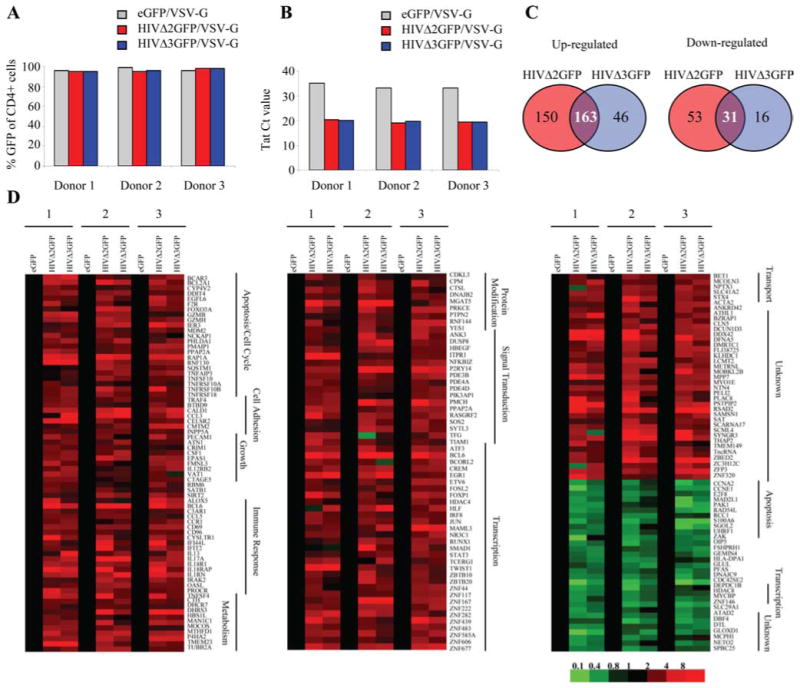
Vpr-independent gene modulation in HIV-1-infected cells. A, Percentage CD4+GFP+ T cells after infection with HIVΔ2GFP or HIVΔ3GFP and sorting using GFP as a marker. B, Detection of Tat expression by quantitative real-time RT-PCR in HIV-1-infected human CD4+ T cells. Cycle threshold (Ct) values are given. C, Venn diagrams illustrating the number of genes modulated in different culture conditions. D, Vpr-independent gene modulation in HIV-1-infected CD4+ T cells. Profiles of up-regulated (red) or down-regulated (green) genes in human CD4+ T cells infected with HIVΔ2GFP or HIVΔ3GFP. The color scale is shown below the panels.
We reasoned that genes similarly regulated by vpr+ and Δvpr HIV-1 viruses may provide an indication of the pathways involved in the apoptosis observed in the cultures infected by these viruses and reported in Fig. 1. There were a number of genes involved in cell cycle, apoptosis, and immune response/activation. One of the genes that was up-regulated in cells infected with HIVΔ2GFP and HIVΔ3GFP viruses was FOXO3a, a downstream effector of PI3K/PTEN/Akt pathway and a key player in embryogenesis, tumorigenesis, and maintenance of differentiation status (55, 56). Egr1, which can increase transcription of PTEN (57), was also found induced. Activated FOXO3a has been shown to control the expression of several proapoptotic genes, including FasL (also known as CD95 ligand), Bcl6 (B cell lymphoma-6), Bim (58–60), and TRAIL/Apo-2 ligand (55, 61). FOXO3a also induces a reversible arrest in the G0 (gap 0) phase of the cell cycle (61–63).
We used real-time RT-PCR to validate our microarray data that demonstrated FOXO3a overexpression in HIV-1-infected cells. First, we analyzed mRNA level of selected genes regulated by FOXO3a: Bcl6, Bim, FasL, GADD45a, and TRAIL (Fig. 5, A and B). Additionally, we measured mRNA expression of TRAIL death receptors DR4 and DR5 and of Fas (Fig. 5A). Real-time RT-PCR results, obtained with RNA from cells from three independent human donors, confirmed our microarray findings: genes encoding FOXO3a and its targets, as well as TRAIL death receptors, were all up-regulated in HIV-1-infected cells. The differences in expression levels between control and HIV-infected cells, observed for all the investigated genes but Fas, were statistically significant (Fig. 5A). Expression of GADD45, a gene involved in DNA repair that is also regulated by FOXO3a, did not change in cells infected with HIVΔ3GFP/VSV-G but was up-regulated in cells infected by HIVΔ2GFP, confirming previous reports that Vpr induces GADD45a (64). We also confirmed by RT-PCR that both HIVΔ2GFP and HIVΔ3GFP induce expression of several key proapoptotic molecules (Egr1, Noxa, and PUMA) and of PTEN, previously described as a key regulator of the PI3K/Akt pathway (65).
FIGURE 5.
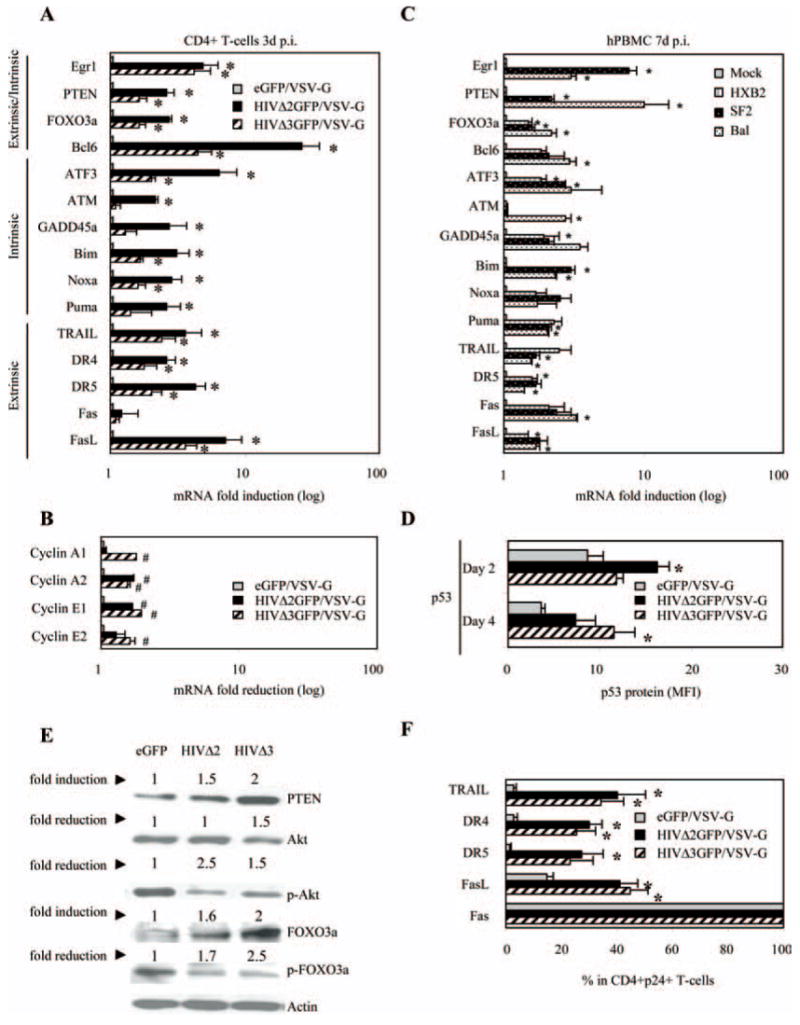
Expression analysis of selected genes modulated by HIV-1 in CD4+ T cells and in total PBMC. mRNA up-regulation of apoptosis-related genes (A) and mRNA down-regulation of selected genes involved in cell cycle regulation (B) were monitored by real-time PCR using RNAs from VSV-G-pseudotyped HIVΔ2GFP or HIVΔ3GFP infected, sorted human CD4+ T lymphocytes. Results are normalized to GAPDH and are expressed as fold induction (A) or fold reduction (B) relative to eGFP control. C, Same as A, but in this case the analysis is conducted in human PBMC infected with wild-type HIV-1 viruses with different tropism. The data represent the means ±SE of three independent experiments. *, p < 0.05 related to induction in expression and #, p < 0.05 related to reduction in expression. D, p53 protein expression was monitored on days 2 and 4 postinfection by intracellular staining. The data are obtained and quantitated with flow cytometric analysis and expressed as MFI. E, Protein accumulation levels for PTEN, Akt, phosphorylated Akt (Ser473), FOXO3a, and phosphorylated FOXO3a (Ser318) were measured on day 3 postinfection by Western blot and quantitated using Kodak image scan. Results are normalized to β-actin and are expressed as fold induction or fold reduction relative to eGFP control. F, TRAIL and TRAIL death receptors (DR4 and DR5), as well as Fas and FasL protein expression, were measured 24 h after sorting by surface or intracellular staining and analyzed by flow cytometry. Mean percentage ± SE of cells expressing each particular protein is reported for CD4+GFP+ T cells. The data represent the means ± SE of three independent experiments. *, p < 0.05 related to induction in expression and #, p < 0.05 related to reduction in expression.
Phosphorylation of FOXO3a by Akt reduces its transcriptional activity by exporting it out of the nucleus where FOXO3a undergoes degradation in favor of cell survival. High levels of PTEN antagonize Akt activity, which favors nuclear retention of FOXO3a and, consequently, activation of transcription of proapoptotic genes that are part of either the extrinsic pathway, such as FasL or TRAIL, or the intrinsic pathway, such as Bim, PUMA, and Noxa. We also observed higher mRNA levels of ATF3, a member of the ATF/CREB family of transcription factors and ATM. Differences in gene expression were observed between the two HIV-1 mutants. While high levels of ATF3 mRNA were found independent of Vpr expression, the induction of ATM was Vpr-specific (Fig. 5, A and C). High expression of ATF3, like FOXO3a, promotes apoptosis and cell cycle arrest in G1/S transition, and the impact of ATF3 on cell cycle arrest depends on reduced protein levels of various cyclins (cyclin A and cyclin E) (66). Interestingly, we a found a significant (p < 0.05) decrease in steady-state mRNA levels of cyclins A and E during infection with the two HIV mutants (Fig. 5B). When expression of the same genes reported in Fig. 5A was investigated in PBMC infected with replicating strains of HIV-1 with different tropisms, such as HXB2, SF2, and Bal, similar patterns of gene expression were detected (Fig. 5C). These results indicate that the data reported in Fig. 5A are not observed only with a pseudotyped, nonreplicating virus, and that these results remain the same when the Env, Vpr, and Nef proteins absent in the psudotyped virus are expressed by the replicating viruses.
When we quantified FOXO3a protein levels we found that there was an increase in the total accumulation in HIV-infected cells but not in the eGFP control (Fig. 5E). Furthermore, the levels of phosphorylated FOXO3a decreased, confirming an accumulation of nuclear FOXO3a that acts as a transcriptional regulator of genes involved in apoptosis. The two FOXO3a regulators, phosphorylated Akt-1 and PTEN, respectively decreased and increased (Fig. 5E), supporting their roles in the accumulation of transcriptionally active FOXO3a. Furthermore, we found an increase in the mean fluorescence intensity (MFI) of p53, a protein critical to cell survival and apoptosis, in cells infected with the HIV-1 viruses but not in cells infected with eGFP (Fig. 5D). FOXO3a has been shown to regulate p53 by increasing its half-life from 20 to 90 min but not by increasing its transcription (67). We did not find an increase in the steady-state accumulation of p53 RNA (data not shown) but the statistically significant higher MFI is consistent with an increase in the p53 half-life. Thus, our results provide evidence of cell death pathway activation and cell cycle deregulation in cells infected with both Vpr-containing and Vpr-lacking pseudotyped HIV-1 viruses, and in these cells FOXO3a may play a significant role in orchestrating apoptosis as well as cell cycle arrest.
TRAIL/TRAIL receptors and Fas/FasL expression in HIV-1 infected CD4+ T cells
The data reported in Fig. 5 indicate that FOXO3a-mediated apoptosis in HIV-1-infected primary CD4+ T cells can result, at least in part, from the up-regulation of the proapoptotic target genes TRAIL or FasL, as the expression of these genes was increased in the HIV-1-infected cultures. We sought to determine whether this increase at the RNA level corresponded to an increase in protein expression of TRAIL, TRAIL receptors, Fas, and FasL in the CD4+ T cell cultures, which may account for higher levels of apoptosis (Fig. 1E–H). Twenty-four hours after sorting, when 95–99% of the cells were still GFP+ in all cultures, we found significant (p < 0.05) increase in percentage of cells expressing TRAIL, TRAIL death receptors DR4 and DR5, and FasL in HIV-1-infected cells (Fig. 5F). There was no difference in Fas protein expression between HIV-1-infected and GFP-infected or noninfected CD4+ T lymphocytes, as 100% of the cells in these cultures were Fas+ as a consequence of the anti-CD3 activation (68). We conclude that both TRAIL and FasL may contribute to CD4+ T cell death during HIV infection. These molecules could also mediate cell death in cells that are not infected by HIV-1, if they express the appropriate receptors.
FOXO3a plays a critical role in the apoptosis of HIV-1-infected CD4+ T cells
Having observed higher protein levels for TRAIL, its death receptors DR4 and DR5, and FasL, we investigated the contribution of FOXO3a, an upstream key regulator of TRAIL, FasL, and Bim to HIV-1-induced apoptosis using siRNA-mediated depletion of FOXO3a. As primary T cells are poorly transfectable, we evaluated FOXO3a induction by HIV-1 in H9 and HeLa cells. We found that FOXO3a RNA levels were induced in HeLa cells and in PBMC after HIV-1 infection, and an increase in apoptosis could be detected as well. The same was not true for H9 cells (Fig. 6A). Furthermore, in HIV-1-infected HeLa cells we could also detect the up-regulation of some of the genes that were found up-regulated in HIV-1-infected CD4+ T cells (data not shown), further supporting the use of this cell line for additional experiments. We transfected HeLa cells with siRNA duplexes directed at FOXO3a or nonspecific siRNA and then transduced the cells with VSV-G-pseudotyped HIVΔ2GFP, HIVΔ3GFP, or eGFP alone as a control. FOXO3a RNA levels could be reduced by the FOXO3a-specific siRNA but not by the control siRNA (Fig. 6B). The same was true for some of the FOXO3a target genes that were found up-regulated after HIV-1 infection (Fig. 6B). We confirmed by Western blot the reduced expression of total FOXO3a protein after specific siRNA treatment and no change in expression of FOXO3a protein in cells treated with nonspecific siRNAs (Fig. 6C). Cyclin A and E, whose expression was reduced in infected cells, increased 2- to 5-fold in infected cells treated with FOXO3a RNAi (Fig. 6C). To evaluate HIV-1-induced apoptosis and its reduction by FOXO3a siRNA, we stained HeLa cells with annexin V and 7AAD. We found that FOXO3a-specific siRNA transfection in HIV-1-infected HeLa cells resulted in a significant (p < 0.05) decrease in apoptosis (Fig. 6D). Apoptosis was not fully abolished, most likely as a consequence of an incomplete knockdown of FOXO3a in these cells by FOXO3a-specific siRNA (Fig. 6C). These experiments support a direct role of FOXO3a in HIV-1-induced apoptosis.
FIGURE 6.
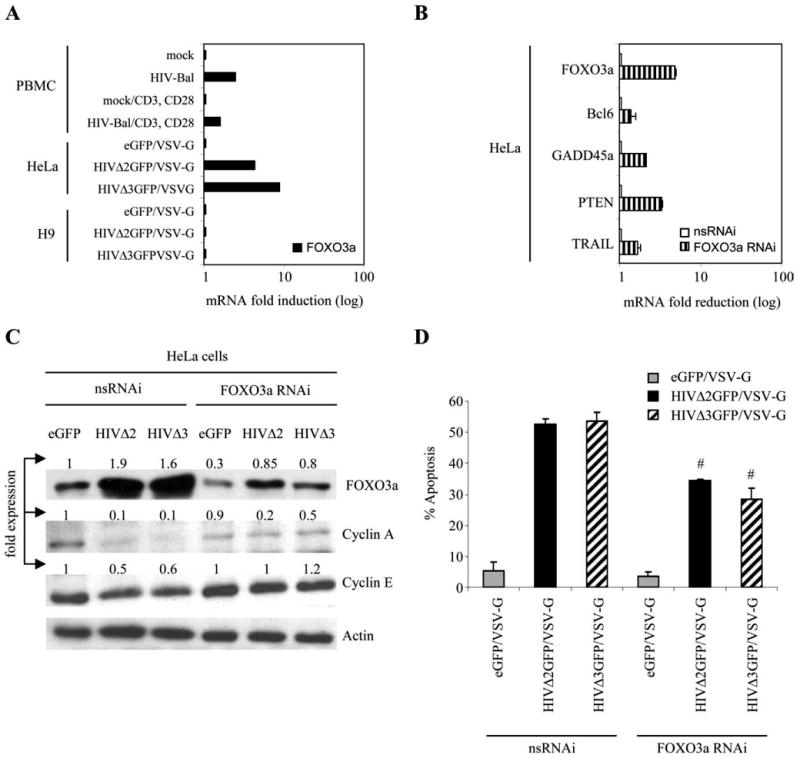
siRNA-mediated knockdown of FOXO3a reduces HIV-1-induced apoptosis. A, Up-regulation of FOXO3a mRNA in human PBMC infected with wild-type HIV-1-Bal 7 days after infection and in HeLa, but not in H9, cells infected with VSV-G-pseudotyped HIVΔ2GFP and HIVΔ3GFP (2 days postinfection) monitored by real-time PCR. Results are normalized to GAPDH and are expressed as fold induction relative to mock or eGFP control. B, mRNA knockdown of FOXO3a and selected target genes of FOXO3a (Bcl6, GADD45a, PTEN, and TRAIL) by real-time PCR in HeLa cells transfected with nonspecific (ns) siRNA or siRNA targeting FOXO3a. Results are normalized to GAPDH and are expressed as fold reduction relative to cells treated with control nonspecific siRNA. Western blot analysis of cell lysates obtained from infected and noninfected cells treated with FOXO3a RNAi and nsRNAi (C), and level of apoptosis (D) in the same cells 48 h following siRNA transduction are shown. The data are expressed as the means ± SE of three independent experiments. #, p < 0.05 with regard to reduction in apoptosis in HIV-1-infected cells treated with siRNA against FOXO3a relative to HIV-1-infected cells treated with nonspecific siRNA.
FOXO3a affects cell cycle progression by binding to the regulatory sequences within the p130 promoter region, leading to an increase in p130 mRNA and protein levels, which consequently induces the entry of cells into a quiescent state (G0 phase of the cell cycle) (62). Our finding that levels of p130 and its phosphorylated forms were increased in HIV-infected cells suggests that FOXO3a may regulate the G0 entry of HIV-1-infected cells.
Egr1 regulates the expression of PTEN and FOXO3a
In the experiments presented in Fig. 5, we showed that Egr1 and PTEN were up-regulated in HIV-infected cells. Egr1 has been reported to increase the expression of PTEN (57, 69), which in turn is known to have an impact on the phosporylation status of Akt and FOXO3a (70, 71). To experimentally investigate the link between Egr1 expression and FOXO3a, we electroporated CD4+ T cells with siRNA against Egr1 and FOXO3a, and then infected them with HIVΔ2GFP/HXB2 (Fig. 7). This treatment reduced the expression of Egr1 to ∼60% of its original level (Fig. 7, A and B), reducing also the amount expressed during HIV infection, which is higher than in noninfected cells (Fig. 7, A and B). Reduction of Egr1 expression resulted in reduced PTEN and FOXO3a expression and in an increase of p-Akt. Treatment with Foxo3a siRNA also reduced the expression of p130, a protein whose expression can be affected by FOXO3a and is expressed in cells in G0 (Fig. 7C), providing further evidence that FOXO3a may play a role in G0 cell cycle entry. Treatment with Egr1 or FOXO3a siRNA reduced apoptosis in HIV-infected primary CD4+ T cells. The reduction was statistically significant, even if only partial. This partial result was not surprising, considering that the siRNA transfection did not result in a complete knockdown of Egr1 or FOXO3a. A similar reduction of apoptosis was achieved after treating the cells with Abs blocking FAS/FASL and TRAIL/DR5-DR4, confirming the involvement of these molecules in HIV-mediated apoptosis (Fig. 7D). The combination of FOXO3a siRNA plus Abs resulted in an even more marked reduction of apoptosis (Fig. 7D, last column), indicating that the partial efficacy of one approach can be compensated by multiple interference in critical players of the apoptotic pathways.
FIGURE 7.
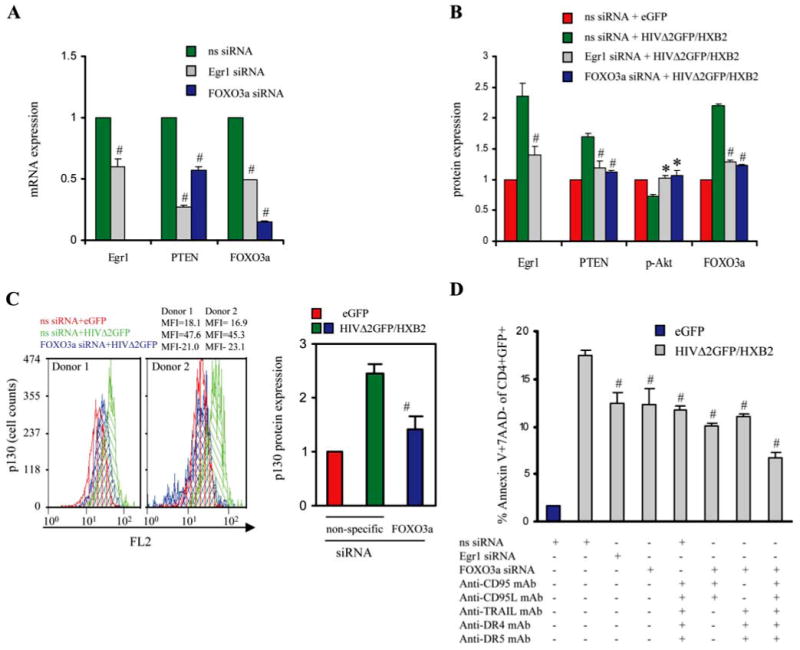
Effects of siRNA-mediated knockdown of FOXO3a and blocking of extrinsic apoptotic pathways in HIV-infected human CD4+ T lymphocytes. A, mRNA expression of Egr1, FOXO3a, and PTEN by real-time PCR in CD4+ T cells electroporated with nonspecific (ns) siRNA or siRNA targeting Egr1 or FOXO3a. Results are normalized to GAPDH and are expressed as fold reduction relative to cells treated with control ns siRNA. B, Protein expression analysis of Egr1, PTEN, p-Akt, and FOXO3a by immunofluorescent staining in CD4+ T cells treated with siRNA targeting Egr1 or FOXO3a and infected with eGFP or HIV-1 pseudotyped with HXB2 envelope on day 2 postinfection. Results are expressed as fold MFI relative to control (eGFP-infected cells treated with nonspecific siRNA). # and *, p < 0.05 decrease or increase, respectively, in mRNA or protein level in cells treated with siRNA against Egr1 or FOXO3a relative to cells treated with nonspecific siRNA. MFI values found in cells of the two donors were used to derive the results reported in B were the following: Egr1 MFI (ns siRNA/eGFP: 10.1, 9.21; ns siRNA/HIVΔ2GFP: 27.5, 22.1; Egr1 siRNA/HIVΔ2GFP: 12.3, 15.2); FOXO3a MFI (ns siRNA/eGFP: 15.5, 15.7; ns siRNA/HIVΔ2GFP: 35.2, 32.1; FOXO3a siRNA/HIVΔ2GFP: 17.5, 19.2; Egr1 siRNA/HIVΔ2GFP: 13.5, 17.9); p-Akt MFI (ns siRNA/eGFP: 57.2, 45.0; ns siRNA/HIVΔ2GFP: 40.4, 34.1; Egr1 siRNA/HIVΔ2GFP: 56.3, 48.0; FOXO3a siRNA/HIVΔ2GFP: 65.1, 45.2); PTEN MFI (ns siRNA/eGFP: 28.6, 29.1; ns siRNA/HIVΔ2GFP: 52.0, 47.5; Egr1 siRNA/HIVΔ2GFP: 31.0, 35.2; FOXO3a siRNA/HIVΔ2GFP: 29.0, 31.7). C, p130 expression by immunofluorescent staining in CD4+ T cells treated with siRNA targeting FOXO3a and infected with eGFP or HIV-1 pseudotyped with HXB2 envelope. The first two panels show the flow cytometric analysis of p130 expression; the third panel reports the results as fold MFI relative to control (eGFP-infected cells treated with nonspecific siRNA). D, Level of apoptosis 48 h after HIV-1 infection in CD4+ T cells transduced with siRNA targeting Egr1 and FOXO3a and/or treated with mAbs blocking extrinsic apoptotic pathway. The data are expressed as the means ± SE of two independent experiments. #, p < 0.05 related to reduction in apoptosis in HIV-1-infected cells treated with siRNA against Egr1 or FOXO3a relative to HIV-1-infected cells treated with nonspecific siRNA.
ATF3 and ATM regulate cyclin A and cyclin E levels in HIV-1-infected CD4+ T lymphocytes
As we observed higher mRNA levels of ATF3 and ATM and it is known that their effect on cell cycle arrest can be mediated by reduced levels of cyclin A and cyclin E (66), which were found significantly decreased during HIV infection, we investigated the effect of ATF3 and ATM on cyclin expression levels using siRNA targeting ATF3 and ATM in primary CD4+ T cells infected with HIVΔ2GFP/HXB2 2 days after infection. Protein levels of ATF3, ATM, and cyclins were evaluated by flow cytometry. The transfection of ATF3 or ATM siRNAs achieved a significant, even if incomplete, reduction of the corresponding RNA levels (Fig. 8A), and this resulted in a reduction of the ATF3 and ATM proteins when the treatment was conducted during HIV infection, despite the fact that HIV does increase the expression of both factors (Fig. 8B). When cyclin A and cyclin E were measured during these experiments we found that their levels increased if ATF3 or ATM siRNAs were present during the HIV infection (Fig. 8C), supporting a role for ATF3 and ATM, besides FOXO3a (Fig. 5B), in the regulation of these cyclins during infection.
FIGURE 8.
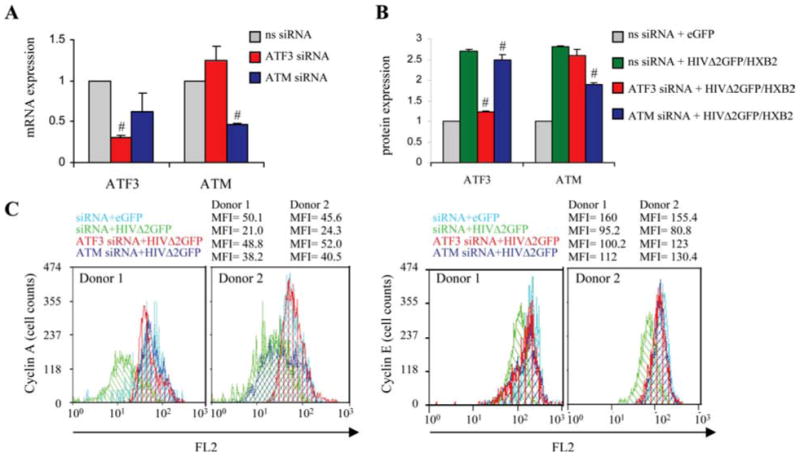
ATF3 and ATM regulate expression of cyclin A and cyclin E in HIV-1-infected human primary CD4+ T lymphocytes. A, mRNA knockdown of ATF3 and ATM by real-time PCR in primary CD4+ T cells electroporated with nonspecific (ns) siRNA or siRNA targeting ATF3 or ATM. Results are normalized to GAPDH and are expressed as fold reduction relative to cells treated with control ns siRNA. B, Protein levels of ATF3 and ATM by flow cytometry in CD4+ T cells treated with siRNA targeting ATF3 or ATM and infected with eGFP or HIVΔ2GFP/HXB2 on day 2 postinfection. #, p < 0.05 related to reduction in mRNA or protein level in cells treated with siRNA against ATF3 or ATM relative to cells treated with ns siRNA. MFI values found in cells of the two donors and used to derive the results reported in B were the following: ATF3 MFI (ns siRNA/eGFP: 12.7, 15.2; ns siRNA/HIVΔ2GFP: 39.0, 33.1; ATF3 siRNA/HIVΔ2GFP: 19.5, 14.9; ATM siRNA/HIVΔ2GFP: 38.3, 32.5); ATM MFI (ns siRNA/eGFP: 4.3, 5.8; ns siRNA/HIVΔ2GFP: 15.0, 13.2; ATM siRNA/HIVΔ2GFP: 8.6, 9.9; ATF3 siRNA/HIVΔ2GFP: 11.7, 14.2). C, Flow cytometric analysis of cyclin A and cyclin E protein levels 2 days after eGFP or HIVΔ2GFP/HXB2 infection in CD4+ T cells from two independent donors transduced with ns siRNA or siRNA targeting ATF3 and ATM.
Tat-mediated activation of Egr1, PTEN, and FOXO3a
We investigated whether Tat could be the protein critical to the induction of some of the proapoptotic genes observed in cells infected with HIVΔ2GFP, HIVΔ3GFP, and wild-type viruses. We conducted real time RT-PCR using RNA obtained from Jurkat cells infected with Adeno-Tat (Fig. 9) and from primary CD4+ T cells from three different donors infected with the retroviral vector Tat-GFP (not shown). We found that genes up-regulated in primary CD4+ T cells infected with HIVΔ2GFP and HIVΔ3GFP were similarly modulated in Jurkat cells expressing Tat (Fig. 9, A and B). Among the genes critical to apoptosis that were induced by Tat expression were Egr1, PTEN, FOXO3a, and genes that are part of both the intrinsic and extrinsic apoptotic pathways. When FOXO3a protein levels were investigated by Western blot, we observed an increased accumulation of FOXO3a and a reduction of phosphorylated FOXO3a (Fig. 9C). Markers of early and late apoptosis could be detected in Jurkat cells on day 1 after Tat trasduction (Fig. 9D). These data support a role of Tat in the activation of FOXO3a and in the induction of its target genes involved in apoptosis.
FIGURE 9.
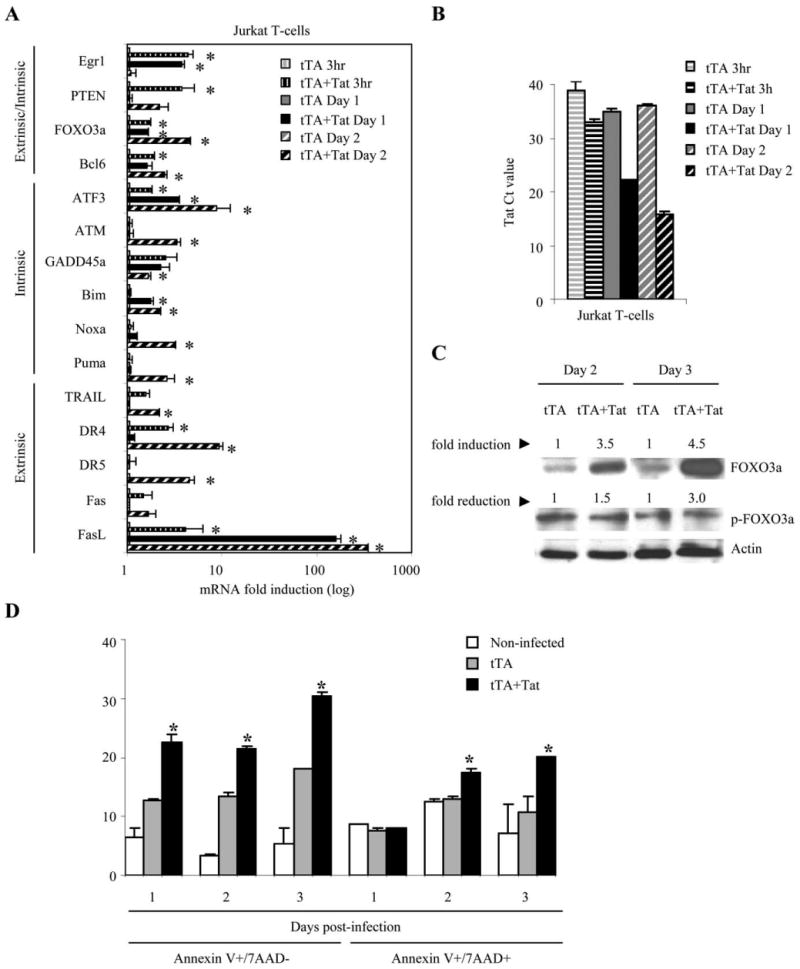
HIV-1 Tat-mediated up-regulation of Egr1-PTEN-FOXO3a and its target genes. A, Jurkat T cells were infected with adenoviruses expressing the tTA or tTA plus the SF2 Tat protein. mRNA levels of FOXO3a and its target genes were monitored by real-time PCR. Results are normalized to GAPDH and are expressed as fold induction relative to control (tTA only). B, Detection of Tat expression by quantitative real-time RT-PCR in Tat-infected Jurkat T cells. Ct values are given. The data represent the means ± SE of two independent experiments. *, p < 0.05 related to induction in expression. C, Protein levels of FOXO3a and phosphorylated FOXO3a (Ser318) by Western blot and quantitated by Kodak image scan. Results are normalized to β-actin and are expressed as fold induction or fold reduction relative to the tTA control. D, Apoptosis in Tat-expressing Jurkat T cells. Levels of early and late apoptosis are expressed as percentage of Jurkat T cells that stain for annexin V only (7AAD–) or annexin V and 7AAD, respectively. The data represent the means ± SE of two experiments. *, p < 0.05 related to increase relative to control (tTA).
In summary, our data indicate that HIV infection of CD4+ T cells leads to the induction of FOXO3a, which modulates genes that are critical to the G0 cell cycle entry observed in the infected cells and are active players in both the extrinsic and the intrinsic pathways of apoptosis.
Discussion
CD4+ T cell depletion over time is the key difference between primate lentiviral infection in species that progress to AIDS and in those who do not. An increase in immunoactivation has been observed in AIDS-susceptible species and has become the focus of how HIV and SIV can progressively deteriorate the immune system in humans and macaques. The molecular pathways engaged in the activated cells that ultimately trigger apoptosis are not fully understood. Furthermore, as some of the HIV and SIV genes are not critical for viral replication and infectivity and these viruses mutate at a high rate, the contribution to disease progression of viral variants that arise in vivo and carry genes with suboptimal function has been called into question. In this paper we have evaluated the impact on cell survival by viruses that carry deletion of Nef, Env, and Vpr, and we investigated how the host cell gene expression is affected by such viruses. Our findings indicated that a virus in which these genes are deleted can still activate different cell death programs in primary CD4+ T cells. Death factors such as FasL and TRAIL produced by the infected cells could affect the survival of noninfected cells when these cells express the appropriate receptors. Fas expression is present in activated T cells with diverse Ag specificities, and these cells could be susceptible to FasL-mediated apoptosis. These findings suggest that even variants with mutated Env, Nef, and Vpr could contribute to apoptosis of infected and noninfected cells as long as they are competent to enter the cell.
Our experiments also showed that the cellular genes that were modulated by pseudotyped viruses were also modulated in PBMC cultures infected with wild-type viral strains with different tropisms. These data are important because they support the role for these cellular genes in HIV-induced cell death after wild-type infection. Furthermore, they exclude the possibility that the presence of the proteins that were deleted in our system countermodulates these genes. Investigation of whether the Tat protein could alone recapitulate our data obtained with mutated and wild-type viruses indicated that indeed Tat can activate the Egr1-PTEN-FOXO3a pathway independent of other HIV-1 proteins.
The microarray and follow-up analyses suggested that genes whose products are involved in DNA repair and in both the extrinsic and the intrinsic apoptotic pathways become activated by HIV-1. The initial signals in this host-pathogen interaction could include DNA damage derived from retroviral integration and/or exposure to inflammatory cytokines. Retroviral integration has been known to lead to double strand break in the host cell DNA, the ends of which are held together by single strand links to viral DNA (72). This damage triggers a very sensitive DNA repair response that is mediated by factors like ATF3 and ATM (73, 74). The DNA repair response seems effective after the infection with the murine retroviral vector expressing GFP or a HIV-based lentiviral vector that only expresses GFP but seems ineffective or too slow after infection with the mutated HIV viruses or the Tat-expressing retroviral vector, suggesting a the role played by the HIV proteins in modulating the DNA repair response. ATF3 and ATM function by inducing cell cycle checkpoints that should permit DNA repair. Apoptosis occurs when the repair is not successful. ATF3 up-regulation has been shown to result in reduced expression of both cyclin A and cyclin E (66, 75), and these events facilitate exit from the cell cycle (66). These two cyclins were down-regulated in HIV-infected T cells, and this event may be important in the G0 entry observed in these populations. Furthermore, their down-regulation could be at least partially reversed by siRNAs targeting either ATF3 or ATM, suggesting that these cyclins may indeed play a role in the cell cycle arrest observed during infection. The same players involved in DNA repair can also trigger apoptosis if the process is too slow. Both ATF3 and ATM increase the stability of p53, which in turn can activate proapoptotic Bcl-2 proteins such as PUMA and Noxa, which we found overexpressed in HIV-1-infected cells, whether Vpr+ or Vpr−.
The expression of a critical regulator of p53, MDM2, was also increased in HIV-infected cells. When p53 stability increases and becomes phosphorylated by ATM or ATF3, p53 increases the transcription of MDM2, but it cannot bind MDM2. When the kinases become inactive after DNA repair, p53 is quickly dephosphorylated and destroyed via ubiquitination by MDM2. Active p53 can transactivate genes such as PTEN, which we found up-regulated in the cultures infected with the two HIV-1 mutants. PTEN, in turn, reduces the phosporylation of Akt1, also observed in our experiments. PTEN is also transcriptionally regulated by Egr1, and this gene was expressed at higher levels in virus-infected or Tat-expressing cells (57, 69). Increased levels of PTEN lead to reduced level of Akt1, which results in reduced phosphorylation of FOXO3a. Unphosphorylated FOXO3a translocates to the nucleus and becomes transcriptionally active (76). The induction of FOXO3a targets such as Bim, TRAIL, and FasL results in the activation of the apoptotic intrinsic (via Bim) and extrinsic pathways (55, 58, 77). The cascade of events was mirrored by the cell populations infected with the two mutant viruses and is illustrated in Fig. 10. Activation of FasL by FOXO3a has been shown to be mediated by c-Jun; the RNA level of this transcriptional regulator was also increased in HIV-infected cells (78).
FIGURE 10.
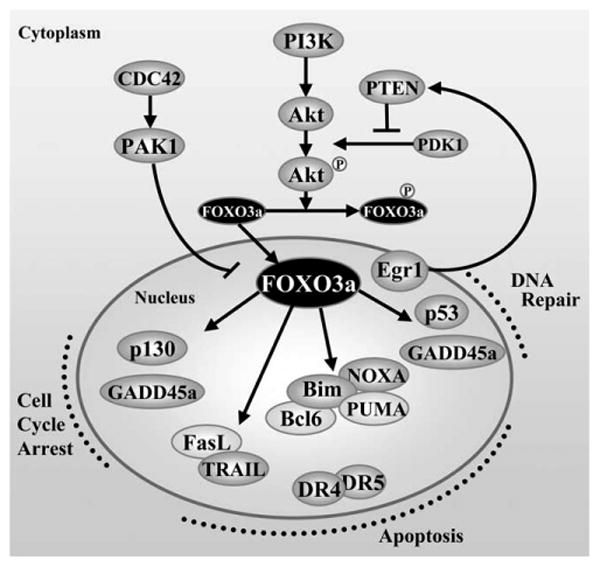
Schematic representation of the regulation of FOXO3a transcription factor and its target genes.
The Egr1-PTEN-FOXO3a induction could be observed in cells expressing only the Tat protein, suggesting that Tat may be the key player in the activation of this pathway. Tat interacts with and is acetylated by p300/CBP. The p300/CBP complex has been described as a transcriptional coactivator of Egr1 (79–82). Tat may enhance the transcriptional activity of p300/CBP by increasing the HAT activity on the Egr1 promoter, as it has been shown for histone H4 and the HIV LTR (83). Inhibition of SIRT1 deacetylase activity by Tat, which was recently reported (84), could also result in increased transcription of Egr1. Interestingly, as investigators have reported that Tat can be found in patients' serum (85) and that this protein can enter cells moving across the cell membrane (27), Tat could play a role in the apoptosis of uninfected cells by activating the Egr1-PTEN-FOXO3a after entering the cells. A recent report indicates that FOXO3a is linked to apoptosis in HIV-infected macrophages (86). A subsequent article reported that the survival of memory CD4+ T cells correlates with the phosphorylated levels of FOXO3a and that the levels of this protein are reduced in HIV-infected individuals and are higher in elite controllers, who control viral replication to undetectable viremia in the absence of therapy (87). Our findings suggest that activation of the Egr1-PTEN-FOXO3a pathway via the Tat protein could be the mechanism by which apoptosis is triggered in HIV-infected and noninfected cells and explain the significant decline of the CD4+ T cell population in HIV-1-infected individuals.
FOXO3a has been shown to be critical to promote DNA repair at the G2 to M checkpoint via the GADD45a protein (88). This protein was up-regulated in cells infected with the Vpr+ virus but not in those infected with the virus without Vpr, suggesting that Vpr may play a role in DNA repair. The induction of GADD45a, observed by others during a Vpr+ HIV infection (64), may explain the longer and more significant G2/M delay observed in cells infected with the Vpr+ virus. However, even Vpr+ cells progress to apoptosis, suggesting that DNA repair does not occur successfully and/or other signals besides DNA repair may be triggered with an overall effect in favor of apoptosis. Cytokines produced intracellularly or present in the environment may be a contributing factor in the progression to apoptosis. Furthermore, another target of FOXO3a, Bcl6, whose expression was increased during HIV infection, could play a role in G2/M delay and apoptosis (59). This effect is usually mediated by a decrease in cyclin D2, which was not observed in our cultures. This may depend on the timing of our analysis, or cyclin D2 down-regulation may be counteracted by other events occurring in the cell. The induction of p130, observed in our cells, can also be mediated by FOXO3a and is critical for the onset of G0 phase of the cell cycle, which was observed in our cultures.
It appears that HIV-1 infection leads to activation of the DNA damage repair pathways and that, possibly because of inefficient repair or exposure to inflammatory cytokines, a large fraction of infected cells undergoes apoptosis. However, a subset of HIV-infected cells remained annexin V− 10 days after infection in our cultures. It would be interesting to know if these are the cells in which DNA repair was completed successfully, leading to the escape from apoptosis, or if this subpopulation is less apt to produce cytokines. Vpr seems to activate additional genes involved in DNA repair, and perhaps this activity is critical to delay apoptosis and favor an increased virus production. As a Vpr− mutant could still induce apoptosis in infected cells, the reduced pathogenesis associated with Vpr-mutated HIV may be more dependent on reduced virus production rather than reduced apoptosis.
The activation of extrinsic apoptotic pathway via FasL and TRAIL may have implications for cell death occurring in noninfected cells. In previous studies we found that production of TRAIL from HIV or SIV immature dendritic cells and macrophages is restricted to cells from AIDS-susceptible species (36). It will be important to evaluate if production of TRAIL and FasL occurs only in CD4+ T cells from AIDS-susceptible and not in nonhuman primates that are natural hosts of primate lentiviruses.
FOXO3a activation, which can be achieved via expression of Tat alone, appears to be at the convergence of pathways involved in both DNA repair and apoptosis. It may be important to investigate what factors or events tip the balance in one of the two directions during HIV infection of CD4+ T cells, and whether FOXO3a levels of uninfected memory CD4+ T cells can be modulated in vivo by the Tat protein present in the patients' serum.
Supplementary Material
Acknowledgments
We thank Sami Kukkonen and Mariana Manrique for critical reading of the manuscript.
Footnotes
This project was supported by National Institutes of Health Grant RO1 AI060398 (to A.A.).
Abbreviations used in this paper: FasL, Fas ligand; ATF3, human activating transcription factor 3; ATM, mutated in ataxia-telangiectasia; Bcl6, B cell lymphoma-6; eGFP, enhanced GFP; Egr1, early growth response protein-1; FOXO3a, Forkhead box transcription factor O class 3a; GADD45, growth arrest and DNA-damage-inducible; HSA, heat-stable Ag; MFI, mean fluorescence intensity; MOI, multiplicity of infection; ns, nonspecific; PTEN, phosphate and tensin homolog deleted on chromosome 10; rh, recombinant human; 7AAD, 7-aminoactinomycin D; siRNA, short interfering RNA; tTA, tet transactivator; VSV-G, vesicular stomatitis virus-G protein; PUMA, p53 up-regulated modulator of apoptosis.
The accession number for the DNA microarray data in GEO (Gene Expression Omnibus) is GSE12963.
The online version of this article contains supplemental material.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Lackner AA, Veazey RS. Current concepts in AIDS pathogenesis: insights from the SIV/macaque model. Annu Rev Med. 2007;58:461–476. doi: 10.1146/annurev.med.58.082405.094316. [DOI] [PubMed] [Google Scholar]
- 2.Weiss RA. HIV and AIDS: looking ahead. Nat Med. 2003;9:887–891. doi: 10.1038/nm0703-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cotton MF, Ikle DN, Rapaport EL, Marschner S, Tseng PO, Kurrle R, Finkel TH. Apoptosis of CD4+ and CD8+ T cells isolated immediately ex vivo correlates with disease severity in human immunodeficiency virus type 1 infection. Pediatr Res. 1997;42:656–664. doi: 10.1203/00006450-199711000-00018. [DOI] [PubMed] [Google Scholar]
- 4.Gougeon ML. Apoptosis as an HIV strategy to escape immune attack. Nat Rev Immunol. 2003;3:392–404. doi: 10.1038/nri1087. [DOI] [PubMed] [Google Scholar]
- 5.Li CJ, Friedman DJ, Wang C, Metelev V, Pardee AB. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science. 1995;268:429–431. doi: 10.1126/science.7716549. [DOI] [PubMed] [Google Scholar]
- 6.Ross TM. Using death to one's advantage: HIV modulation of apoptosis. Leukemia. 2001;15:332–341. doi: 10.1038/sj.leu.2402028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunham RM, Cervasi B, Brenchley JM, Albrecht H, Weintrob A, Sumpter B, Engram J, Gordon S, Klatt NR, Frank I, et al. CD127 and CD25 expression defines CD4+ T cell subsets that are differentially depleted during HIV infection. J Immunol. 2008;180:5582–5592. doi: 10.4049/jimmunol.180.8.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silvestri G, Sodora DL, Koup RA, Paiardini M, O'Neil SP, McClure HM, Staprans SI, Feinberg MB. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–452. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]
- 9.Sumpter B, Dunham R, Gordon S, Engram J, Hennessy M, Kinter A, Paiardini M, Cervasi B, Klatt N, McClure H, et al. Correlates of preserved CD4+ T cell homeostasis during natural, nonpathogenic simian immunodeficiency virus infection of sooty mangabeys: implications for AIDS pathogenesis. J Immunol. 2007;178:1680–1691. doi: 10.4049/jimmunol.178.3.1680. [DOI] [PubMed] [Google Scholar]
- 10.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 11.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 12.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 13.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 14.Adrain C, Martin SJ. The mitochondrial apoptosome: a killer unleashed by the cytochrome seas. Trends Biochem Sci. 2001;26:390–397. doi: 10.1016/s0968-0004(01)01844-8. [DOI] [PubMed] [Google Scholar]
- 15.Huang DC, Strasser A. BH3-only proteins: essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- 16.Vander Heiden MG, Thompson CB. Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nature Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- 17.Selliah N, Finkel TH. Biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001;8:127–136. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- 18.Biard-Piechaczyk M, Robert-Hebmann V, Richard V, Roland J, Hipskind RA, Devaux C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120) Virology. 2000;268:329–344. doi: 10.1006/viro.1999.0151. [DOI] [PubMed] [Google Scholar]
- 19.Cicala C, Arthos J, Rubbert A, Selig S, Wildt K, Cohen OJ, Fauci AS. HIV-1 envelope induces activation of caspase-3 and cleavage of focal adhesion kinase in primary human CD4+ T cells. Proc Natl Acad Sci USA. 2000;97:1178–1183. doi: 10.1073/pnas.97.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinkelein M, Sopper S, Jassoy C. Contact of human immunodeficiency virus type 1-infected and uninfected CD4+ T lymphocytes is highly cytolytic for both cells. J Virol. 1995;69:6925–6931. doi: 10.1128/jvi.69.11.6925-6931.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masci AM, Paz FL, Borriello A, Cassano S, Della Pietra V, Stoiber H, Matarese G, Della Ragione F, Zappacosta S, Racioppi L. Effects of human immunodeficiency virus type 1 on CD4 lymphocyte subset activation. Eur J Immunol. 1999;29:1879–1889. doi: 10.1002/(SICI)1521-4141(199906)29:06<1879::AID-IMMU1879>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Nardelli B, Gonzalez CJ, Schechter M, Valentine FT. CD4+ blood lymphocytes are rapidly killed in vitro by contact with autologous human immunodeficiency virus-infected cells. Proc Natl Acad Sci USA. 1995;92:7312–7316. doi: 10.1073/pnas.92.16.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Este JA, Cabrera C, Blanco J, Gutierrez A, Bridger G, Henson G, Clotet B, Schols D, De Clercq E. Shift of clinical human immunodeficiency virus type 1 isolates from X4 to R5 and prevention of emergence of the syncytium-inducing phenotype by blockade of CXCR4. J Virol. 1999;73:5577–5585. doi: 10.1128/jvi.73.7.5577-5585.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez G, Llano A, Esgleas M, Clotet B, Este JA, Martinez MA. Purifying selection of CCR5-tropic human immunodeficiency virus type 1 variants in AIDS subjects that have developed syncytium-inducing, CXCR4-tropic viruses. J Gen Virol. 2006;87:1285–1294. doi: 10.1099/vir.0.81722-0. [DOI] [PubMed] [Google Scholar]
- 25.Locher CP, Witt SA, Kassel R, Dowell NL, Fujimura S, Levy JA. Differential effects of R5 and X4 human immunodeficiency virus type 1 infection on CD4+ cell proliferation and activation. J Gen Virol. 2005;86:1171–1179. doi: 10.1099/vir.0.80674-0. [DOI] [PubMed] [Google Scholar]
- 26.New DR, Ma M, Epstein LG, Nath A, Gelbard HA. Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J Neurovirol. 1997;3:168–173. doi: 10.3109/13550289709015806. [DOI] [PubMed] [Google Scholar]
- 27.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 28.Li CJ, Wang C, Friedman DJ, Pardee AB. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:5461–5464. doi: 10.1073/pnas.92.12.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swingler S, Mann A, Jacque J, Brichacek B, Sasseville VG, Williams K, Lackner AA, Janoff EN, Wang R, Fisher D, Stevenson M. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med. 1999;5:997–1003. doi: 10.1038/12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swingler S, Brichacek B, Jacque JM, Ulich C, Zhou J, Stevenson M. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature. 2003;424:213–219. doi: 10.1038/nature01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmons A, Aluvihare V, McMichael A. Nef triggers a transcriptional program in T cells imitating single-signal T cell activation and inducing HIV virulence mediators. Immunity. 2001;14:763–777. doi: 10.1016/s1074-7613(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 32.de Noronha CM, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, Greene WC. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science. 2001;294:1105–1108. doi: 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- 33.Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Somasundaran M, Sharkey M, Brichacek B, Luzuriaga K, Emerman M, Sullivan JL, Stevenson M. Evidence for a cytopathogenicity determinant in HIV-1 Vpr. Proc Natl Acad Sci USA. 2002;99:9503–9508. doi: 10.1073/pnas.142313699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lum JJ, Cohen OJ, Nie Z, Weaver JG, Gomez TS, Yao XJ, Lynch D, Pilon AA, Hawley N, Kim JE, et al. Vpr R77Q is associated with long-term nonprogressive HIV infection and impaired induction of apoptosis. J Clin Invest. 2003;111:1547–1554. doi: 10.1172/JCI16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim N, Dabrowska A, Jenner RG, Aldovini A. Human and simian immunodeficiency virus-mediated upregulation of the apoptotic factor TRAIL occurs in antigen-presenting cells from AIDS-susceptible but not from AIDS-resistant species. J Virol. 2007;81:7584–7597. doi: 10.1128/JVI.02616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izmailova E, B FMN, Huang Q, Makori N, Miller CJ, Young RA, Aldovini A. HIV Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat Med. 2003;9:191–197. doi: 10.1038/nm822. [DOI] [PubMed] [Google Scholar]
- 38.Viscidi RP, Mayur K, Lederman HM, Frankel AD. Inhibition of antigen-induced lymphocyte proliferation by Tat protein from HIV-1. Science. 1989;246:1606–1608. doi: 10.1126/science.2556795. [DOI] [PubMed] [Google Scholar]
- 39.Zagury D, Lachgar A, Chams V, Fall LS, Bernard J, Zagury JF, Bizzini B, Gringeri A, Santagostino E, Rappaport J, et al. Interferon α and Tat involvement in the immunosuppression of uninfected T cells and C-C chemokine decline in AIDS. Proc Natl Acad Sci USA. 1998;95:3851–3856. doi: 10.1073/pnas.95.7.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badley AD, Dockrell DH, Algeciras A, Ziesmer S, Landay A, Lederman MM, Connick E, Kessler H, Kuritzkes D, Lynch DH, et al. In vivo analysis of Fas/FasL interactions in HIV-infected patients. J Clin Invest. 1998;102:79–87. doi: 10.1172/JCI2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katsikis PD, Garcia-Ojeda ME, Torres-Roca JF, Tijoe IM, Smith CA, Herzenberg LA. Interleukin-1β converting enzyme-like protease involvement in Fas-induced and activation-induced peripheral blood T cell apoptosis in HIV infection: TNF-related apoptosis-inducing ligand can mediate activation-induced T cell death in HIV infection. J Exp Med. 1997;186:1365–1372. doi: 10.1084/jem.186.8.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeremias I, Herr I, Boehler T, Debatin KM. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur J Immunol. 1998;28:143–152. doi: 10.1002/(SICI)1521-4141(199801)28:01<143::AID-IMMU143>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 43.Miura Y, Misawa N, Maeda N, Inagaki Y, Tanaka Y, Ito M, Kayagaki N, Yamamoto N, Yagita H, Mizusawa H, Koyanagi Y. Critical contribution of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to apoptosis of human CD4+ T cells in HIV-1-infected hu-PBL-NOD-SCID mice. J Exp Med. 2001;193:651–660. doi: 10.1084/jem.193.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–1475. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 45.Herbeuval JP, Boasso A, Grivel JC, Hardy AW, Anderson SA, Dolan MJ, Chougnet C, Lifson JD, Shearer GM. TNF-related apoptosis-inducing ligand (TRAIL) in HIV-1-infected patients and its in vitro production by antigen-presenting cells. Blood. 2005;105:2458–2464. doi: 10.1182/blood-2004-08-3058. [DOI] [PubMed] [Google Scholar]
- 46.Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ, Yagita H, Lifson JD, Shearer GM. CD4+ T-cell death induced by infectious and noninfectious HIV-1: role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood. 2005;106:3524–3531. doi: 10.1182/blood-2005-03-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boise LH, Thompson CB. Hierarchical control of lymphocyte survival. Science. 1996;274:67–68. doi: 10.1126/science.274.5284.67. [DOI] [PubMed] [Google Scholar]
- 48.Groux H, Torpier G, Monte D, Mouton Y, Capron A, Ameisen JC. Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. J Exp Med. 1992;175:331–340. doi: 10.1084/jem.175.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RP, Miedema F. Programmed death of T cells in HIV-1 infection. Science. 1992;257:217–219. doi: 10.1126/science.1352911. [DOI] [PubMed] [Google Scholar]
- 50.Chartier C, Degryse E, Gantzer M, Dieterle A, Pavirani A, Mehtali M. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mologni D, Citterio P, Menzaghi B, Zanone Poma B, Riva C, Broggini V, Sinicco A, Milazzo L, Adorni F, Rusconi S, et al. Vpr and HIV-1 disease progression: R77Q mutation is associated with long-term control of HIV-1 infection in different groups of patients. AIDS. 2006;20:567–574. doi: 10.1097/01.aids.0000210611.60459.0e. [DOI] [PubMed] [Google Scholar]
- 52.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 53.Vilar E, Salazar R, Perez-Garcia J, Cortes J, Oberg K, Tabernero J. Chemotherapy and role of the proliferation marker Ki-67 in digestive neuroendocrine tumors. Endocr Relat Cancer. 2007;14:221–232. doi: 10.1677/ERC-06-0074. [DOI] [PubMed] [Google Scholar]
- 54.Sakai K, Dimas J, Lenardo MJ. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc Natl Acad Sci USA. 2006;103:3369–3374. doi: 10.1073/pnas.0509417103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression: implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–47937. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 56.Caserta S, Zamoyska R. Memories are made of this: synergy of T cell receptor and cytokine signals in CD4+ central memory cell survival. Trends Immunol. 2007;28:245–248. doi: 10.1016/j.it.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 57.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nature Cell Biol. 2001;3:1124–1128. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 58.Barthelemy C, Henderson CE, Pettmann B. Foxo3a induces motoneuron death through the Fas pathway in cooperation with JNK. BMC Neurosci. 2004;5:48. doi: 10.1186/1471-2202-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez de Mattos S, Essafi A, Soeiro I, Pietersen AM, Birkenkamp KU, Edwards CS, Martino A, Nelson BH, Francis JM, Jones MC, et al. FoxO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Mol Cell Biol. 2004;24:10058–10071. doi: 10.1128/MCB.24.22.10058-10071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Obexer P, Geiger K, Ambros PF, Meister B, Ausserlechner MJ. FKHRL1-mediated expression of Noxa and Bim induces apoptosis via the mitochondria in neuroblastoma cells. Cell Death Differ. 2007;14:534–547. doi: 10.1038/sj.cdd.4402017. [DOI] [PubMed] [Google Scholar]
- 61.Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ, Lam EW, Burgering BM. Control of cell cycle exit and entry by protein kinase B-regulated Forkhead transcription factors. Mol Cell Biol. 2002;22:2025–2036. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 64.Andersen JL, Zimmerman ES, DeHart JL, Murala S, Ardon O, Blackett J, Chen J, Planelles V. ATR and GADD45alpha mediate HIV-1 Vpr-induced apoptosis. Cell Death Differ. 2005;12:326–334. doi: 10.1038/sj.cdd.4401565. [DOI] [PubMed] [Google Scholar]
- 65.Hartmann W, Digon-Sontgerath B, Koch A, Waha A, Endl E, Dani I, Denkhaus D, Goodyer CG, Sorensen N, Wiestler OD, Pietsch T. Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin Cancer Res. 2006;12:3019–3027. doi: 10.1158/1078-0432.CCR-05-2187. [DOI] [PubMed] [Google Scholar]
- 66.Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem. 2006;281:10473–10481. doi: 10.1074/jbc.M509278200. [DOI] [PubMed] [Google Scholar]
- 67.You H, Yamamoto K, Mak TW. Regulation of transactivation-independent proapoptotic activity of p53 by FOXO3a. Proc Natl Acad Sci USA. 2006;103:9051–9056. doi: 10.1073/pnas.0600889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moulian N, Bidault J, Planche C, Berrih-Aknin S. Two signaling pathways can increase fas expression in human thymocytes. Blood. 1998;92:1297–1307. [PubMed] [Google Scholar]
- 69.Baron V, Adamson ED, Calogero A, Ragona G, Mercola D. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFβ1, PTEN, p53, and fibronectin. Cancer Gene Ther. 2006;13:115–124. doi: 10.1038/sj.cgt.7700896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wan X, Helman LJ. Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene. 2003;22:8205–8211. doi: 10.1038/sj.onc.1206878. [DOI] [PubMed] [Google Scholar]
- 72.Skalka AM, Katz RA. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005;12(Suppl 1):971–978. doi: 10.1038/sj.cdd.4401573. [DOI] [PubMed] [Google Scholar]
- 73.Fan F, Jin S, Amundson SA, Tong T, Fan W, Zhao H, Zhu X, Mazzacurati L, Li X, Petrik KL, et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene. 2002;21:7488–7496. doi: 10.1038/sj.onc.1205896. [DOI] [PubMed] [Google Scholar]
- 74.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 75.James CG, Woods A, Underhill TM, Beier F. The transcription factor ATF3 is upregulated during chondrocyte differentiation and represses cyclin D1 and A gene transcription. BMC Mol Biol. 2006;7:30. doi: 10.1186/1471-2199-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 77.Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 78.Suhara T, Kim HS, Kirshenbaum LA, Walsh K. Suppression of Akt signaling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol. 2002;22:680–691. doi: 10.1128/MCB.22.2.680-691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hottiger MO, Nabel GJ. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J Virol. 1998;72:8252–8256. doi: 10.1128/jvi.72.10.8252-8256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marzio G, Tyagi M, Gutierrez MI, Giacca M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc Natl Acad Sci USA. 1998;95:13519–13524. doi: 10.1073/pnas.95.23.13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ott M, Schnolzer M, Garnica J, Fischle W, Emiliani S, Rackwitz HR, Verdin E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr Biol. 1999;9:1489–1492. doi: 10.1016/s0960-9822(00)80120-7. [DOI] [PubMed] [Google Scholar]
- 82.Silverman ES, Du J, Williams AJ, Wadgaonkar R, Drazen JM, Collins T. cAMP-response-element-binding-protein-binding protein (CBP) and p300 are transcriptional co-activators of early growth response factor-1 (Egr-1) Biochem J. 1998;336(Pt 1):183–189. doi: 10.1042/bj3360183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng L, Wang D, de la Fuente C, Wang L, Li H, Lee CG, Donnelly R, Wade JD, Lambert P, Kashanchi F. Enhancement of the p300 HAT activity by HIV-1 Tat on chromatin DNA. Virology. 2001;289:312–326. doi: 10.1006/viro.2001.1129. [DOI] [PubMed] [Google Scholar]
- 84.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe. 2008;3:158–167. doi: 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 86.Cui M, Huang Y, Zhao Y, Zheng J. Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages. J Immunol. 2008;180:898–906. doi: 10.4049/jimmunol.180.2.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van Grevenynghe J, Procopio FA, He Z, Chomont N, Riou C, Zhang Y, Gimmig S, Boucher G, Wilkinson P, Shi Y, et al. Transcription factor FOXO3a controls the persistence of memory CD4+ T cells during HIV infection. Nat Med. 2008;14:266–274. doi: 10.1038/nm1728. [DOI] [PubMed] [Google Scholar]
- 88.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the Forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.