

Abstract
LightCycler technology combines rapid-cycle polymerase chain reaction with real-time fluorescent monitoring and melting curve analysis. Since its introduction in 1997, it is now used in many areas of molecular pathology, including oncology (solid tumors and hematopathology), inherited disease, and infectious disease. By monitoring product accumulation during rapid amplification, quantitative polymerase chain reaction in a closed-tube system is possible in 15 to 30 minutes. Furthermore, melting curve analysis of probes and/or amplicons provides genotyping and even haplotyping. Novel mutations are identified by unexpected melting temperature or curve shape changes. Melting probe designs include adjacent hybridization probes, single labeled probes, unlabeled probes, and snapback primers. High-resolution melting allows mutation scanning by detecting all heterozygous changes. This review describes the major advances throughout the last 15 years regarding LightCycler technology and its application in clinical laboratories.
From the first report of the LightCycler as a real-time polymerase chain reaction (PCR) instrument1 clinical laboratories quickly realized its potential as a diagnostic tool. Quantitative applications important for infectious disease and oncology applications were followed by genotyping assays that took advantage of accurate temperature control to melt DNA amplicons or probe/amplicon duplexes. The first Food and Drug Administration-cleared molecular genetic assays were developed for this platform, specifically genotyping single base variants in F5 and F2. According to a College of American Pathologists survey,2 ∼30% of clinical laboratories report using the LightCycler for these two assays. This article will review the history of LightCycler technology, focusing on those applications widely incorporated into molecular diagnostics. In addition we will review recent developments that may impact future clinical testing.
Real-time PCR instruments simultaneously amplify and detect, eliminating the need to open tubes containing PCR amplicon and therefore reducing the risk of future contamination. Fluorescent dyes or probes allow continuous monitoring as template DNA is amplified.3 By monitoring fluorescence every cycle, the amount of original target can be calculated. By monitoring fluorescence as the temperature changes, genotyping and heterozygote scanning can be performed by melting analysis, often removing the need for downstream analysis.
The first two platforms developed for real-time PCR were the LightCycler (Roche, Indianapolis, IN) and the ABI 7700 (Applied Biosystems, Foster City, CA). The instruments were as different as the groups that created them. At the time, ABI was the undisputed corporate leader of PCR technology and the 7700 was a large 96-well laser-based instrument focused on throughput. The LightCycler arose out of academic (University of Utah), reference laboratory (Associated Regional and University Pathologists), and small company (Idaho Technology, Salt Lake City, UT) collaboration. The LightCycler was first commercialized by Idaho Technology as a small, 24-sample capillary instrument focused on speed. In 1997, the LightCycler was licensed to Boehringer Mannheim (Indianapolis, IN) and Boehringer Mannheim was purchased by Roche (Indianapolis, IN) that same year. The widely known 32-capillary Roche LightCycler was released in 1998. By analogy to the automotive industry, if the LightCycler was a sports car, the 7700 was a bus. A bus can carry more people than a sports car, but a sports car can get a few people there faster.
The LightCycler was introduced with SYBR Green I as a generic DNA dye and dual hybridization probes (HybProbes) for probe-specific monitoring. The ABI 7700 was introduced with hydrolysis probes (TaqMan). These three chemistries are compared in Figure 1. Most research applications use SYBR Green I to monitor real-time PCR. As a generic dye for double-stranded DNA, SYBR Green I can detect any target without probes. Fluorescence increases exponentially during PCR producing S-shaped logistic curves similar to bacterial growth.4,5 Although melting analysis can usually distinguish different PCR products,6 probes with additional sequence specificity may be preferred for clinical applications.
Figure 1.
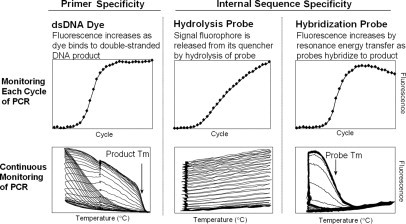
Monitoring PCR in real time using DNA dyes, hydrolysis probes, and hybridization probes. The top row shows data collected once each PCR cycle, and the bottom row shows data collected continuously (five times per second) during all PCR cycles. Adapted from Wittwer and Kusukawa4 with permission of the publisher.
Hybridization probes change fluorescence on hybridization.3 Fluorescence increases during PCR as the amount of PCR product increases. Genotyping by melting analysis is possible because different alleles form probe duplexes of different stabilities.7 Multiple alleles can be distinguished in a single analysis color. Hydrolysis probes are labeled with both a fluorescent reporter and a quencher, usually at opposite ends of the probe.8 During PCR, annealed probes are hydrolyzed by the 5′-3′ exonuclease activity of the polymerase, separating the fluorophore from the quencher and increasing fluorescence.9 Because fluorescence results from probe hydrolysis (not probe hybridization), they are referred to as hydrolysis probes.10 Multiplexing is possible by using more than one probe, each with a fluorophore of a different color. For example, a biallelic single base change can be genotyped with two hydrolysis probes, each complementary to one allele.11
In 1998, the ABI 7700 and the Roche LightCycler commanded distinct niches. Throughout time, cross fertilization between platforms occurred and many other manufacturers entered the field. ABI adopted SYBR Green I and hydrolysis probes became an option on the LightCycler. Melting curves on the LightCycler became known as dissociation curves on ABI instruments. A 96/384 microtiter format LightCycler appeared and a 1536-well version will appear in 2009. Fast protocols were introduced on plate-based instruments to challenge the rapid-cycle PCR of the capillary LightCyclers. Characteristics of the different LightCycler versions are summarized in Table 1.
Table 1.
Characteristics of Different LightCycler Versions
| LightCycler version |
|||
|---|---|---|---|
| Characteristic | 1.0 to 1.5 | 2.0 | LC 480 |
| Format | Capillaries | Capillaries | Microtiter plate |
| Number of samples | 32 | 32 | 96 or 384 |
| Sample volume (μl) | 5 to 20 | 5 to 100 | 5 to 50 |
| Excitation channels | 1 | 1 | 5 |
| Emission channels | 3 | 6 | 6 |
| Time for 30 cycles (minutes) | 10 to 30 | 10 to 60 | 40 to 90 |
| High resolution melting | No | No | Yes |
Rapid-Cycle PCR
Rapid-cycle PCR actually predates the LightCycler. It is defined as 30 cycles completed in 10 to 30 minutes.12 Ten-minute PCR was first demonstrated in capillaries with their high surface area to volume ratio, reaction volumes of 5 to 20 μl, and air thermal cycling.13 With accurate temperature control, annealing and denaturation times of 0 second (temperature spikes) increase specificity and yield.14 Rapid PCR has been recently reviewed.15 For many, it was the appearance of the LightCycler that popularized rapid cycling methods. A rapid-cycle PCR protocol (40 cycles in 15 to 20 minutes) on the capillary LightCycler is shown in Figure 2. When combined with fluorescence acquisition each cycle, real-time detection and quantification is enabled. With the addition of melting analysis, product and probe melting curves provide scanning and genotyping capabilities.
Figure 2.
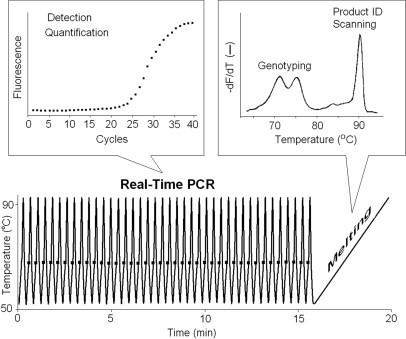
Rapid-cycle, real-time PCR. Momentary denaturation and annealing allows amplification in 10 to 20 minutes. Fluorescence is acquired once each cycle and is used for detection and quantification. Melting analysis is performed immediately after PCR and is used for genotyping, product identification, or heterozygote scanning. The real-time data were obtained on a Roche LightCycler 1.5 by amplifying a 250-bp fragment of exon 2 of PIGA from human genomic DNA in the presence of 1× LCGreen Plus dye. After a 5-second denaturation at 95°C, 45 cycles of 95°C for 0 second, 60°C for 0 second, and 72°C for 2 seconds with a 2°C/second ramp between annealing and extension temperatures was performed. Fluorescence was acquired at the end of each extension step. Temperature cycling required just longer than 15 minutes, whereas melting analysis at 0.2°C/second required another 4 minutes.
Even though the LightCycler was introduced with rapid-cycle PCR, not all reactions are performed so rapidly today. Depending on the DNA target and isolation method, a predenaturation step of 5 to 10 seconds may be required. When higher reaction volumes are needed to increase sensitivity, larger (100 μl) capillaries can be used on the LightCycler 2.0, but slower cycling times are required (Table 1). Additional temperature/time steps are necessary if uracil-DNA glycosylase is used for decontamination and/or heat-activated polymerases are used to increase specificity. These added steps may take longer than the time required for rapid-cycle PCR. When high-precision quantification is necessary (for example to detect copy number changes) care must be taken to ensure complete template denaturation.16 Furthermore, the primer concentration must be high enough to ensure complete annealing each cycle when high PCR efficiency is required. Although some may elect not to use rapid-cycle PCR, the capillary LightCycler provides this option.
Genotyping
The first LightCycler genotyping assay described was for the factor V Leiden mutation.7 In this assay, a primer near to the mutation was labeled with a fluorophore (Cy5) attached three bases internal to its 3′end. A probe covering the region of the Leiden mutation was labeled with fluorescein on the end nearest the Cy5-labeled primer. At temperatures that allow probe hybridization, the two fluorophores are brought into close proximity and fluorescence resonance energy transfer occurs, generating long wavelength fluorescence. At low temperature, the probe hybridizes to both alleles. By slowly increasing the temperature, the probe first denatures from the mismatched allele, then at higher temperatures denatures from the perfectly matched allele, producing the characteristic melting curves for homozygous mutations, heterozygotes, and homozygous wild types. The negative derivative of the melting curves allows easy visualization (Figure 3). In general, the probe can be complementary to either the normal allele or variant allele.
Figure 3.
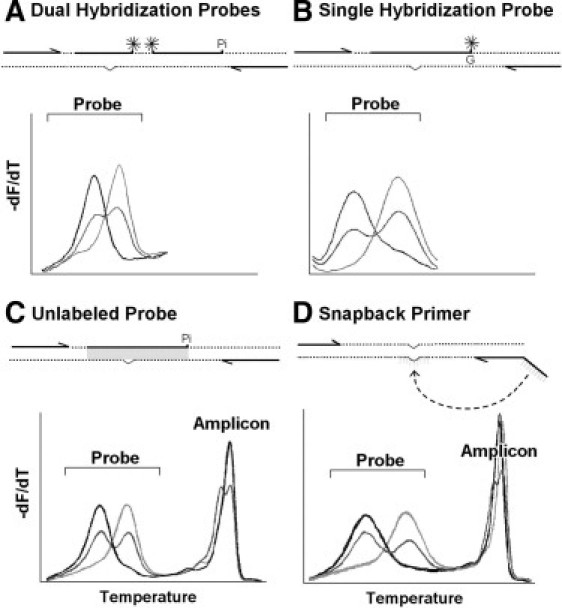
Genotyping by melting analysis using dual hybridization probes (A), single hybridization probes (B), unlabeled probes (C), and snapback primers (D). Within each panel, a conceptual diagram of the assay is on the top and resulting derivative melting curves are on the bottom. Dotted lines indicate the DNA template, arrows represent primers, thick line segments indicate probes, Pi indicates 3′-blocking with phosphate, asterisks indicate covalent fluorescent dyes, and a shallow v in the template indicates the position of variation. Three samples of each genotype are shown in each panel, including matched homozygous wild-type (light gray), heterozygous (dark gray), and mismatched homozygous variant (black). A: The dual hybridization probe design was introduced with the original LightCycler.3,17 Two adjacent probes are each labeled with one fluorophore, selected so that they form a resonance energy transfer pair. Long wavelength fluorescence is generated when both the anchor and mutation probe are hybridized. The probe with an exposed 3′-end is blocked with phosphate to prevent extension. B: The single hybridization probe design includes only one probe with a single fluorescent label.28 It may be 3′-labeled (as shown) or 5′-labeled with the 3′-end blocked. The G indicates a deoxyguanosine base, either in the template or the probe that influences fluorescence. Depending on the configuration, either quenching or dequenching of fluorescence occurs on hybridization. No PCR product or amplicon melting curves are detected with hybridization probes. C: The unlabeled probe design requires a 3′-blocked probe without any covalently attached fluorophore.32 Fluorescence is generated by a saturating DNA dye (shaded area) that only fluoresces when bound to duplex DNA. Asymmetric PCR produces both full-length PCR product (amplicon) and product/probe duplexes (probe). D: The snapback primer design only requires two primers, one with a 5′ extension that is complementary to its own extension product.39 Asymmetric PCR overproduces one strand, so that both full-length PCR product (amplicon) and an excess of one strand are formed. The excess single strand snaps back on itself (dotted line) so that its complementary regions (light gray lines) anneal to form a hairpin stem. Melting of the hairpin results in probe melting peaks. Both unlabeled probe and snapback primer designs typically have amplicon melting peaks (that can be use for heterozygote scanning) as well as probe melting peaks (that are used for genotyping).
Other assays soon followed, demonstrating the versatility of the technology. The use of dual hybridization probes became common, including a higher melting temperature Tm anchor probe and a lower Tm reporter probe that covers the variant of interest (Figure 3A). With dual-hybridization probes, both labeled oligonucleotides are end labeled, whereas the primer system required an internal label to allow primer extension. Multiplex genotyping was accomplished by either Tm, by color, or both. An assay for hereditary hemochromatosis was described distinguishing the HFE C282Y, H63D and S65L mutant and normal alleles with unique Tms for all four possible alleles.17 Later, multiplexed assays with different fluorophores used multiple channels of the LightCycler for MTHFR (C677T and A1298C)18 and HFE (C282Y, H63D, and S65C).19 With only minor changes, both assays are currently used in clinical laboratories. The HFE assay demonstrates the ability to differentiate two mutations using one probe to cover both the S65C and H63D variants. Because the probe is a perfect match to the H63D mutation, the S65C allele with a wild-type base at the H63D position results in two mismatches of the probe. Higher multiplexing with novel designs has been described in the literature, for example, an assay for TPMT that identifies and differentiates eight mutations using a combination of fluorescence resonance energy transfer and fluorescent quenching probes.20
Identifying Novel Variants
Probe Tms are determined by GC content, nearest neighbor effects, and the number, position, and base change of mismatches. Many of the variant possibilities covered by a probe will have unique Tms. Novel variants with unexpected melting temperatures differing from the targeted mutation began to be identified. For example, the 20209C>T variant, found in African-Americans was identified by the factor II (prothrombin) assay21 and may have clinical significance in thrombophilia.22 Similarly, the A20218G mutation was identified.23
Because different samples vary slightly in absolute Tm, relying on the Tm difference between normal and variant alleles (ΔTm) is preferable for identifying new variants.24,25 An unexpected F5 1689G>A heterozygote was initially interpreted as a F5 Leiden heterozygote by absolute Tm, but was later correctly identified by ΔTm.26 F5 Leiden heterozygotes usually have a ΔTm of 8.4 ± 0.2°C. In the sample with the unexpected 1689G>A variant, two bases away from the Leiden mutation, the ΔTm was 7.6°C. The ΔTm difference of 0.8°C was much higher than usual, leading to correct identification by sequencing. This F5 variant and others identified by hybridization probes are likely deficiency alleles rather than activated protein C (APC) resistance alleles.27 Additional variants have been identified in the same way, including a relatively common HFE variant (c.189T>C) resulting in a silent H63H variant. Although this is a silent base change, it may interfere with correct genotyping by other methods.25
The reporter probe covering the variant of interest can be designed to be a perfect match to the normal allele or to the variant. In the scenario in which the probe is matched to the normal allele, a new variant results in a single mismatch that is different in base identity, position, and/or nearest neighbor stability. In the scenario in which the probe is matched to the expected variant, a novel variant results in two mismatches—one with the normal base and one with the novel base. The two mismatches will further destabilize the probe, reducing the Tm. By using the probe matched to the variant of interest, potential false-positives are minimized.
Reducing Complexity
The design of melting assays can be simplified by using single rather than dual hybridization probes (Figure 3B). The fluorophore on the probe is quenched by nearby guanine nucleotides in the target28 or in the probe.29 As the hybridization state changes, so does the extent of quenching and the fluorescence intensity is modified. A single-labeled probe assay (SimpleProbe, Roche Applied Science) is available that detects the three variants recommended by the Food and Drug Administration for warfarin sensitivity susceptibility (Idaho Technology). In this design, the two variants, *2 and *3 of the CYP2C9 gene and a promoter variant in the VKORC1 gene are amplified separately (nonmultiplexed) but under the same reaction conditions. The simplicity and speed of this assay makes it feasible for point of care testing.30,31 Single hybridization probes can be used on any LightCycler, although the fluorescence to background ratio is less than with dual hybridization probes.
To eliminate labeled probes altogether, an unlabeled probe can be used if a saturating DNA binding dye is included and the probe is blocked at its 3′-end (Figure 3C).32 As the probe denatures, the fluorescence decreases. Unlabeled probe designs have been implemented in the clinical laboratory for F5 and F2 assays.33,34 Unlabeled probe genotyping has also been reported for RET,35 HSV typing,36 and common variants of CFTR,34 ACVRL1 and ENG.37 Although unlabeled probes can be used on capillary LightCyclers, genotypes are better resolved on high-resolution melting instruments such as the LightScanner (Idaho Technology) and the LightCycler 480. The LightScanner requires separate amplification on a conventional block cycler, but includes software with exponential background subtraction specifically for unlabeled probes.38
A new variant of unlabeled probes attaches the probe as a 5′-tail to one of the primers.39 Such snapback primers form intramolecular hairpins (similar to the more complicated Scorpion primers) and allow genotyping by melting. With snapback primers, only two unlabeled oligonucleotides are necessary and 3′-blocking is not required (Figure 3D). The hairpins are stabilized so that small regions can be probed. The hairpin Tm is linearly related to the stem length (6 to 28 bp) and inversely related to the log of the loop size (17 to 135 bases). F5 Leiden and several CFTR variants have been genotyped with snapback primers. Snapback primer genotyping provides the specificity of a probe with only two primers that are free of special covalent labels. Single hybridization probes, unlabeled probes, and snapback primers require asymmetric PCR (usually 5:1 to 10:1) to accumulate strands complementary to the probe and reduce the competition with full-length PCR product. Optimization may require adjustment of the primer ratios and increasing the number of cycles to 40 to 50.
Known benign variants may be located very close to mutations of interest, complicating the selection of probes for melting analysis. Such variants can be masked by designing probes that include a mismatched base, a universal base, or a deletion at the site of the variation to be ignored. As a result, all benign alleles have the same probe stability and only mutations of interest are reflected in the melting curve, greatly simplifying analysis.40 Deletions are generally preferred because a mismatched base may not be equally stable with all masked alleles and universal bases (such as 5-nitroindole) carry an additional cost.
Genotyping by melting analysis becomes even simpler if probes are eliminated altogether. Allele-specific PCR can provide biallelic genotyping by product melting analysis with SYBR Green I using three primers, at least one of which includes a Tm-shifting tail.41 Perhaps the ultimate in simplification is to use only two unmodified PCR primers for genotyping by high-resolution melting with the saturating dye LCGreen, a double-stranded DNA binding dye that detects heteroduplexes.42 With small PCR products, homozygous variants are distinguished by Tm and heterozygotes are identified by melting curve shape. The method has been applied to many common clinical single-base variants43 and compared against dual-hybridization probes44 and unlabeled probes.45 However, many small insertions/deletions and some single base changes can be difficult to distinguish from wild type when homozygous. For example, bp neutral single base changes with nearest neighbor symmetry are predicted to have identical Tms. Even these can sometimes be resolved with internal temperature controls.46 Another comprehensive solution is quantitative heteroduplex analysis, in which a known genotype is mixed with unknown samples before PCR.47 The accuracy of genotyping by PCR product melting is directly related to instrument precision.48 Capillary LightCyclers are not recommended for high-resolution melting.49 For clinical applications, the added sequence specificity of a probe may be preferred. However, for basic and clinical research, the simplicity of genotyping by PCR product melting is a strong incentive and the method is finding greater acceptance. Perhaps the clinical requirement for probes will decrease in the future, especially as high-resolution instruments become more widely available.
Molecular Haplotyping by Melting
Genotyping usually identifies individual variants. However, the combination of variants on the same or opposite chromosomes may need to be determined. In research, the population frequencies of the mutations occurring together are often used to determine the haplotype. For clinical testing, family members may be tested to assign the chromosome phase based on Mendelian inheritance. If family members are unavailable or difficult to collect, molecular haplotyping can be used. If the loci are close together, one hybridization probe across both loci may allow haplotyping by melting. For example, two biallelic single base changes will often result in four probe Tms correlating with each possible haplotype. If the loci are too far apart to be covered with one probe, noncontinuously binding probes can be used.50 For example, one hybridization reporter probe was used to cover two loci of the adrenergic β2 receptor with the intervening sequence looped out (∼50 bp). Melting curve analysis distinguished all four possible combinations, establishing the haplotype. Criteria for the design of noncontinuous probes have been published.51
Mutation Scanning by Melting
High-resolution melting of PCR products can be used as a mutation scanning technique to identify any heterozygous variant between the PCR primers. It is the only closed-tube scanning method available and appears competitive with dHPLC and other options in terms of price, sensitivity, and specificity. Once a variant is found, sequencing the specific exon(s) typically identifies the variant. Common variants can usually be identified by the shape of the melting curve52 or with unlabeled probes.37 Most homozygous variants are also identified, although small homozygous deletions/insertions may be missed unless the samples are mixed with a wild type to create an artificial heterozygote. The first high-resolution melting instrument (HR-1, Idaho Technology) analyzed samples that were amplified in LightCycler capillary tubes. The LightCycler 480 integrates amplification and high-resolution melting in a plate format.
High-resolution melting has been used clinically to detect somatic changes in select exons of oncogenes such as EGFR,53 KRAS,54 PDGFRA,55 KIT,56 BRAF,57 and TP53.58 In tumors with mixed populations of tumor and normal cells, the sensitivity of scanning (<10%) is superior to sequencing (∼20%).59 In genetics, the method has been applied to BRCA1/2,60,61 cystic fibrosis,52,62 hereditary hemorrhagic telangiectasia,37 hemophilia,63,64 and Charcot-Marie-Tooth disease,65 and several others. In sum, 19 genetic studies using DNA extracted from human blood59 had an overall sensitivity for heterozygote detection of 99.3% (n = 839) and a specificity of 98.8% (n = 2659). The overall sensitivity and specificity of 18 clinical oncology studies59 using tumor DNA (mostly from formalin fixed, paraffin-embedded tissue) is somewhat lower at 96.9% (n = 428) and 97.1% (n = 3080), respectively. Clinical laboratories are beginning to use scanning by high-resolution melting. The method holds promise for reducing the costs and complexity of analyzing complete genes.
Simultaneous PCR product-scanning and probe-melting analysis is possible from the same melting curve.34,66 At low temperature, specific genotyping is obtained by melting unlabeled probes or the hairpin of snapback primers, whereas at high temperature the full PCR product is scanned for variants. Such a combined analysis, in combination with masking common polymorphisms, can be used to efficiently identify mutations in multiple endocrine neoplasia type II.35
Quantification
Quantification by real-time PCR is extraordinarily sensitive with a broad dynamic range. It is an ideal tool for DNA quantification. RNA can also be quantified if it is first converted to DNA with a reverse transcriptase. Real-time quantification has found broad use in research and diagnostics. The principles behind real-time PCR have been recently reviewed5 and guidelines for experimental design and reporting established.10 Real-time quantification is used in oncology, infectious disease, and genetics.
Oncology
Cancer correlates with many genetic and epigenetic changes. These changes can be monitored by real-time PCR and include translocations, inversions, duplications, deletions, and small sequence variants including methylation. For example, BCR-ABL1 quantification monitors minimal residual disease and therapy of chronic myelogenous leukemia. Both dual-hybridization probes67 and hydrolysis probes68 have been used for BCR-ABL1 quantification on the LightCycler. In either case, real-time PCR is used to quantify RNA transcripts of the fusion gene BCR-ABL1, a somatic alteration in chronic myelogenous leukemia. Quantification of a reference transcript (such as the G6PDH gene) is usually included to normalize BCR-ABL1 results between samples for relative quantification. Results can be compared to previous results if serial samples are tested by the same laboratory. Melting analysis of either the PCR product or the probe may be used to confirm detection of the correct product. The reported sensitivity for commercial assays is 1 × 10−5 K562 cells (a BCR-ABL1-positive cell line) diluted in BCR-ABL1-negative cells and does not vary significantly with probe design or instrument used.69 Other targets for monitoring minimal residual disease in hematological malignancies include chromosomal rearrangements such as t(14:18) and inv(16), fusion transcripts such as PML-RAR α and RUNX1-ETO, expression of WT1, and mutations in NPM1, FLT3, and JAK2. In solid tumors, detection of mutations in c-KIT, EGFR, and BRAF has therapeutic and prognostic significance. In breast cancer, expression arrays have established prognostic groups that can be identified by quantifying a reduced set of transcripts from paraffin-embedded tissue by real-time PCR.70 Quantitative real-time PCR remains a key technique for identifying circulating nucleic acid tumor markers and detecting fetal DNA in maternal serum.
DNA methylation as an epigenetic modification is important in tumor biology and can be assessed by several real-time methods. For example, bisulfite treatment and PCR convert unmethylated C:G pairs to A:T pairs so that the amplicon melting temperature is directly related to the degree of methylation of the target. Methylation analysis by melting was introduced on the LightCycler with SYBR Green I71 and can be used to diagnose imprinting disorders, such as the Angelman and Prader-Willi syndromes.72
Microbiology
Real-time PCR has revolutionized microbiology testing. Extensive development at the Mayo Clinic by Dr. Franklin Cockerill and others73,74 is well documented in previous reviews. Particularly for organisms that are difficult or slow to culture, real-time PCR is a leap forward in time to result and sensitivity over pre-existing assays. For example, monitoring the cytopathic effect of viruses in shell vial cultures is expensive, time consuming, and requires considerable expertise. Real-time assays for herpes simplex virus, varicella-zoster virus, cytomegalovirus, Epstein-Barr virus, enterovirus, polyomavirus, parvovirus, respiratory viruses, poxviruses, and others are more sensitive than their culture counterparts and can be performed in <1 hour in a capillary LightCycler. Quantification has proven clinical utility for cytomegalovirus, Epstein-Barr virus, BK virus, hepatitis B and C, and human immunodeficiency virus. Real-time analysis can identify mycobacteria in hours instead of the weeks required for culture. For example, dual-hybridization probes have been used to detect 33 species of mycobacteria using the 16S rRNA gene.75
LightCycler technology has been applied to many bacteria, fungi, and parasites. Targeting a single pathogen of clinical importance can immediately guide therapy. For example, appropriate antibiotic therapy can follow identification of group A streptococcus in the throat or group B streptococcus from vaginal swabs. Another example is detection of methicillin-resistant Staphylococcus aureus from the nasopharyx that may warrant isolation procedures on admission to the hospital. Melting analysis is often used to differentiate between species or subspecies, for example, Bordetella pertussis and parapertussis.76 LightCycler technology is also used for rapid detection of bioterrorism agents, pathogens in food (Salmonella, Listeria, Edcherichia coli O157), food spoilage, and genetically modified organism quantification. Fungal agents that have been successfully targeted include Aspergillus, Candida, and Pneumocystis. Parasitic targets include Plasmodia, Leishmania, Toxoplasma, and protozoan pathogens in stool.
The LightCycler SeptiFast test is CE-IVD marked and identifies the 25 most common pathogens known to cause sepsis, including 10 Gram-negative bacteria, 9 Gram-positive bacteria, and 6 fungal agents. Without prior incubation or culture, whole blood (1.5 ml) is lysed, extracted, and amplified on a LightCycler 2.0 instrument in 100-μl capillaries. Results are available in less than 6 hours. A multicopy target (the internal transcribed spacer between the ribosomal 16S and 23S of bacteria or the 18S and 5.8S of fungi) is amplified for increased sensitivity. The different targets are distinguished using multiple sets of dual hybridization probes by a combination of both color and Tm. In initial studies, comparison against culture looks promising.77
Genetics
Detecting copy number variants, eg, deletions and duplications of chromosomal regions, genes, or partial genes are increasingly important in genetics. Whole genome scans by comparative genomic hybridization or single base variant arrays have recently become available, and multiplexed ligation product amplification can selectively target many exons in a gene. The 1:2 ratios for deletions and the 3:2 ratios of duplications are challenging to detect by any method. When evidence for a deletion or duplication is suspected, a second method to confirm the result is wise. For example, comparative genomic hybridization arrays that detect large chromosomal rearrangements are often confirmed by fluorescent in situ hybridization probes. However, readily available probes may be inadequate for small deletions or duplications. In these instances, quantitative real-time PCR can be used for confirmation.78 Advantages are rapid primer design and optimization with low costs to confirm any region of the genome within acceptable clinical turn-around times. Real-time quantification, properly performed, can reliably detect copy number variants.79
Relative quantification can also be performed by melting curve analysis. For example, HER2/neu (ERBB2) gene dosage can be quantified by competitive PCR.80 Competitive transcripts with a single base change from the target gene were designed for ERBB2 as well as for β-globin as a control. Melting analysis differentiated between the competitor and the target transcripts and derivative melting curves were used to determine relative peak areas or peak heights. Compared to real-time analysis the methods were similar in their accuracy and precision.81 As another example, melting curve analysis of natural single base variants along chromosome 21 can be exploited for trisomy 21 determination.82 Allelic ratios of 0.5 or 2.0 indicate trisomy, compared to the normal 1:1 ratio.
In summary, LightCycler technology has enabled routine clinical testing in many areas of molecular diagnostics, including inherited diseases, infectious diseases, and oncology. The LightCycler introduced real-time display, rapid cycling, SYBR Green I, hybridization probes, and melting analysis to real-time PCR. Based on rapid-cycle PCR, capillary LightCyclers can complete PCR in 15 to 30 minutes, providing rapid turn around in critical clinical settings.83,84,85,86 In situations in which high throughput is more important than turn around time, the LC480 allows amplification on 96- or 384-well plates. In addition to monitoring fluorescence once each cycle, fluorescence can be monitored continuously as the temperature changes so that hybridization is assessed. Both PCR product hybridization with SYBR Green I and probe hybridization for genotyping can be monitored. As melting technology became better, high-resolution melting analysis was introduced as the latest method for product analysis. Labeled probes were no longer necessary for genotyping and entire PCR products could be scanned for single base changes. By combining amplification and analysis in an open platform, the LightCycler enabled in-house laboratory development at a critical time in the evolution of molecular diagnostics.
Acknowledgements
We thank Jackie McCowen-Rose, B.S., for her editing assistance; and Gudrun Reed, M.S., for the figures.
Footnotes
Light Cycler technology was developed under federal, state, and industrial grants, including the following NIH STTR awards: GM051647, GM058983, GM072419, GM073396 and GM082116.
The AMP Award for Excellence in Molecular Diagnostics is given by the Association for Molecular Pathology to recognize lifetime, pioneering, and special achievements related to molecular diagnostics and molecular medicine. Carl T. Wittwer, recipient of the 2008 AMP Award for Excellence in Molecular Diagnostics, delivered a lecture entitled “Rapid Cycle PCR, Real Time Analysis, and Hi-Res Melting” on October 31, 2008 at the annual meeting of the Association for Molecular Pathology in Grapevine, TX.
Aspects of rapid-cycle PCR, LightCycler technology, and melting analysis were developed at the University of Utah, licensed to Idaho Technology, and sublicensed to Roche Applied Science. C.T.W. has an equity interest in Idaho Technology.
References
- 1.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques. 1997;22:176–181. doi: 10.2144/97221pf02. [DOI] [PubMed] [Google Scholar]
- 2.Participant Summary Report: MGL-B. ACMG/CAP Molecular Genetics Committee; 2006. pp. 4–6. [Google Scholar]
- 3.Wittwer CT, Herrmann MG, Moss AA, Rasmussen RP. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques. 1997;22:130–138. doi: 10.2144/97221bi01. [DOI] [PubMed] [Google Scholar]
- 4.Wittwer CT, Kusukawa N. Real-time PCR. In: Persing DH, Tenover FC, Versalovic J, Tang YW, Unger ER, Relman DA, White TJ, editors. Diagnostic Molecular Microbiology; Principles and Applications. ASM Press; Washington DC: 2004. pp. 71–84. [Google Scholar]
- 5.Wittwer CT, Kusukawa N. Real-time PCR and melting analysis. In: Persing DH, Tenover FC, Hayden R, Nolte F, Tang YW, Belkum AV, editors. Molecular Microbiology: Diagnostic Principles and Practice. ed 2. ASM Press; Washington, DC: 2009. (in press) [Google Scholar]
- 6.Ririe KM, Rasmussen RP, Wittwer CT. Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem. 1997;245:154–160. doi: 10.1006/abio.1996.9916. [DOI] [PubMed] [Google Scholar]
- 7.Lay MJ, Wittwer CT. Real-time fluorescence genotyping of factor V Leiden during rapid-cycle PCR. Clin Chem. 1997;43:2262–2267. [PubMed] [Google Scholar]
- 8.Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl. 1995;4:357–362. doi: 10.1101/gr.4.6.357. [DOI] [PubMed] [Google Scholar]
- 9.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 10.Bustin S, Garson J, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl M, Shipley G, Vandesompele J, Wittwer C: The MIQE guidelines: minimal information for publication of quantitative real-time PCR experiments. Clin Chem (in press) [DOI] [PubMed]
- 11.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–149. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 12.Wittwer CT, Reed GB, Ririe KM. Rapid cycle DNA amplification. In: Mulls K, Ferre F, Gibbs R, editors. The Polymerase Chain Reaction. Springer-Verlag; Deerfield Beach: 1994. pp. 174–181. [Google Scholar]
- 13.Wittwer CT, Fillmore GC, Garling DJ. Minimizing the time required for DNA amplification by efficient heat transfer to small samples. Anal Biochem. 1990;186:328–331. doi: 10.1016/0003-2697(90)90090-v. [DOI] [PubMed] [Google Scholar]
- 14.Wittwer CT, Garling DJ. Rapid cycle DNA amplification: time and temperature optimization. Biotechniques. 1991;10:76–83. [PubMed] [Google Scholar]
- 15.Wittwer CT, Rasmussen RP, Ririe KM. Rapid PCR and melting analysis. In: Bustin SA, editor. The PCR Revolution. Cambridge University Press; Cambridge: 2009. (in press) [Google Scholar]
- 16.Wilhelm J, Hahn M, Pingoud A. Influence of DNA target melting behavior on real-time PCR quantification. Clin Chem. 2000;46:1738–1743. [PubMed] [Google Scholar]
- 17.Bernard PS, Ajioka RS, Kushner JP, Wittwer CT. Homogeneous multiplex genotyping of hemochromatosis mutations with fluorescent hybridization probes. Am J Pathol. 1998;153:1055–1061. doi: 10.1016/s0002-9440(10)65650-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura S, Aoshima T, Ikeda M, Sekido Y, Shimokata K, Niwa T. Simultaneous detection of methylenetetrahydrofolate reductase gene polymorphisms, C677T and A1298C, by melting curve analysis with LightCycler. Anal Biochem. 2002;306:340–343. doi: 10.1006/abio.2002.5709. [DOI] [PubMed] [Google Scholar]
- 19.Phillips M, Meadows CA, Huang MY, Millson A, Lyon E. Simultaneous detection of C282Y and H63D hemochromatosis mutations by dual-color probes. Mol Diagn. 2000;5:107–116. doi: 10.1007/BF03262029. [DOI] [PubMed] [Google Scholar]
- 20.Schütz E, von Ahsen N, Oellerich M. Genotyping of eight thiopurine methyltransferase mutations: three-color multiplexing, “two-color/shared” anchor, and fluorescence-quenching hybridization probe assays based on thermodynamic nearest-neighbor probe design. Clin Chem. 2000;46:1728–1737. [PubMed] [Google Scholar]
- 21.Warshawsky I, Hren C, Sercia L, Shadrach B, Deitcher SR, Newton E, Kottke-Marchant K. Detection of a novel point mutation of the prothrombin gene at position 20209. Diagn Mol Pathol. 2002;11:152–156. doi: 10.1097/00019606-200209000-00005. [DOI] [PubMed] [Google Scholar]
- 22.van der Putten HH, Spaargaren-van Riel CC, Bertina RM, Vos HL. Functional analysis of two prothrombin 3′-untranslated region variants: the C20209T variant, mainly found among African-Americans, and the C20209A variant. J Thromb Haemost. 2006;4:2285–2287. doi: 10.1111/j.1538-7836.2006.02102.x. [DOI] [PubMed] [Google Scholar]
- 23.Tag CG, Schifflers MC, Mohnen M, Gressner AM, Weiskirchen R. Atypical melting curve resulting from genetic variation in the 3′ untranslated region at position 20218 in the prothrombin gene analyzed with the LightCycler factor II (prothrombin) G20210A assay. Clin Chem. 2005;51:1560–1561. doi: 10.1373/clinchem.2005.050021. [DOI] [PubMed] [Google Scholar]
- 24.Lyon E. Mutation detection using fluorescent hybridization probes and melting curve analysis. Expert Rev Mol Diagn. 2001;1:92–101. doi: 10.1586/14737159.1.1.92. [DOI] [PubMed] [Google Scholar]
- 25.Lyon E. Discovering rare variants by use of melting temperature shifts seen in melting curve analysis. Clin Chem. 2005;51:1331–1332. doi: 10.1373/clinchem.2005.051177. [DOI] [PubMed] [Google Scholar]
- 26.Lyon E, Millson A, Phan T, Wittwer CT. Detection and identification of base alterations within the region of factor V Leiden by fluorescent melting curves. Mol Diagn. 1998;3:203–210. doi: 10.154/MODI00300203. [DOI] [PubMed] [Google Scholar]
- 27.Mahadevan MS, Benson PV. Factor V null mutation affecting the Roche LightCycler factor V Leiden assay. Clin Chem. 2005;51:1533–1535. doi: 10.1373/clinchem.2005.050351. [DOI] [PubMed] [Google Scholar]
- 28.Crockett AO, Wittwer CT. Fluorescein-labeled oligonucleotides for real-time PCR: using the inherent quenching of deoxyguanosine nucleotides. Anal Biochem. 2001;290:89–97. doi: 10.1006/abio.2000.4957. [DOI] [PubMed] [Google Scholar]
- 29.Vaughn CP, Elenitoba-Johnson KS. Hybridization-induced dequenching of fluorescein-labeled oligonucleotides: a novel strategy for PCR detection and genotyping. Am J Pathol. 2003;163:29–35. doi: 10.1016/S0002-9440(10)63627-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haug KB, Sharikabad MN, Kringen MK, Narum S, Sjaatil ST, Johansen PW, Kierulf P, Seljeflot I, Arnesen H, Brors O. Warfarin dose and INR related to genotypes of CYP2C9 and VKORC1 in patients with myocardial infarction. Thromb J. 2008;6:7. doi: 10.1186/1477-9560-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson JL, Horne BD, Stevens SM, Grove AS, Barton S, Nicholas ZP, Kahn SF, May HT, Samuelson KM, Muhlestein JB, Carlquist JF. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation. 2007;116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312. [DOI] [PubMed] [Google Scholar]
- 32.Zhou L, Myers AN, Vandersteen JG, Wang L, Wittwer CT. Closed-tube genotyping with unlabeled oligonucleotide probes and a saturating DNA dye. Clin Chem. 2004;50:1328–1335. doi: 10.1373/clinchem.2004.034322. [DOI] [PubMed] [Google Scholar]
- 33.Chou LS, Meadows C, Wittwer CT, Lyon E. Unlabeled oligonucleotide probes modified with locked nucleic acids for improved mismatch discrimination in genotyping by melting analysis. Biotechniques. 2005;39:644–648. doi: 10.2144/000112050. [DOI] [PubMed] [Google Scholar]
- 34.Zhou L, Wang L, Palais R, Pryor R, Wittwer CT. High-resolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution. Clin Chem. 2005;51:1770–1777. doi: 10.1373/clinchem.2005.054924. [DOI] [PubMed] [Google Scholar]
- 35.Margraf RL, Mao R, Highsmith WE, Holtegaard LM, Wittwer CT. RET proto-oncogene genotyping using unlabeled probes, the masking technique, and amplicon high-resolution melting analysis. J Mol Diagn. 2007;9:184–196. doi: 10.2353/jmoldx.2007.060091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dames S, Pattison DC, Bromley LK, Wittwer CT, Voelkerding KV. Unlabeled probes for the detection and typing of herpes simplex virus. Clin Chem. 2007;53:1847–1854. doi: 10.1373/clinchem.2007.090761. [DOI] [PubMed] [Google Scholar]
- 37.Vandersteen JG, Bayrak-Toydemir P, Palais RA, Wittwer CT. Identifying common genetic variants by high-resolution melting. Clin Chem. 2007;53:1191–1198. doi: 10.1373/clinchem.2007.085407. [DOI] [PubMed] [Google Scholar]
- 38.Erali M, Palais R, Wittwer CT. SNP genotyping by unlabeled probe melting analysis. In: Seitz O, Marx A, editors. Molecular Beacons: Signalling Nucleic Acid Probes, Methods and Protocols. Humana Press; Totowa: 2008. pp. 199–206. [DOI] [PubMed] [Google Scholar]
- 39.Zhou L, Errigo RJ, Lu H, Poritz MA, Seipp MT, Wittwer CT. Snapback primer genotyping with saturating DNA dye and melting analysis. Clin Chem. 2008;54:1648–1656. doi: 10.1373/clinchem.2008.107615. [DOI] [PubMed] [Google Scholar]
- 40.Margraf RL, Mao R, Wittwer CT. Masking selected sequence variation by incorporating mismatches into melting analysis probes. Hum Mutat. 2006;27:269–278. doi: 10.1002/humu.20290. [DOI] [PubMed] [Google Scholar]
- 41.Germer S, Higuchi R. Single-tube genotyping without oligonucleotide probes. Genome Res. 1999;9:72–78. [PMC free article] [PubMed] [Google Scholar]
- 42.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 43.Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, Wittwer C. Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem. 2004;50:1156–1164. doi: 10.1373/clinchem.2004.032136. [DOI] [PubMed] [Google Scholar]
- 44.Liew M, Nelson L, Margraf R, Mitchell S, Erali M, Mao R, Lyon E, Wittwer C. Genotyping of human platelet antigens 1 to 6 and 15 by high-resolution amplicon melting and conventional hybridization probes. J Mol Diagn. 2006;8:97–104. doi: 10.2353/jmoldx.2006.050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liew M, Seipp M, Durtschi J, Margraf RL, Dames S, Erali M, Voelkerding K, Wittwer C. Closed-tube SNP genotyping without labeled probes/a comparison between unlabeled probe and amplicon melting. Am J Clin Pathol. 2007;127:341–348. doi: 10.1309/N7RARXH3623AVKDV. [DOI] [PubMed] [Google Scholar]
- 46.Gundry CN, Dobrowolski SF, Martin YR, Robbins TC, Nay LM, Boyd N, Coyne T, Wall MD, Wittwer CT, Teng DH. Base-pair neutral homozygotes can be discriminated by calibrated high-resolution melting of small amplicons. Nucleic Acids Res. 2008;36:3401–3408. doi: 10.1093/nar/gkn204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palais RA, Liew MA, Wittwer CT. Quantitative heteroduplex analysis for single nucleotide polymorphism genotyping. Anal Biochem. 2005;346:167–175. doi: 10.1016/j.ab.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 48.Herrmann MG, Durtschi JD, Bromley LK, Wittwer CT, Voelkerding KV. Amplicon DNA melting analysis for mutation scanning and genotyping: cross-platform comparison of instruments and dyes. Clin Chem. 2006;52:494–503. doi: 10.1373/clinchem.2005.063438. [DOI] [PubMed] [Google Scholar]
- 49.von Ahsen N, Oellerich M, Schutz E. Limitations of genotyping based on amplicon melting temperature. Clin Chem. 2001;47:1331–1332. [PubMed] [Google Scholar]
- 50.Pont-Kingdon G, Lyon E. Direct molecular haplotyping by melting curve analysis of hybridization probes: beta 2-adrenergic receptor haplotypes as an example. Nucleic Acids Res. 2005;33:e89. doi: 10.1093/nar/gni090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pont-Kingdon G, Margraf RL, Sumner K, Millson A, Lyon E, Schutz E. Design and application of noncontinuously binding probes used for haplotyping and genotyping. Clin Chem. 2008;54:990–999. doi: 10.1373/clinchem.2007.100487. [DOI] [PubMed] [Google Scholar]
- 52.Montgomery J, Wittwer CT, Kent JO, Zhou L. Scanning the cystic fibrosis transmembrane conductance regulator gene using high-resolution DNA melting analysis. Clin Chem. 2007;53:1891–1898. doi: 10.1373/clinchem.2007.092361. [DOI] [PubMed] [Google Scholar]
- 53.Takano T, Ohe Y, Tsuta K, Fukui T, Sakamoto H, Yoshida T, Tateishi U, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Matsuno Y, Furuta K, Tamura T. Epidermal growth factor receptor mutation detection using high-resolution melting analysis predicts outcomes in patients with advanced non small cell lung cancer treated with gefitinib. Clin Cancer Res. 2007;13:5385–5390. doi: 10.1158/1078-0432.CCR-07-0627. [DOI] [PubMed] [Google Scholar]
- 54.Simi L, Pratesi N, Vignoli M, Sestini R, Cianchi F, Valanzano R, Nobili S, Mini E, Pazzagli M, Orlando C. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 2008;130:247–253. doi: 10.1309/LWDY1AXHXUULNVHQ. [DOI] [PubMed] [Google Scholar]
- 55.Holden JA, Willmore-Payne C, Coppola D, Garrett CR, Layfield LJ. High-resolution melting amplicon analysis as a method to detect c-kit and platelet-derived growth factor receptor alpha activating mutations in gastrointestinal stromal tumors. Am J Clin Pathol. 2007;128:230–238. doi: 10.1309/7TEH56K6WWXENNQY. [DOI] [PubMed] [Google Scholar]
- 56.Willmore C, Holden JA, Zhou L, Tripp S, Wittwer CT, Layfield LJ. Detection of c-kit-activating mutations in gastrointestinal stromal tumors by high-resolution amplicon melting analysis. Am J Clin Pathol. 2004;122:206–216. doi: 10.1309/4E6U-YBY6-2N2F-CA6N. [DOI] [PubMed] [Google Scholar]
- 57.Willmore-Payne C, Holden JA, Tripp S, Layfield LJ. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol. 2005;36:486–493. doi: 10.1016/j.humpath.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 58.Bastien R, Lewis TB, Hawkes JE, Quackenbush JF, Robbins TC, Palazzo J, Perou CM, Bernard PS. High-throughput amplicon scanning of the TP53 gene in breast cancer using high-resolution fluorescent melting curve analyses and automatic mutation calling. Hum Mutat. 2008;29:757–764. doi: 10.1002/humu.20726. [DOI] [PubMed] [Google Scholar]
- 59.Farrar JS, Reed GH, Wittwer CT. High resolution melting curve analysis for molecular diagnostics. In: Patrinos GP, Ansorge W, editors. Molecular Diagnostics. ed 2. Elsevier; Burlington: 2009. (in press) [Google Scholar]
- 60.De Juan I, Esteban E, Palanca S, Barragan E, Bolufer P: High-resolution melting analysis for rapid screening of BRCA1 and BRCA2 Spanish mutations. Breast Cancer Res Treat doi 10.1007/s10549-008-0073-7 [DOI] [PubMed]
- 61.De Leeneer K, Coene I, Poppe B, De Paepe A, Claes K. Rapid and sensitive detection of BRCA1/2 mutations in a diagnostic setting: comparison of two high-resolution melting platforms. Clin Chem. 2008;54:982–989. doi: 10.1373/clinchem.2007.098764. [DOI] [PubMed] [Google Scholar]
- 62.Audrezet MP, Dabricot A, Le Marechal C, Ferec C. Validation of high-resolution DNA melting analysis for mutation scanning of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. J Mol Diagn. 2008;10:424–434. doi: 10.2353/jmoldx.2008.080056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laurie AD, Smith MP, George PM. Detection of factor VIII gene mutations by high-resolution melting analysis. Clin Chem. 2007;53:2211–2214. doi: 10.1373/clinchem.2007.093781. [DOI] [PubMed] [Google Scholar]
- 64.Lin SY, Su YN, Hung CC, Tsay W, Chiou SS, Chang CT, Ho HN, Lee CN. Mutation spectrum of 122 hemophilia A families from Taiwanese population by LD-PCR, DHPLC, multiplex PCR and evaluating the clinical application of HRM. BMC Med Genet. 2008;9:53. doi: 10.1186/1471-2350-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kennerson ML, Warburton T, Nelis E, Brewer M, Polly P, De Jonghe P, Timmerman V, Nicholson GA. Mutation scanning the GJB1 gene with high-resolution melting analysis: implications for mutation scanning of genes for Charcot-Marie-Tooth disease. Clin Chem. 2007;53:349–352. doi: 10.1373/clinchem.2006.080010. [DOI] [PubMed] [Google Scholar]
- 66.Montgomery J, Wittwer CT, Palais RA, Zhou L. Simultaneous mutation scanning and genotyping by high-resolution DNA melting analysis. Nat Prot. 2007;2:59–66. doi: 10.1038/nprot.2007.10. [DOI] [PubMed] [Google Scholar]
- 67.Bolufer P, Sanz GF, Barragan E, Sanz MA, Cervera J, Lerma E, Senent L, Moreno I, Planelles MD. Rapid quantitative detection of BCR-ABL transcripts in chronic myeloid leukemia patients by real-time reverse transcriptase polymerase-chain reaction using fluorescently labeled probes. Haematologica. 2000;85:1248–1254. [PubMed] [Google Scholar]
- 68.Kreuzer KA, Lass U, Bohn A, Landt O, Schmidt CA. LightCycler technology for the quantitation of bcr/abl fusion transcripts. Cancer Res. 1999;59:3171–3174. [PubMed] [Google Scholar]
- 69.Zhang T, Grenier S, Nwachukwu B, Wei C, Lipton JH, Kamel-Reid S. Inter-laboratory comparison of chronic myeloid leukemia minimal residual disease monitoring: summary and recommendations. J Mol Diagn. 2007;9:421–430. doi: 10.2353/jmoldx.2007.060134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mullins M, Perreard L, Quackenbush JF, Gauthier N, Bayer S, Ellis M, Parker J, Perou CM, Szabo A, Bernard PS. Agreement in breast cancer classification between microarray and quantitative reverse transcription PCR from fresh-frozen and formalin-fixed, paraffin-embedded tissues. Clin Chem. 2007;53:1273–1279. doi: 10.1373/clinchem.2006.083725. [DOI] [PubMed] [Google Scholar]
- 71.Worm J, Aggerholm A, Guldberg P. In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem. 2001;47:1183–1189. [PubMed] [Google Scholar]
- 72.Procter M, Chou LS, Tang W, Jama M, Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem. 2006;52:1276–1283. doi: 10.1373/clinchem.2006.067603. [DOI] [PubMed] [Google Scholar]
- 73.Cockerill FR., III Application of rapid-cycle real-time polymerase chain reaction for diagnostic testing in the clinical microbiology laboratory. Arch Pathol Lab Med. 2003;127:1112–1120. doi: 10.5858/2003-127-1112-AORRPC. [DOI] [PubMed] [Google Scholar]
- 74.Espy MJ, Uhl JR, Sloan LM, Buckwalter SP, Jones MF, Vetter EA, Yao JD, Wengenack NL, Rosenblatt JE, Cockerill FR, III, Smith TF. Real-time PCR in clinical microbiology: applications for routine laboratory testing. Clin Microbiol Rev. 2006;19:165–256. doi: 10.1128/CMR.19.1.165-256.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lachnik J, Ackermann B, Bohrssen A, Maass S, Diephaus C, Puncken A, Stermann M, Bange FC. Rapid-cycle PCR and fluorimetry for detection of mycobacteria. J Clin Microbiol. 2002;40:3364–3373. doi: 10.1128/JCM.40.9.3364-3373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sloan LM, Hopkins MK, Mitchell PS, Vetter EA, Rosenblatt JE, Harmsen WS, Cockerill FR, Patel R. Multiplex LightCycler PCR assay for detection and differentiation of Bordetella pertussis and Bordetella parapertussis in nasopharyngeal specimens. J Clin Microbiol. 2002;40:96–100. doi: 10.1128/JCM.40.1.96-100.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mancini N, Clerici D, Diotti R, Perotti M, Ghidoli N, De Marco D, Pizzorno B, Emrich T, Burioni R, Ciceri F, Clementi M. Molecular diagnosis of sepsis in neutropenic patients with haematological malignancies. J Med Microbiol. 2008;57:601–604. doi: 10.1099/jmm.0.47732-0. [DOI] [PubMed] [Google Scholar]
- 78.Vaughn CP, Lyon E, Samowitz WS. Confirmation of single exon deletions in MLH1 and MSH2 using quantitative polymerase chain reaction. J Mol Diagn. 2008;10:355–360. doi: 10.2353/jmoldx.2008.080021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schneider M, Joncourt F, Sanz J, von Kanel T, Gallati S. Detection of exon deletions within an entire gene (CFTR) by relative quantification on the LightCycler. Clin Chem. 2006;52:2005–2012. doi: 10.1373/clinchem.2005.065136. [DOI] [PubMed] [Google Scholar]
- 80.Lyon E, Millson A, Lowery MC, Woods R, Wittwer CT. Quantification of HER2/neu gene amplification by competitive PCR using fluorescent melting curve analysis. Clin Chem. 2001;47:844–851. [PubMed] [Google Scholar]
- 81.Millson A, Suli A, Hartung L, Kunitake S, Bennett A, Nordberg MC, Hanna W, Wittwer CT, Seth A, Lyon E. Comparison of two quantitative polymerase chain reaction methods for detecting HER2/neu amplification. J Mol Diagn. 2003;5:184–190. doi: 10.1016/S1525-1578(10)60471-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pont-Kingdon G, Lyon E. Rapid detection of aneuploidy (trisomy 21) by allele quantification combined with melting curves analysis of single-nucleotide polymorphism loci. Clin Chem. 2003;49:1087–1094. doi: 10.1373/49.7.1087. [DOI] [PubMed] [Google Scholar]
- 83.Meuer S, Wittwer CT, Nakaguawara K. Rapid Cycle Real Time PCR—Methods and Applications. Springer; Berlin: 2001. [Google Scholar]
- 84.Wittwer C, Hahn M, Kaul K. Rapid Cycle Real Time PCR—Methods and Applications: Quantification. Springer; Berlin: 2004. [Google Scholar]
- 85.Dietmaier W, Wittwer C, Sivasubramanian N. Rapid Cycle Real Time PCR—Methods and Applications: Genetics and Oncology. Springer; Berlin: 2002. [Google Scholar]
- 86.Reischl U, Wittwer CT, Cockerill F. Rapid Cycle Real Time PCR—Methods and Applications: Microiology and Food Analysis. Springer; Berlin: 2002. [Google Scholar]