

Abstract
High-resolution melting (HRM) analysis is a novel tool for analysis of promoter methylation. The aim of the present study was to establish and validate HRM analysis for detection of promoter methylation on archival formalin-fixed paraffin-embedded tissues from colorectal cancer patients. We first evaluated HRM assays for O6-methylguanine-DNA methyltransferase (MGMT) and adenomatous polyposis coli (APC) promoter methylation on a methylated DNA dilution matrix and DNA extracted from eight fresh or formalin-fixed paraffin-embedded human cancer cell lines. Then we used these assays for the analysis of MGMT and APC promoter methylation in a subset of archival formalin-fixed paraffin-embedded colorectal tumor specimens. All samples with promoter methylation of MGMT or APC and randomly selected samples without promoter methylation were analyzed twice. All results generated by HRM were validated with MGMT and APC MethyLight assays. APC and MGMT promoter methylation data were consistent and reproducible throughout the dilutions and all three replicates in the methylated DNA dilution matrix and between two experiments in clinical samples. There was high concordance between HRM and MethyLight results. HRM for APC promoter methylation revealed consistent results between fresh and formalin-fixed paraffin-embedded human cancer cell line DNA. The methylation status in archival tumor specimens from patients with colorectal cancer can therefore be determined with high quality by HRM. The ability to analyze archival tissues greatly facilitates further research and its clinical implementation.
Promoter hypermethylation is one of the hallmarks of carcinogenesis associated with transcriptional silencing and loss of expression of genes encoding for diverse cellular pathways.1 Most of the evidence exists for tumor suppressor genes.2,3 DNA-methylation-based assays are promising tools for detection of biomarkers for early cancer diagnosis, risk assessment, and response to therapy.1,4 As a result, a variety of methods to detect aberrant DNA methylation in cancer patients have been developed.5
The most popular approaches rely on treatment of genomic DNA with sodium bisulfite,6 which converts cytosine into uracil while 5-methyl cytosine remains unmodified, thus allowing identification of cytosine methylation status following PCR amplification.5 Among the PCR-based methods, methylation-specific PCR is the most widely used technique for detection of methylation. This assay uses primers specific for methylated, bisulfite-modified DNA.7 One major drawback of methylation-specific PCR is that it provides only qualitative information regarding the methylation status of the analyzed sequence and, therefore, cannot distinguish low versus high levels of methylation. Quantitative measurement of methylation is important because low levels of methylation (below the threshold of transcriptional silencing) may not be biologically important.8,9 Also, quantification of promoter methylation may enable early detection of cancer and early metastatic spread.10
To enhance specificity and sensitivity of DNA-methylation based assays, alternative techniques have been developed and widely used in basic research. These include fluorescence-based real-time PCR analysis (MethyLight), as a quantitative and high throughput technology, and bisulfite sequencing of multiple clones. However, they are neither cost-effective nor fast enough to be implemented for routine clinical diagnosis.11
Thus, there is a clear need for development of a more reliable method for DNA methylation assays. A new approach, high-resolution melt curve analysis (HRM), has recently been reported, and is based on the “melting” properties of DNA in solution.12 Originally it was developed for single-nucleotide polymorphism genotyping.13 The principle of this method is that bisulfite-treated DNA templates with different contents of methylcytosine can be resolved by melting analysis due to differences in melting temperatures.14 HRM offers several advantages over the widely used MethyLight assay.12 The use of probes in MethyLight assays increases the costs of experiments. Also, quantitative MethyLight requires normalization against a reference gene, which needs to be run for each sample. HRM, by contrast, does not require expensive probes, and no reference gene for normalization, making the experiment relatively simple and cost-effective. Another important aspect is that HRM scans all of the CpGs flanked by the primers binding to the target sequence, regardless of the methylation status of CpGs in the primer-binding site, while MethyLight detects methylation of CpG sites covered by the primers and probes. This enables HRM to distinguish heterogeneous from homogeneous methylation by the shape of the melting curve. This factor is of importance because methylation patterns at promoter CpG islands are typically not homogeneous.15,16
Archival tissues represent an enormous source for testing of clinically important issues and most previously mentioned methods have been tested for their performance on formalin-fixed paraffin-embedded (FFPE) tissues. The aim of this study was to validate HRM for methylation status detection on archival FFPE tissues from colorectal cancer patients. All results generated by HRM were validated with MethyLight assays. As a proof of principle we demonstrated HRM using assays for O6-methylguanine-DNA methyltransferase (MGMT) and adenomatous polyposis coli (APC) promoters in a methylated DNA dilution matrix and DNA isolated from fresh and FFPE human cancer cell lines. In a second step, HRM assays were validated in a clinical setting using archival FFPE colorectal tumor specimens. To our knowledge this is the first report on the use of HRM for detection of the promoter methylation status on FFPE tissues.
Materials and Methods
Controls and Patient Samples
CpGenome Universal Methylated DNA (Chemicon, Millipore Billerica, MA, USA) was used as 100% methylated control DNA. DNA extracted from peripheral blood mononuclear cells of normal individuals was used as unmethylated control DNA. Methylation standards were constructed by diluting 100% methylated bisulfite-modified control DNA in a pool of normal bisulfite-modified DNA at ratios of 50%, 25%, 10%, 5%, 1%, and 0.1%. These standards were included in each experimental run.
In addition, the following eight human cancer cell lines were used for validation experiments: human breast cancer cell lines MCF-7, MDA-MB-231, SKBR3, T47D, and MDA-MB-453, and human prostate cancer cell lines DU145, LNCAP, and PC3. All cell lines were obtained from the American Tissue and Cell Collection (Manassas, VA, USA) and were cultured according to the supplier's instructions. We used cell lines obtained either directly from cultures or after formalin-fixation and paraffin-embedding, as adapted from the published protocol by Kerstens et al.17
Surgically resected tissues were collected from 66 colorectal carcinoma patients (48 male and 18 female) at the Surgical Department of the Danube Hospital, Vienna, Austria. Median age at surgery was 67 years (range, 41 to 87 years). The cancers were classified according to the UICC TNM guidelines.18 Seventeen patients were UICC I, Dukes A (25.8%), 11 patients were UICC II, Dukes B (16.7%), 20 patients were UICC III, Dukes C (30.3%), 12 patients were UICC IV, Dukes D (18.2%), and 6 patients (9.1%) had local relapses. Additionally, 9 normal tissue samples were analyzed. The study was approved by the local Ethics Committee.
Extraction of Genomic DNA
Healthy volunteers' peripheral blood mononuclear cell DNA and DNA from cultured cancer cell lines was isolated using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) according to the supplier's recommendation.
For patient tumor samples, an appropriate paraffin block containing tumor tissue was selected for analysis after reviewing the H&E-stained slides. An area of tumor on the H&E-stained slide was identified on a corresponding unstained slide and circled with an indelible fine-tipped pen. DNA was isolated from material scraped from the unstained slide as previously described.19 Genomic DNA from FFPE cancer cell lines was isolated using the same protocol as for patient tumor samples. DNA quantity was assessed spectrophotometrically and quality of genomic DNA was confirmed by agarose gel electrophoresis.
Sodium Bisulfite Modification
One μg of genomic DNA was subjected to bisulfite conversion with the EpiTect Bisulfite kit (Qiagen, Hilden, Germany) according to the manufacturer's instruction. The eluted DNA (40 μl volume) was used for the HRM and MethyLight analysis.
MethyLight Assay
The MethyLight assays for MGMT and APC have been described previously.11,20,21 Briefly, PCR was performed on the LightCycler 480 (Roche Applied Science, Mannheim, Germany) in a 20-μl volume containing: 1 × LightCycler 480 Probes Master (Roche Applied Science), 500 nmol/L of each primer, 200 nmol/L of probe, and 50 ng bisulfite-treated DNA. Each reaction was performed in triplicate. The cycling conditions were as follows: mono color hydrolysis probes detection format, 1 cycle of 95°C for 10 minutes, 45 cycles of 95°C for 10 seconds, 60°C for 30 seconds, and 72° for 1 second. COL2A1 was used to normalize for the amount of input DNA.
HRM Analysis
PCR amplification and HRM were performed on the LC 480 (Roche Applied Science) as adapted from the published protocol by Wojdacz and Dobrovic.12 The primers were designed as outlined by Wojdacz and Dobrovic. They included not more than 1 to 2 CpG sites and were placed at or adjacent to the 5′ end. For MGMT published primer sequences were used12 and sequences for APC (GenBank Accession U02509) were as following: APC Fw 5′- AAGTAGTTGTGTAATTCGTTGGAT-3′ and APC Rv 5′-CACCTCCATTCTATCTCCAATA-3′ (149 bp).
PCR was performed in a 20-μl volume containing: 1 × LightCycler 480 High-Resolution Melting Master mix (Roche Applied Science), 200 nmol/L of each primer for MGMT and 500 nmol/L for APC, and 50 ng bisulfite treated DNA template, with 3 mmol/L final MgCl2 for MGMT and 4 mmol/L final MgCl2 for APC. Each reaction was performed in triplicate. The cycling conditions were as follows: SYBR Green 1 detection format; 1 cycle of 95°C for 10 minutes, 50 cycles of 95°C for 10 seconds, a touch down of 64°C to 58°C for 10 seconds (1°C/cycle), and 72°C for 20 seconds; followed by an HRM step of 95°C for 1 minute, 40°C for 1 minute, 74°C for 5 seconds, and continuous acquisition to 90°C at 25 acquisitions per 1°C. Each plate included multiple water blanks and peripheral blood mononuclear cell DNA from healthy individuals was used as a negative control. A standard curve with known methylation ratios was included in each assay and the resulting equation was used to deduce the methylation ratio of each tumor sample. The resulting relative signal values (%) reflect the proportion of nonmethylated alleles and methylated alleles.
HRM data were analyzed using the Gene Scanning Software (Roche Applied Science). The melting curves are normalized by calculation of two normalization regions before and after the major fluorescence decrease representing the melting of the PCR product. This algorithm allows the direct comparison of the samples that have different starting fluorescence levels. Output plots are in the form of normalized temperature-shifted melting curves that show the decrease in fluorescence against increasing temperature.
Results
DNA Dilution Matrix
To test the sensitivity of the HRM assays, we used the standard dilution series as described in the Materials and Methods section. Both HRM assays (APC and MGMT) were able to detect reproducibly 1% methylated DNA in a background of unmethylated DNA (Figure 1, A and B). 1% of methylation was reliably and reproducibly detected also by MethyLight (data not shown).
Figure 1.
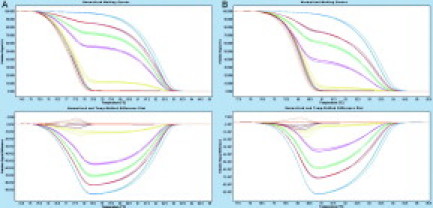
Sensitivity of HRM assays for MGMT (A) and APC (B) methylation. Assays were run at annealing temperatures ranging from 64°C to 58°C using a touchdown protocol. Data were analyzed using software modules “Gene Scanning” and “Difference Plot.” Methylation standards are displayed as triplicates. Standards 100% blue lines, 50% red lines, 25% green lines, 10% pink lines, 1% yellow lines, 0.1% brown lines, and 0% orange lines.
To test interassay variability of the HRM assay, we compared the methylation results obtained from exactly the same set of bisulfite-converted methylation standards tested as four independent assays on four different days. Figure 2A shows the corresponding standard curve for APC including the SD values. The mean correlation coefficient (R2) of the four standard curves was 0.9984 (range 0.9971 to 0.9991; SD 0.0009). These data show small and reproducible run-to-run HRM variations. We also tested if bisulfite treatments performed on different days influenced HRM results. Therefore, we compared methylation standards prepared from four separate bisulfite treatments analyzed in the same HRM assays. Figure 2B illustrates the resulting standard curve including the SD values. The mean correlation coefficient (R2) of the four standard curves was 0.9982 (range 0.9963 to 0.9994; SD 0.0014). These data indicate good reproducibility in standards of different bisulfite treatments. For MGMT, both bisulfite-to-bisulfite variation and run-to-run variation gave comparable results to APC (data not shown).
Figure 2.
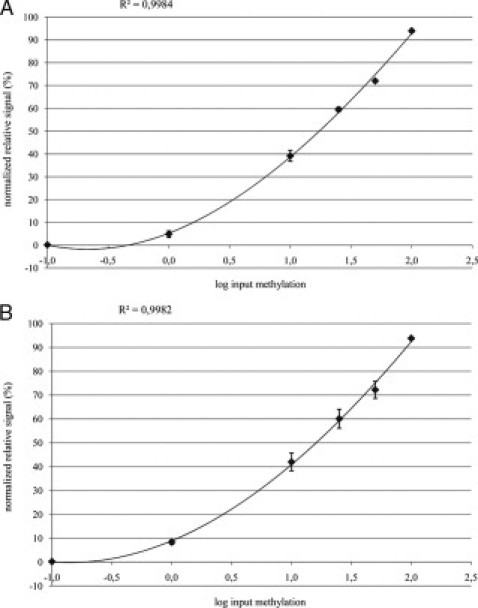
Reproducibility analysis of the APC HRM assay. A: Interassay variability for the APC HRM assay. The same set of bisulfite-converted methylation standards were subjected to four independent HRM assays performed on different days. B: Influence of bisulfite treatment on methylation results. The bisulfite treatment was performed on four different days and bisulfite-treated methylation standards were subjected to the same APC HRM assay.
Cancer Cell Lines
DNA extracted directly from cultured and FFPE cell lines was used to evaluate the impact of formalin-fixation and paraffin-embedding on detection of promoter hypermethylation. Further, to test the reproducibility of the HRM assays on FFPE DNA, we performed six independent experiments. We could demonstrate that both HRM and MethyLight assays can reliably detect promoter hypermethylation in FFPE cell line DNA. Figure 3A demonstrates the concordance of the methylation status detected by the APC HRM assay between fresh and FFPE cell line DNA. In Figure 3B consistent results are shown for the APC MethyLight assay. Figure 3A also shows similar reproducibility of the APC HRM for fresh and FFPE cell line DNA with a mean SD of ± 5.0 (range 0.0 to 11.9) for fresh and ± 5.1 (range 0.1 to 14.7) for FFPE DNA.
Figure 3.
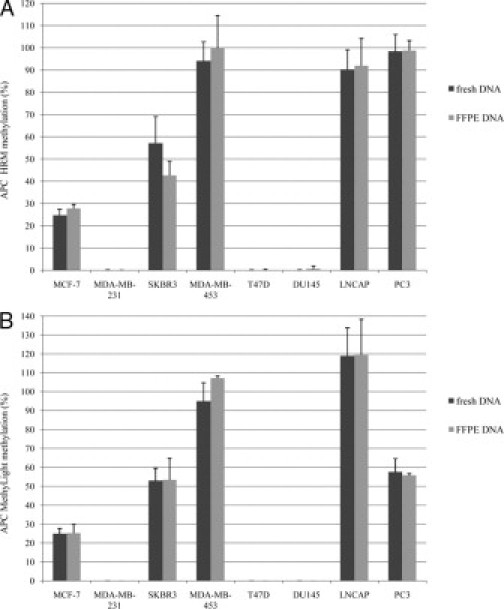
Interassay variability for the APC HRM assay (A) and MethyLight assay (B) using fresh and FFPE cancer cell line DNA from eight breast and prostate cancer cell lines. The bars represent the mean of six independent experiments and the SD.
Results for MGMT were comparable (data not shown).
Analysis of FFPE Specimens
We applied HRM for MGMT and APC promoter methylation to analyze 66 archival FFPE colorectal tumor specimens. In 28 (42.4%) out of 66 cases, MGMT methylation was identified. Methylation of the APC promoter was detected in 22 (33.3%) out of 66 colorectal cancer samples. For MGMT the mean methylation level was 28.1 (range 6.0 to 67.5) and for APC the mean methylation level was 28.1 (range 5.2 to 82.1). Nine normal archival tissue samples analyzed showed no methylation for both the MGMT and APC promoter. Figure 4 represents APC HRM results of four representative samples. In addition, MGMT and APC promoter methylation was analyzed by MethyLight in all 66 samples. There was a high concordance between HRM and MethyLight results. Table 1 shows the overall concordance for MGMT of 91% and APC of 98.5%. MGMT promoter methylation was detected in six cases by HRM, where the methylation was not detected by MethyLight. The APC promoter methylation was detected in one case by HRM, but not with MethyLight. The melting curves of those discrepant cases indicated that there was low level of methylation detected by HRM.
Figure 4.
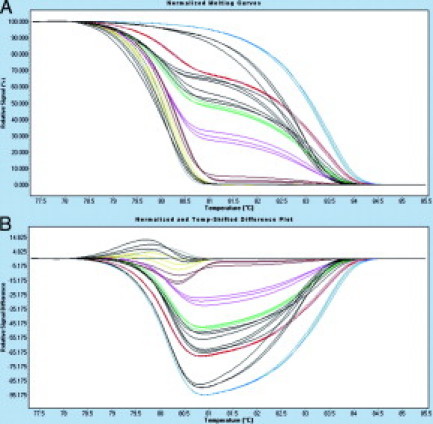
A:APC HRM curves for methylation standards containing varying amounts of methylated DNA (colored lines) and four samples (three methylated and one unmethylated) represented by black lines. B: Difference plot for the data represented in (A). Melting curves were normalized to the 0% standard. Methylation standards and samples are displayed as triplicates. Standards 100% blue lines, 50% red lines, 25% green lines, 10% pink lines, 1% yellow lines, 0.1% brown lines, and 0% orange lines.
Table 1.
Methylation Detection of 66 FFPE Colorectal Tissue Samples Using HRM versus MethyLight
|
MGMT MethyLight |
APC MethyLight |
|||
|---|---|---|---|---|
| UM | M | UM | M | |
| HRM | ||||
| UM | 38 | 0 | 43 | 0 |
| M | 6 | 22 | 1 | 22 |
The analysis of all cases was repeated twice by HRM and randomly selected cases were analyzed twice by MethyLight. This included bisulfite treatment and subsequent methylation assay, to exclude false positives due to incomplete bisulfite conversion. HRM and MethyLight results between these two different experiments were reproducible for all samples.
We also tested whether varying amounts of bisulfite-treated target DNA influences HRM results. We found no differences in methylation ratios when using two different DNA amounts (200 ng versus 50 ng).
Discussion
FFPE tissue samples are the largest source of clinical material from normal controls and diseased tissues, and their use is of inestimable research and clinical value. We demonstrate here for the first time, the utility of quantitative HRM for promoter methylation of two distinct markers on FFPE colorectal tissues. The design of this experiment was based on the recently reported work by Wojdacz and Dobrovic.12 Their study was among the first to demonstrate the applicability of HRM for promoter methylation analysis.12,22 Both White et al22 and Wojdacz and Dobrovic have demonstrated the advantages of quantitative HRM for assessment of methylation over the currently used methods, mainly methylation-specific PCR and its quantitative adaptation MethyLight. In summary, the advantages are scanning of all CpGs included in the primer sequence, determination of the heterogeneity of the methylation by the shape of the curves, independence from expensive probes used for MethyLight and no need for reference assay.12 The study by Wojdacz and Dobrovic was performed using cell lines and patient samples. Description of the clinical samples analyzed in their study is lacking some information and it can only be assumed that fresh frozen samples were used. As stated above, the validation of HRM for analysis of FFPE tumor tissue is important and we show here that the same assay quality as shown by Wojdacz and Dobrovic can be maintained in the analysis of FFPE tissues.
To keep the same assay quality throughout the analysis of FFPE tissues, we have first reproduced the data published by Wojdacz and Dobrovic, using CpGenome Universal Methylated DNA diluted in unmethylated DNA from peripheral blood mononuclear cells of normal controls. As we show here, we were able to reproducibly detect promoter methylation of both MGMT and APC when 1% of methylated DNA was mixed with unmethylated DNA (Figure 1). We have achieved better reproducibility of results when using the touchdown protocol. It has been shown previously that touchdown PCR improves the amplification efficiency for methylated DNA.23 Importantly, we show here that our data were reproducible through three distinct replicates even in the mix with the low amount of methylated DNA. For certain applications, eg, detection of rare events, the sensitivity can be enhanced through the elevation of the annealing temperature, as already described by Wojdacz and Dobrovic.12 In contrast, when low-level methylation is detected in tumor tissues, the biological significance is still not established. This remains to be studied, independent of the method used for detection of methylation.
According to the study performed by Ogino et al21 we have further tested the precision and reproducibility of the bisulfite treatment, as well as interassay variability of the HRM assay. Our data demonstrate that our results were stable and reproducible. The analysis of the reproducibility of methylation analysis in FFPE tissues is restricted by the lack of models reliably reflecting the archival tissue sampling. First we verified the results by replicate analysis of tissue samples and there was no inconsistence. In an attempt to evaluate the influence of formalin fixation and paraffin embedding on methylation analysis, we demonstrated that the reproducibility of methylation results was comparable between fresh and FFPE cell line DNA.
Methylation of APC and the MGMT promoter region in colorectal cancer tissue has been known and documented in several independent studies.24,25,26,27,28 Most of the studies have used the methylation-specific PCR for detection of the promoter methylation. Some quantitative approaches have been used as well, like COBRA or MethyLight.12,26 Wojdacz and Dobrovic have validated the HRM based detection of MGMT promoter methylation in comparison with the MethyLight based detection.12 Even though the direct comparison of these two methods is restricted due to technical differences, particularly in clinical samples, they achieved at least the same detection sensitivity using HRM based assays, when compared with MethyLight for analysis of promoter methylation in dilution models. In addition to technical differences, the heterogeneity of analyzed cohorts makes the comparison of results with published methylation rates for both APC and MGMT promoter regions in colorectal cancers difficult. However, the quality of data generated in our study can be indirectly compared with the quality of data obtained in the study performed by Wojdacz and Dobrovic. When analyzing DNA extracted from FFPE tissue, there is a risk of lower analysis quality since DNA is fragmented, a fact that may influence both bisulfite treatment and further PCR.21 Our data show that normalized melting curves and the corresponding difference plot from both FFPE colorectal cancer tissue and methylated DNA dilution matrix demonstrate the same quality, comparable to melting curve shapes previously shown by Wojdacz and Dobrovic.12 To underpin the value of HRM based methylation analysis we concomitantly performed the analysis by MethyLight. The low number of inconsistent results was likely due to the low level of methylation detected by HRM. The biological significance of such low level methylation, however, was not the scope of the present study.
The quantification of methylation in the tissue obtained from clinical specimens, regardless of the type of stored tissue (ie, fresh frozen or FFPE), is difficult to interpret. One reason is that not much is known about the heterogeneity of the methylation within tumor tissue, including intracellular and intercellular heterogeneity. Another reason is that most of the data are generated, as in our case, by microdissection after H&E staining. Some proportion of normal tissue mixed with tumor tissue cannot be avoided, and it varies from case to case and from study to study, even when performed very precisely, making the standardization difficult. In the study performed by Wojdacz and Dobrovic it is not clear which way the tumor tissue has been verified and if microdissection has been performed.12 We also show here in several normal controls that no promoter methylation could be determined in the promoter regions of MGMT or APC in normal controls.
There are several applications for the use of the present technique on FFPE tissues. Identification of the methylation profiles in primary tumors that are associated with metastasis would not only elucidate those epigenetic events involved with disease progression but may aid in the development of a genomic prediction marker panel for patient outcome that can readily be assessed from paraffin-embedded tissue specimens.29,30 Thus, one application may be the risk stratification of patients based on methylation status of specific markers. For such an application the cutoff of 1%, as used in our study, is sufficient for discrimination. The same assay can be adapted and used to detect low amounts of methylated cells within the tumor, or even to detect low numbers of tumor cells in the background of non-tumor cells in lymph nodes and other organs. Data indicates that lymph node metastasis is a critical benchmark in cancer disease and is often the earliest sign of tumor progression.31,32 Once the protocol has been adapted for such an application, as mentioned above, even the lower cut off levels might be used for selection of patients with low tumor burden.
Our report adds substantial information on the HRM-based DNA methylation analysis and demonstrates its applicability for analysis of FFPE tissues. Such validations are of high importance to demonstrate robustness of the assay, facilitating its establishment as a research tool and possibly a clinical test.
Acknowledgements
We thank Gerhard Mühlbauer (Roche Applied Science, Vienna) for the excellent technical assistance.
Footnotes
Supported by the Austrian National Bank Fund (grant 12680 to N.D.).
References
- 1.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 2.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 3.Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;(14 Spec No 1):R65–R76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 4.Cote RJ, Laird PW, Datar RH. Promoter hypermethylation: a new therapeutic target emerges in urothelial cancer. J Clin Oncol. 2005;23:2879–2881. doi: 10.1200/JCO.2005.11.923. [DOI] [PubMed] [Google Scholar]
- 5.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 6.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cameron EE, Baylin SB, Herman JG. p15(INK4B) CpG island methylation in primary acute leukemia is heterogeneous and suggests density as a critical factor for transcriptional silencing. Blood. 1999;94:2445–2451. [PubMed] [Google Scholar]
- 9.Hsieh CL. Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994;14:5487–5494. doi: 10.1128/mcb.14.8.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taback B, Giuliano AE, Lai R, Hansen N, Singer FR, Pantel K, Hoon DS. Epigenetic analysis of body fluids and tumor tissues: application of a comprehensive molecular assessment for early-stage breast cancer patients. Ann NY Acad Sci. 2006;1075:211–221. doi: 10.1196/annals.1368.029. [DOI] [PubMed] [Google Scholar]
- 11.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wojdacz TK, Dobrovic A. Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007;35(6):e41. doi: 10.1093/nar/gkm013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wittwer CT, Reed GH, Gundry CN, Vandersteen JG, Pryor RJ. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853–860. doi: 10.1373/49.6.853. [DOI] [PubMed] [Google Scholar]
- 14.Dahl C, Guldberg P. High-resolution melting for accurate assessment of DNA methylation. Clin Chem. 2007;53:1877–1878. doi: 10.1373/clinchem.2007.094854. [DOI] [PubMed] [Google Scholar]
- 15.Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de La Chapelle A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci USA. 2001;98:591–596. doi: 10.1073/pnas.011528698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione-S-transferase gene (GSTP1) in prostate cancer cells. Oncogene. 2002;21:1048–1061. doi: 10.1038/sj.onc.1205153. [DOI] [PubMed] [Google Scholar]
- 17.Kerstens HM, Robben JC, Poddighe PJ, Melchers WJ, Boonstra H, de Wilde PC, Macville MV, Hanselaar AG. AgarCyto: a novel cell-processing method for multiple molecular diagnostic analyses of the uterine cervix. J Histochem Cytochem. 2000;48:709–718. doi: 10.1177/002215540004800515. [DOI] [PubMed] [Google Scholar]
- 18.International Union Against Cancer . TNM Classification of Malignant Tumors. ed 5. Springer; New York: 1997. pp. 75–80. [Google Scholar]
- 19.Wu L, Patten N, Yamashiro CT, Chui B. Extraction and amplification of DNA from formalin-fixed, paraffin-embedded tissues. Appl Immunohistochem Mol Morphol. 2002;10:269–274. doi: 10.1097/00129039-200209000-00015. [DOI] [PubMed] [Google Scholar]
- 20.Widschwendter M, Siegmund KD, Muller HM, Fiegl H, Marth C, Muller-Holzner E, Jones PA, Laird PW. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004;64:3807–3813. doi: 10.1158/0008-5472.CAN-03-3852. [DOI] [PubMed] [Google Scholar]
- 21.Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn. 2006;8:209–217. doi: 10.2353/jmoldx.2006.050135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White HE, Hall VJ, Cross NC. Methylation-sensitive high-resolution melting-curve analysis of the SNRPN gene as a diagnostic screen for Prader-Willi and Angelman syndromes. Clin Chem. 2007;53:1960–1962. doi: 10.1373/clinchem.2007.093351. [DOI] [PubMed] [Google Scholar]
- 23.Shen L, Guo Y, Chen X, Ahmed S, Issa JP. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. Biotechniques. 2007;42:48. doi: 10.2144/000112312. 50, 52 passim. [DOI] [PubMed] [Google Scholar]
- 24.Arnold CN, Goel A, Niedzwiecki D, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR. APC promoter hypermethylation contributes to the loss of APC expression in colorectal cancers with allelic loss on 5q. Cancer Biol Ther. 2004;3:960–964. doi: 10.4161/cbt.3.10.1113. [DOI] [PubMed] [Google Scholar]
- 25.Prall F, Weirich V, Ostwald C. Phenotypes of invasion in sporadic colorectal carcinomas related to aberrations of the adenomatous polyposis coli (APC) gene. Histopathology. 2007;50:318–330. doi: 10.1111/j.1365-2559.2007.02609.x. [DOI] [PubMed] [Google Scholar]
- 26.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, Buckmeier J, Alberts DS, Hamilton SR, Issa JP. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 27.Suehiro Y, Wong CW, Chirieac LR, Kondo Y, Shen L, Webb CR, Chan YW, Chan AS, Chan TL, Wu TT, Rashid A, Hamanaka Y, Hinoda Y, Shannon RL, Wang X, Morris J, Issa JP, Yuen ST, Leung SY, Hamilton SR. Epigenetic-genetic interactions in the APC/WNT. RAS/RAF, and P53 pathways in colorectal carcinoma. Clin Cancer Res. 2008;14:2560–2569. doi: 10.1158/1078-0432.CCR-07-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto K, Shimizu Y, Suehiro Y, Okayama N, Hashimoto S, Okada T, Hiura M, Ueno K, Hazama S, Higaki S, Hamanaka Y, Oka M, Sakaida I, Hinoda Y. Hypermethylation status of APC inversely correlates with the presence of submucosal invasion in laterally spreading colorectal tumors. Mol Carcinog. 2008;47:1–8. doi: 10.1002/mc.20363. [DOI] [PubMed] [Google Scholar]
- 29.Bastian PJ, Ellinger J, Wellmann A, Wernert N, Heukamp LC, Muller SC, von Ruecker A. Diagnostic and prognostic information in prostate cancer with the help of a small set of hypermethylated gene loci. Clin Cancer Res. 2005;11:4097–4106. doi: 10.1158/1078-0432.CCR-04-1832. [DOI] [PubMed] [Google Scholar]
- 30.Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, Glockner S, Piantadosi S, Gabrielson E, Pridham G, Pelosky K, Belinsky SA, Yang SC, Baylin SB, Herman JG. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med. 2008;358:1118–1128. doi: 10.1056/NEJMoa0706550. [DOI] [PubMed] [Google Scholar]
- 31.Cote RJ, Peterson HF, Chaiwun B, Gelber RD, Goldhirsch A, Castiglione-Gertsch M, Gusterson B, Neville AM. Role of immunohistochemical detection of lymph-node metastases in management of breast cancer. International Breast Cancer Study Group. Lancet. 1999;354:896–900. doi: 10.1016/s0140-6736(98)11104-2. [DOI] [PubMed] [Google Scholar]
- 32.Pagliarulo V, Hawes D, Brands FH, Groshen S, Cai J, Stein JP, Lieskovsky G, Skinner DG, Cote RJ. Detection of occult lymph node metastases in locally advanced node-negative prostate cancer. J Clin Oncol. 2006;24:2735–2742. doi: 10.1200/JCO.2005.05.4767. [DOI] [PubMed] [Google Scholar]