

Abstract
In monogenic disorders, the functional evaluation of rare, unclassified variants helps to assess their pathogenic relevance and can improve differential diagnosis and predictive testing. We characterized six rare APC variants in patients with familial adenomatous polyposis at the mRNA level. APC variants c.531 + 5G>C and c.532-8G>A in intron 4, c.1409-2_1409delAGG in intron 10, c.1548G>A in exon 11, and a large duplication of exons 10 and 11 result in a premature stop codon attributable to aberrant transcripts whereas the variant c.1742A>G leads to the in-frame deletion of exon 13 and results in the removal of a functional motif. Mutation c.1548G>A was detected in the index patient but not in his affected father, suggesting mutational mosaicism. A literature review shows that most of the rare APC variants detected by routine diagnostics and further analyzed at the transcript level were evaluated as pathogenic. The majority of rare APC variants, particularly those located close to exon-intron boundaries, could be classified as pathogenic because of aberrant splicing. Our study shows that the characterization of rare variants at the mRNA level is crucial for the evaluation of pathogenicity and underlying mutational mechanisms, and could lead to better treatment modalities.
Determining the pathogenicity of a germline mutation in an inherited disease is crucial for diagnostics and counseling and is particularly relevant for predictive testing in persons at risk. Generally, nonsense and frameshift mutations, large exonic deletions, and mutations in the conserved splice sequences in a gene are considered as pathogenic. In contrast, the pathological significance of rare variants predicted to result in amino acid substitutions (missense mutations), small in-frame deletions, or even same-sense mutations (silent variants) is often unclear. Similarly, the functional effect of DNA variants at less conserved positions in introns is difficult to predict. As a consequence, a growing number of single bp substitutions identified in routine diagnostics are denoted as unclassified variants (UVs) or variants of uncertain significance, respectively.
Based on functional analysis, it has become evident in recent years that a substantial proportion of so-called UVs, particularly those located close to exon-intron boundaries, affect splicing because of the disruption of putative regulatory elements such as exonic splicing enhancer and silencer motifs, or composite exonic regulatory elements of splicing.1,2,3,4,5,6,7,8 Aberrant splicing as the underlying mechanism of presumed missense or silent variants has been detected in a number of genes involved in cancer predisposition such as MLH1, MSH2, BRCA1, BRCA2, RB1, NF1, and ATM.9,10,11,12,13,14,15,16,17
Familial adenomatous polyposis (FAP) (MIM no. 175100) is an autosomal-dominant precancerous condition characterized by the appearance of numerous colorectal adenomas, which, if not detected early and removed, invariably result in colorectal cancer. In classic FAP, patients develop hundreds to thousands of adenomatous polyps during the second decade of life.18 The mild phenotype (attenuated FAP) is etiologically heterogeneous and poorly defined; usually the presence of less than 100 colorectal adenomas and an advanced age at onset of both polyposis and colorectal cancer are used as diagnostic criteria.19,20,21,22
FAP is caused by germline mutations in the tumor suppressor gene APC on chromosome 5.21,23 The gene consists of 15 exons, exon 15 encompassing approximately three-quarters of the coding sequence. To date, more than 900 different APC germline mutations have been identified in FAP patients [see Human Gene Mutation Database (HGMD, www.hgmd.org) and references therein]. The vast majority of mutations identified in FAP patients are predicted to result in truncated proteins because of either nonsense or frameshift mutations, or lead to exon skipping because of mutations in the highly conserved splice sequences.24,25,26,27 In addition, large genomic deletions were found in ∼7 to 12% of FAP patients.28,29,30 Biallelic mutations in the base excision repair gene MUTYH (MIM no. 604933) contribute to a subset of APC mutation-negative patients.31,32,33
To date, only a few APC missense or silent mutations in the coding sequence and unique variants in less-conserved intronic sequences close to the splice site have been reported in FAP families. The vast majority have been found in exons 1 to 14 and the adjacent intronic sequences. The functional relevance of these substitutions is difficult to evaluate; only a small number were characterized by mRNA or segregation studies. A silent substitution in exon 14 (c.1869G>T;p.Arg623) has been reported to induce exon skipping because of changes in exonic splice enhancer sites.34 In a previous study, we characterized five apparent missense or silent mutations and five rare variants in less conserved intronic sequences at mRNA level and have demonstrated that all but one of these variants lead to exon skipping and may consequently be classified as pathogenic.35 Here, we present results of mRNA analysis on another six APC variants, five of which are novel, including a large genomic duplication, and provide a summary of all APC variants characterized at the RNA level published to date.
Materials and Methods
Patients
Since 1991, blood samples from 1431 apparently unrelated patients with the clinical diagnosis of either classic or attenuated FAP have been referred to the Institute of Human Genetics, University of Bonn, for mutation analysis in the APC and/or MUTYH gene. If a rare APC variant was identified during routine mutation screening in the absence of a concurrent pathogenic APC or biallelic MUTYH mutation, we obtained fresh blood samples for mRNA analysis from each of the patients who were available and gave informed consent. Clinical information on polyposis disease in the patients and their families was obtained during genetic counseling sessions and from medical records.
Rare APC Variants
A genetic alteration of the APC gene was considered as rare variant if it i) represented a single-base substitution in the coding sequence or at a less-conserved intronic splice-site position (+/−∼20 bp), ii) was observed only a few times (one to three times) in the whole sample of 1431 patients, and iii) did not occur together with a clearly pathogenic APC or biallelic MUTYH mutation.
Detection of Germline Mutations on Genomic DNA
Genomic DNA was extracted from peripheral ethylenediaminetetraacetic acid-anticoagulated blood samples according to the standard salting-out procedure. Screening for APC germline point mutations was performed by examination of genomic DNA using the protein truncation test for exon 15 and denaturing high pressure liquid chromatography (WAVE, Transgenomic Glasgow, United Kingdom) for exons 1 to 14 and the first 500 bp of exon 15 as described.35 Polymerase chain reaction (PCR) fragments showing aberrant patterns by either method were sequenced on an ABI 3100 automated sequencer (Applied Biosystems, Darmstadt, Germany) using the cycle-sequencing procedure and the BigDye terminator kit version 1.1 (Applied Biosystems). The cDNA bases were numbered according to the APC reference sequence in GenBank NM_000038.2, where +1 corresponds to the A of the ATG translation initiation codon.
Screening for large genomic deletions or duplications was performed using MLPA (multiplex ligation-dependent probe amplification). The MLPA test kit (SALSA P043 APC exon deletion test kit; MRC Holland, Amsterdam, The Netherlands) contains 23 paired probes from the APC region to examine three fragments of the promoter region, exons 1 to 14, and five fragments of exon 15 (including the two hotspot mutations at codon 1061 and 1309), in addition to 11 control probes from other chromosomal regions. Screening for large deletions or duplications was performed according to the manufacturer's protocol. Data were analyzed by use of GeneMapper, version 4.0 software (Applied Biosystems) and gene dosage was calculated with the Coffalyser V4 program (MRC Holland).
APC Transcript Analysis
Fresh venous blood samples (2.5 ml) were collected into PAXgene blood RNA tubes (Becton Dickinson, Heidelberg, Germany) containing RNA stabilizing solution. Total RNA was extracted by use of the PAXgene blood RNA kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. First strand cDNA was synthesized from 2 to 3 μg of total RNA by random hexamer-primed reverse transcription with the SuperScript first strand system for reverse transcriptase (RT)-PCR (Invitrogen GmbH, Karlsruhe, Germany) according to the manufacturer's protocol. RT-PCR fragments were obtained according to standard PCR protocols by use of different primers to generate the appropriate fragments. RT-PCR products were separated on 2% agarose gel and visualized with ethidium bromide. Gels containing different RT-PCR fragments were examined on an UV imaging system (Bio-Rad, Hercules, CA). Individual bands were excised from the gel and eluted by use of the High Pure PCR product purification kit (Roche Diagnostics GmbH, Mannheim, Germany). Eluted DNA was reamplified with the same pairs of primers and sequenced as described above.
Calculation of Splicing Efficiencies
Splicing efficiencies in the normal and mutant sequences were calculated by use of the splice prediction program of the Berkeley Drosophila Genome Project.
Results and Discussion
In the context of molecular APC diagnostics in 1431 unrelated FAP patients, we found pathogenic APC mutations in 784 families and biallelic MUTYH mutations in another 101 families (mutation detection rate 62%). In the remaining 546 mutation-negative polyposis patients we successively identified 11 different rare heterozygous single-base substitutions in exons 1 to 14 (predicted as missense or silent mutations), 15 different rare heterozygous UVs at less-conserved intronic splice-site positions, and two large duplications. In our previous study, five of the exonic and five of the intronic UVs could be characterized at the transcript level.35 In the meantime, another six patients carrying a heterozygous rare variant agreed to mRNA analysis (Table 1).
Table 1.
Characterization of the Six Pathogenic APC Variants
| FAP no. | APC mutation | Exon/intron | Predicted effect | Influence on normal splice efficiency (BDGP) | RNA processing | Phenotype |
|---|---|---|---|---|---|---|
| 1159* | c.531 + 5G>C | Intron 4 | 0.98 to 0.25 | Deletion of exon 4, premature stop codon | Attenuated | |
| 1398 | c.532 − 8G>A | Intron 4 | 0.45 to <0.01 new SA site: 0.98 | Aberrant transcript, premature stop codon | Classic | |
| 1476 | c.1409-2_1409 delAGG | Intron 10 | Destruction of SA site | Activation of two cryptic SA sites | Two aberrant transcripts, premature stop codon | Attenuated |
| 0005 | c.1548G>A | Exon 11 | p.Lys516 | 1.00 to 0.84 | Deletion of exon 11, premature stop codon | Classic |
| 1172 | c.1742A>G | Exon 13 | p.Lys581Arg | 0.92 to 0.78 | Deletion of exon 13 | Attenuated |
| 1199 | dup exon 10 to 11 | Exon 10 to 11 | Duplication of exon 10 to 11, premature stop codon | Classic |
SA, splice acceptor site; BDGP, Berkeley Drosophila Genome Project (splice prediction program).
Also reported by Moisio et al.36
Variant c.531 + 5G>C in Intron 4
The intronic variant c.531 + 5G>C in a less-conserved region close to the splice donor site of exon 4 (Figure 1A) was identified in a patient diagnosed with FAP at 47 years of age (FAP 1159). According to the splice prediction program the G>C substitution results in a reduction of splicing efficiency from 0.98 to 0.25.
Figure 1.
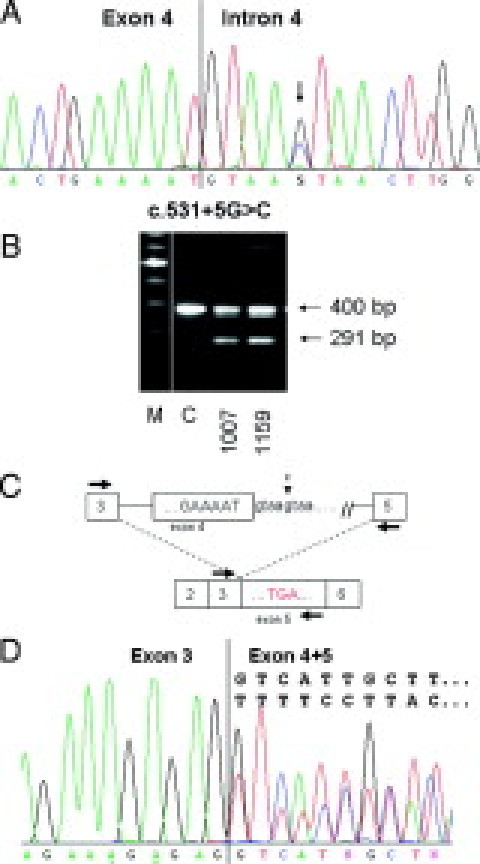
Characterization of variant c.531 + 5G>C in intron 4 of the APC gene (patient 1159). A: Sequencing pattern of genomic DNA reveals the heterozygous substitution G>C. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 3F and in exon 5R in patient 1159, in patient 1007 with the mutation c.531 + 1G>A, and a control (C). C: Diagram representing the mutation in genomic DNA (top) and the resulting aberrant splicing and premature stop codon analyzed by RT-PCR (bottom; primers indicated as arrows; boxes with numbers denote individual exons; the mutation and surrounding sequences are indicated). D: Sequencing pattern of the entire RT-PCR product showing the heterozygous deletion of exon 4.
mRNA analysis demonstrated that the variant leads to skipping of exon 4 of the APC gene: examination of the RT-PCR product obtained with primers localized in exon 3 (forward) and 5 (reverse) on agarose gel revealed, apart from the normal fragment of 400 bp, an additional fragment of ∼300 bp (Figure 1, B and C). Sequencing of the whole RT-PCR product showed a heterozygous deletion of the entire exon 4, resulting in a fragment of 291 bp (Figure 1D). Thus, the mutation was designated as: c.531 + 5G>C;r.423_531del; p.Arg144SerfsX8.
As expected, sequencing of the excised and reamplified full-length fragment revealed the normal fragment including exon 4. However, it still contains a minor fraction of the exon 4-deleted product, probably attributable to some heteroduplex formation between the full length and deleted sequence. Sequencing of the short gel fragment clearly demonstrated the complete lack of exon 4 beside a slight contamination with an additional sequence that does not derive from the APC gene but rather seems to result from reamplification of unspecific PCR products (data not shown).
The intensity of the 291-bp fragment in relation to the normal 400-bp fragment was comparable to that observed for mutations at highly conserved positions +1 or −1 of splice sites, or for mutation c.423G>T at the first position of exon 4. For the latter variant, we demonstrated that exon 4 was almost completely deleted in the mutant allele.35 Based on this comparison we concluded that the variant c.531 + 5G>C led to an (almost) complete deletion of exon 4 and hence can be considered as pathogenic. These findings are consistent with a previous report in which the mutation was proven to affect splicing in a family with an attenuated phenotype.36
Substitution c.532-8G>A in Intron 4
The intronic variant c.532-8G>A in a nonconserved region close to the splice acceptor site of exon 5 (Figure 2A) was detected in a patient diagnosed with classic FAP at age 20 (FAP 1398). The family encompasses eight affected persons in three generations. The affected sister of the index patient also carried the variant. According to the splice prediction program this G>A substitution introduced a new AG sequence just before the normal splice acceptor site at a splicing efficiency of 0.98, whereas the efficiency of the normal splice acceptor site was reduced from 0.45 to <0.01. The new splice site was predicted to result in the inclusion of six intronic nucleotides in the mRNA and a premature stop codon because of the in-frame TAG sequence in the three last included nucleotides.
Figure 2.
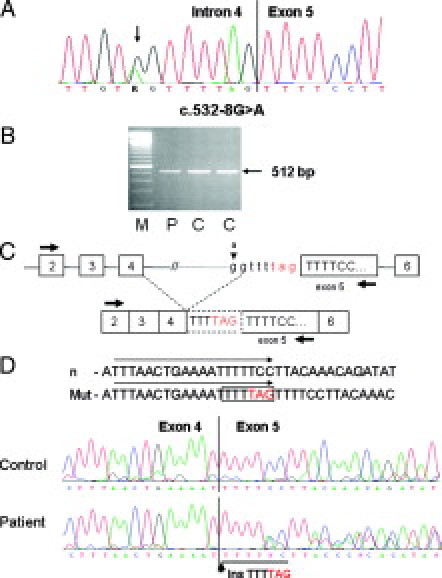
Characterization of the variant c.532-8G>A in intron 4 of the APC gene (patient 1398). A: The sequencing pattern of genomic DNA reveals the heterozygous substitution G>A. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 2F and in exon 5R in the patient (P) and in two control samples (C). C: Diagram representing the mutation in genomic DNA and the aberrant splicing analyzed by RT-PCR (primers indicated as arrows) resulting in a premature stop codon (TAG). D: Sequencing pattern of the RT-PCR product showing the heterozygous insertion of six intronic nucleotides in the patient's sample. The sequences of the normal and mutant forward primer are marked by arrows.
The RT-PCR product obtained with primers localized in exon 2 (forward) and exon 5 or 7 (reverse) revealed no additional fragment on agarose gel (Figure 2B and 2C). However, sequencing of the entire RT-PCR product demonstrated that the predicted aberrant splice product was in fact present at the mRNA level (Figure 2D). The mutation should thus be designated as: c.532-8G>A;r.531_532insTTTTAG;p.Ser179X. The mutation led to complete aberrant splicing as shown by use of the polymorphic site c.1458T/C in exon 11 of the APC gene: reverse sequencing of RT-PCR products obtained with either a normal or a mutant forward primer localized at the mutation site and a reverse primer in exon 13 exclusively demonstrated the T-allele in the normal sequence and the C-allele in the mutant sequence (not shown).
Variant c.1409-2_1409delAGG in the Splice Acceptor Site of Exon 11
The variant c.1409-2_1409delAGG was detected in a patient who was diagnosed with rectal carcinoma and adenomatous polyposis at age 68 (FAP 1476). This variant can be regarded as pathogenic per se because it destroys the normal splice acceptor site of exon 11 (Figure 3A). Instead of the regular splice acceptor site, activation of two cryptic splice acceptor sites was indicated by the splice prediction program: one was localized in exon 11 leading to loss of the first 11 nucleotides. The second splice acceptor site was localized in intron 10 leading to an insertion of 34 intronic nucleotides.
Figure 3.
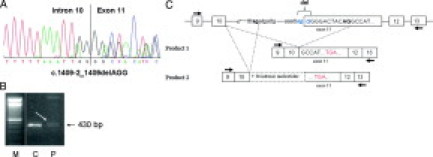
Characterization of variant c.1409-2_1409delAGG (patient 1476). A: Sequencing pattern of genomic DNA reveals the mutation in the splice acceptor site of exon 11. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 9F and in exon 13R in patient 1476 (P) and in a control (C), indicated by a white arrow. C: Diagram representing the mutation on genomic DNA and the two aberrant transcripts detected by RT-PCR leading to premature stop codons. Arrows indicate primer positions.
On agarose gel, RT-PCR products obtained with primers localized in exon 9 (forward) and 13 (reverse) showed a faint diffuse smear close to the normal fragment of 430 bp (Figure 3, B). Both aberrant transcripts (r.1409_1419del and r.1409-36_1409-3ins;1409delG) were barely detected, apart from the normal transcript, by sequencing the entire RT-PCR product (not shown).
Allele-specific RT-PCR with two forward primers designed to specifically amplify the two expected mutant transcripts demonstrated the presence of the two splice products. Reverse sequencing of the RT-PCR products confirmed the 11-bp deletion in the r.1409_1419del product, whereas the sequence of the fragment obtained by use of the second mutation-specific primer was not analyzable (not shown).
“Silent” Mutation c.1548G>A in Exon 11
According to the splice prediction program (the Berkeley Drosophila Genome Project) this apparently silent substitution c.1548G>A;p.Lys516 at the last position of exon 11 (FAP 5) (Figure 4A) resulted in a moderate reduction of splicing efficiency, from 1.00 to 0.84. However, mRNA analysis demonstrated that the variant leads to complete skipping of exon 11 of the APC gene (Figure 4C and 4E): examination of the RT-PCR product obtained with primers localized in exon 9 (forward) and 13 (reverse) on agarose gel revealed, apart from the normal fragment of ∼400 bp (corresponding to the expected 430 bp), an additional fragment of ∼300 bp (Figure 4B and 4E). Sequencing of the 430-bp fragment showed at nucleotide position 1548 only the normal G allele (Figure 4C). Moreover, the patient was heterozygous (C/T) at the polymorphic site C/T at nucleotide position 1458 (codon 486) on genomic DNA, but only allele C was present in the RT-PCR fragment of 430 bp. In the short fragment, the entire exon 11 was missing, resulting in a 290-bp product. Thus, substitution c.1548G>A did not result in the predicted silent variant p.Lys516 but rather in a deletion of exon 11 and a premature stop codon in exon 12. Hence, this mutation can be classified as clearly pathogenic. The correct nomenclature of the mutation is: c.1548G>A;r.1409_1548del;p.Gly471TyrfsX19. This mutation was detected in a patient with classic FAP (FAP no. 5) in accordance with the known genotype-phenotype correlation. He was diagnosed with rectal bleeding at the age of 20 years. Approximately 140 adenomas were removed in three sessions of rectoscopy. The patient underwent total proctocolectomy with ileo pouch anal anostomosis at age 21.
Figure 4.
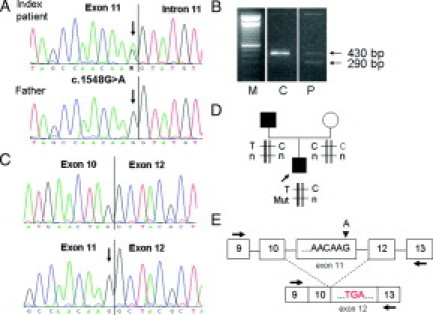
Characterization of variant c.1548G>A in exon 11 of the APC gene (patient 5). A: Sequencing pattern of genomic DNA reveals the heterozygous substitution G>A, arrows, in the index patient but not in his affected father. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 9F and in exon 13R (P) and a control (C). C: Sequencing pattern of the 290-bp and 430-bp fragments excised from the gel showing the deletion of exon 11 in the short fragment and the complete lack of the mutant allele in the full-length fragment. Arrow indicates the position of the mutation. D: Haplotype analysis in family 5 shows that the mutation in the index patient occurred in the paternal haplotype. T, C, the two alleles of the SNP at nucleotide position 1458 (codon 486); Mut, mutation (G>A, arrow) at nucleotide position 1548; and n, normal sequence (G). E: Schematic diagram representing the mutation in genomic DNA and the aberrant splicing variant detected by RT-PCR leading to a premature stop codon. Arrows indicate primer positions, arrowhead indicates location of the G>A mutation.
Interestingly, the mutation was not present in the peripheral blood sample of the patient's father, who had been diagnosed at the age of 45 years with a rather attenuated FAP with ∼90 adenomas being removed during the following 4 years. Now, at age 68, the father still has not undergone colectomy but has regular colonoscopies with several adenomas removed in every session. The patient's healthy mother does not carry the variant either. Thus, it is likely that the mutation was present as a somatic mosaic in the patient's father and that it did not occur de novo in the index patient. This hypothesis is supported by results of linkage analysis with the intragenic SNP at codon 486 in exon 11 (Figure 4D). Sequencing of the normal fragment showed only allele C. Hence, the mutation must have occurred in the paternal allele with T at nucleotide position 1458.
“Missense” Mutation c.1742A>G in Exon 13 of the APC Gene
The single base substitution c.1742A>G (FAP 1172) localized at the second last position of exon 13 (Figure 5A) was predicted to change the triplet AAG (lysine) in codon 581 into AGG (arginine). According to the splice prediction program (the Berkeley Drosophila Genome Project), this substitution reduces the splicing efficiency from 0.92 to 0.78. Examination of the RT-PCR product obtained with a primer localized in exon 11 (forward) and in exon 15A (reverse) revealed a shorter fragment of ∼500 bp at similar intensity as the normal fragment of ∼600 bp (corresponding to the expected 618 bp) (Figure 5B and 5C). Sequencing of the small fragment revealed a loss of the entire exon 13, resulting in a 501-bp product. Sequencing of the large fragment showed at nucleotide position 1742 predominantly the normal allele A and only a very small amount of the mutant allele G (Figure 5D). Thus, loss of exon 13 occurred most likely in the mutant allele.
Figure 5.
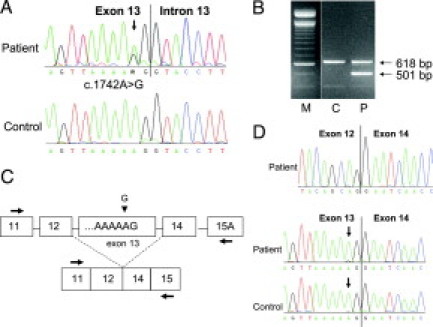
Characterization of variant c.1742A>G in exon 13 of the APC gene (patient 1172). A: Sequencing pattern of genomic DNA reveals the heterozygous substitution A>G. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 11F and in exon 15A-R in the patient (P) and a control (C). C: Diagram representing the normal and aberrant transcript. Arrows indicate primer positions. D: Sequencing pattern of the 501-bp and 618-bp fragments excised from the gel showing the deletion of exon 13 in the short fragment (above) and the (almost) complete lack of the mutant allele (arrow) in the full-length fragment.
These results demonstrate that variant 1742A>G does not result in the predicted amino acid change (p.Lys581Arg) but rather in reduction of the splicing efficiency of the splice donor site of exon 13 and in an almost complete deletion of exon 13 at mRNA level. Loss of exon 13 is not associated with a change in the reading frame. The variant is thus designated as c.1742A>G;r.1627_1743del;p.543_581del. The mutation can be considered as pathogenic because it results in removal of a complete heptad repeat motif from the N-terminal region of APC.21
Mutation c.1742A>G was detected in a patient (FAP no. 1172) with fairly attenuated FAP. He was diagnosed with multiple adenomas in the entire colorectum at age 34. The attenuated phenotype might be explained by the fact that a small proportion of the mutant allele was maintained in the full-length transcript, preserving a residual activity. Moreover, the transcript with the in-frame deletion of exon 13 may also maintain some residual function.
Large Genomic Duplication of Exons 10 to 11
Although large APC deletions encompassing one exon up to the entire gene are frequently detected in FAP,28,29,37 to date, only one duplication of exon 4 has been published and characterized at the mRNA level.38 We have identified a large genomic duplication by MLPA in two apparently unrelated patients with a classic FAP phenotype (FAP nos. 1199 and 1487). The patients carried a duplication of exons 10, 10a, and 11 (Figure 6A). In family 1199 the duplication was transmitted to both affected children of the index patient.
Figure 6.
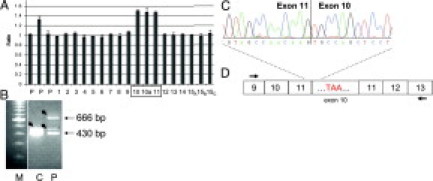
Characterization of the duplication of exons 10 to 11 of the APC gene (patient 1199) detected by MLPA. A: Normalized peak areas showing the duplication of exons 10, 10a, and 11. B: Agarose gel showing the RT-PCR product obtained with primers localized in exon 9F and 13R in the patient (P) and a control (C). The faint bands above the main bands represent the products containing the alternatively spliced exon 10a (small arrows). C: Partial sequence showing the junction of exon 11 and exon 10 in the duplicated fragment (666 bp) excised from the agarose gel (without exon 10a). D: Diagram representing the order of the exons and the resulting premature stop codon in the duplicated exon 10. Arrows indicate primer positions.
From one of these patients, mRNA was available. By RT-PCR with primers localized in exon 9 (forward) and exon 13 (reverse), we demonstrated that the duplicated exons were expressed at normal intensity (Figure 6B). Sequencing of the larger RT-PCR product extracted from agarose gel showed that exons 10 and 11 were correctly spliced and duplicated in the order of the exons: 9-10-11-10-11-12 (Figure 6C and 6D). The alternatively spliced exon 10a was not detected in the duplicated RT-PCR product. This duplication was predicted to result in a frameshift leading to a premature stop codon within the first part of the duplicated exon 10. The designation of this mutation is: c.1313-?_1549+ ?dup;r.1313_1549dup;p.Ala517CysfsX16.
Conclusions
Identification of the genetic basis in patients with adenomatous polyposis is required for differential diagnosis between FAP and MUTYH-associated polyposis or even hereditary non-polyposis colorectal cancer in some families and is particularly important for predictive testing in persons at risk. To date, ∼40 different rare missense or silent APC mutations have been published or listed in mutation databases (HGMD and references therein).39 However, only a minority was characterized at mRNA level or by other functional assays.17,34,35,36,40,41,42,43,44
Although criteria for the assessment of the pathogenicity of UVs have been established (de novo appearance, change of amino acid polarity or size, occurrence in an evolutionarily conserved sequence across species, absence in healthy controls, co-segregation with the phenotype in pedigrees, LOH or loss of protein expression in tumor tissue),45,46 these features are too uncertain to allow sufficient prediction of functional consequences. The prediction power of in silico programs such as the Berkeley Drosophila Genome Project or ESEfinder is limited. Although they may provide hints for possible effects on splicing, the prediction may differ from that demonstrated in vivo by examination of mRNA.10,12,35
We analyzed six rare APC variants at the RNA level and could demonstrate aberrant transcripts in all cases. Five of the six variants result in premature stop codons because of an aberrant out-of-frame transcript, whereas the in-frame deletion of exon 13 is assumed to be pathogenic on the basis of missing functional motifs. Two patients (FAP nos. 1476 and 1172) presented with an unexpected mild (attenuated) phenotype according to the site of mutation. These phenotypes may be explained, at least in part, by incomplete aberrant splicing, undiscovered mosaicism, or residual function in case of the in-frame mutation. Our results obtained in family FAP 5 suggest somatic APC mosaicism, the mutation would not have been detected if we had performed mutation analysis in the affected father.
The high frequency of obvious pathogenic mutations among the examined variants, both in the present and our previous study, possibly indicates some kind of selection bias. However, because all available patients were contacted, this possibility seems to be of no great importance. Interestingly, our findings correspond with previous studies and in silico analyses. Table 2 summarizes the results of a literature review with respect to rare APC variants examined by functional analyses indicating a high frequency of pathogenic mutations among rare APC variants, the majority attributable to aberrant splicing.17,34,35,36,40,41,42,43,44 Although a publication bias cannot be excluded, these considerations are in line with the current analyses of systematic data on human genetic variation, which suggest that a considerable fraction of (de novo) rare missense mutations may have deleterious effects.47,48 In a recent comprehensive evaluation, Azzopardi and colleagues49 found rare (<2%) nonsynonymous APC mutations significantly overrepresented in mutation-negative FAP patients compared with carriers of pathogenic mutations and normal controls.
Table 2.
Summary of Published Rare APC Variants Characterized by Functional Analysis
| Variant | Site | Predicted effect | Method | Result | Interpretation | Reference |
|---|---|---|---|---|---|---|
| c.423-6del8ins13 | Intron 3 | PTT* (RNA-based) | Aberrant splicing intron 3 | Pathogenic | 40 | |
| c.423-5A>G | Intron 3 | Transcript analysis (mRNA) | Deletion of exon 4 | Pathogenic | 35 | |
| c.423G>T | Exon 4 | p.Arg141Ser | Transcript analysis (mRNA) | Deletion of exon 4 | Pathogenic | 35 |
| c.531 + 5G>C | Intron 4 | Transcript analysis (mRNA) | Deletion of exon 4 | Pathogenic | 36 | |
| c.531 + 5_531 + 8delGTAA | Intron 4 | Transcript analysis (mRNA) | Deletion of exon 4 | Pathogenic | 35 | |
| c.834G>C | Exon 7 | p.Gln278 | Transcript analysis (mRNA) | Deletion of 11 bp, premature stop codon | Pathogenic | 40 |
| c.835-17A>G | Intron 7 | Transcript analysis (mRNA) | Insertion of 16 bp, premature stop codon | Pathogenic | 41 | |
| c.835-7T>G | Intron 7 | Transcript analysis (mRNA) | Insertion of 6 bp, premature stop codon | Pathogenic | 40 | |
| c.1312 + 3A>G | Intron 9 | Transcript analysis (mRNA) | Deletion of exon 9 | Pathogenic | 35 | |
| c.1312 + 5G>A | Intron 9 | Transcript analysis (mRNA) | Deletion of exon 9 | Pathogenic | 35 | |
| c.1312 + 5G>T | Intron 9 | Transcript analysis (mRNA) | Deletion of exon 9 | Pathogenic | 42 | |
| c.1419G>A | Exon 11 | p.Gln473 | Transcript analysis (mRNA) | Normal transcript | Nonpathogenic | 41 |
| c.1869G>T | Exon 14 | p.Arg623 | Transcript analysis (mRNA) | Deletion of exon 14 | Pathogenic | 34 |
| c.1956C>T | Exon 14 | p.His652 | Transcript analysis (mRNA) | Deletion of exon 14 | Pathogenic | 35 |
| c.1957A>G | Exon 14 | p.Arg653Gly | Transcript analysis (mRNA) | Deletion of exon 14 | Pathogenic | 35 |
| c.1957A>C | Exon 14 | p.Arg653 | Transcript analysis (mRNA) | Deletion of exon 14 | Pathogenic | 35 |
| c.1958 + 3A>G | Intron 14 | Transcript analysis (mRNA) | Deletion of exon 14 | Pathogenic | 35 | |
| c.1959G>A | Exon 15A | p.Arg653 | Transcript analysis (mRNA) | Normal transcript | Nonpathogenic | 35 |
| c.3077A>G | Exon 15E/F | p.Asn1026Ser | ß-catenin binding analysis | Reduced affinity for ß-catenin | Pathogenic | 43 |
| c.3871C>G | Exon 15G | p.Gln1291Glu | PTT* | Truncation† | Pathogenic | 44 |
| c.7504G>A | Exon 15T | p.Gly2502Ser | Transcript analysis (mRNA) | Normal transcript | Nonpathogenic | 17 |
| c.7862C>G | Exon 15 | p.Ser2621Cys | Transcript analysis (mRNA) | Normal transcript | Nonpathogenic | 17 |
Protein truncation test.
The reason for the truncated product was not identified.
Despite these statistical observations the pathogenicity and mutational mechanism of a certain variant can only be predicted by functional assays. As soon as routine APC diagnostics will include extended intron sequencing, a greater variety of rare APC variants will be discovered. Classification of these variants as pathogenic or neutral will remain a challenge.
Acknowledgements
We thank the patients and their physicians for participating in the study.
Footnotes
Supported by the German Cancer Aid (grant 106244).
References
- 1.Gorlov IP, Gorlova OY, Frazier ML, Amos CI. Missense mutations in hMLH1 and hMSH2 are associated with exonic splicing enhancers. Am J Hum Genet. 2003;73:1157–1161. doi: 10.1086/378819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caputi M, Kendzior RJ, Jr, Beemon KL. A nonsense mutation in the fibrillin-1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer. Genes Dev. 2002;16:1754–1759. doi: 10.1101/gad.997502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pagani F, Buratti E, Stuani C, Baralle FE. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J Biol Chem. 2003;278:26580–26588. doi: 10.1074/jbc.M212813200. [DOI] [PubMed] [Google Scholar]
- 4.Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, Casals T, Baralle FE. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003;12:1111–1120. doi: 10.1093/hmg/ddg131. [DOI] [PubMed] [Google Scholar]
- 5.Pagani F, Stuani C, Zuccato E, Kornblihtt AR, Baralle FE. Promoter architecture modulates CFTR exon 9 skipping. J Biol Chem. 2003;278:1511–1517. doi: 10.1074/jbc.M209676200. [DOI] [PubMed] [Google Scholar]
- 6.Sironi M, Menozzi G, Riva L, Cagliani R, Comi GP, Bresolin N, Giorda R, Pozzoli U. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004;32:1783–1791. doi: 10.1093/nar/gkh341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 8.Fu XD. Towards a splicing code. Cell. 2004;119:736–738. doi: 10.1016/j.cell.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 9.Zhang K, Nowak I, Rushlow D, Gallie BL, Lohmann DR. Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Hum Mutat. 2008;29:475–484. doi: 10.1002/humu.20664. [DOI] [PubMed] [Google Scholar]
- 10.Auclair J, Busine MP, Navarro C, Ruano E, Montmain G, Desseigne F, Saurin JC, Lasset C, Bonadona V, Giraud S, Puisieux A, Wang Q. Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mutations on aberrant splicing. Hum Mutat. 2006;27:145–154. doi: 10.1002/humu.20280. [DOI] [PubMed] [Google Scholar]
- 11.Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, Sengteller M, Henn W, Buettner R, Propping P, Mangold E. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Genet. 2006;119:9–22. doi: 10.1007/s00439-005-0107-8. [DOI] [PubMed] [Google Scholar]
- 12.Campos B, Diez O, Domenech M, Baena M, Balmana J, Sanz J, Ramirez A, Alonso C, Baiget M. RNA analysis of eight BRCA1 and BRCA2 unclassified variants identified in breast/ovarian cancer families from Spain. Hum Mutat. 2003;22:337. doi: 10.1002/humu.9176. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Swaminathan S, Martin BK, Sharan SK. Aberrant splicing induced by missense mutations in BRCA1: clues from a humanized mouse model. Hum Mol Genet. 2003;12:2121–2131. doi: 10.1093/hmg/ddg222. [DOI] [PubMed] [Google Scholar]
- 14.Liu HX, Cartegni L, Zhang MQ, Krainer AR. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet. 2001;27:55–58. doi: 10.1038/83762. [DOI] [PubMed] [Google Scholar]
- 15.Ars E, Serra E, Garcia J, Kruyer H, Gaona A, Lazaro C, Estivill X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum Mol Genet. 2000;9:237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- 16.Teraoka SN, Telatar M, Becker-Catania S, Liang T, Onengut S, Tolun A, Chessa L, Sanal O, Bernatowska E, Gatti RA, Concannon P. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am J Hum Genet. 1999;64:1617–1631. doi: 10.1086/302418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharp A, Pichert G, Lucassen A, Eccles D. RNA analysis reveals splicing mutations and loss of expression defects in MLH1 and BRCA1. Hum Mutat. 2004;24:272. doi: 10.1002/humu.9267. [DOI] [PubMed] [Google Scholar]
- 18.Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 19.Knudsen AL, Bisgaard ML, Bulow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2:43–55. doi: 10.1023/a:1023286520725. [DOI] [PubMed] [Google Scholar]
- 20.Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet. 1998;62:1290–1301. doi: 10.1086/301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, Sargeant L, Krapcho K, Wolff E, Burt R, Hughes JP, Warrington J, McPherson J, Wasmuth J, Le Paslier D, Abderrahim H, Cohen D, Leppert M, White R. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 22.Spirio L, Olschwang S, Groden J, Robertson M, Samowitz W, Joslyn G, Gelbert L, Thliveris A, Carlson M, Otterud B, Lynch H, Watson P, Lynch P, Laurent-Puig P, Burt R, Hughes JP, Thomas G, Leppert M, White R. Alleles of the APC gene: an attenuated form of familial polyposis. Cell. 1993;75:951–957. doi: 10.1016/0092-8674(93)90538-2. [DOI] [PubMed] [Google Scholar]
- 23.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D, Finniear R, Markham A, Groffen J, Boguski MS, Altschul SF, Horii A, Ando H, Miyoshi Y, Miki Y, Nishisho I, Nakamura Y. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 24.van der Luijt RB, Khan PM, Vasen HF, Tops CM, van Leeuwen-Cornelisse IS, Wijnen JT, van der Klift HM, Plug RJ, Griffioen G, Fodde R. Molecular analysis of the APC gene in 105 Dutch kindreds with familial adenomatous polyposis: 67 germline mutations identified by DGGE, PTT, and Southern analysis. Hum Mutat. 1997;9:7–16. doi: 10.1002/(SICI)1098-1004(1997)9:1<7::AID-HUMU2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 25.Wallis YL, Morton DG, McKeown CM, Macdonald F. Molecular analysis of the APC gene in 205 families: extended genotype-phenotype correlations in FAP and evidence for the role of APC amino acid changes in colorectal cancer predisposition. J Med Genet. 1999;36:14–20. [PMC free article] [PubMed] [Google Scholar]
- 26.Friedl W, Lamberti C. Hereditäre Tumorerkrankungen. Springer-Verlag; Heidelberg: 2001. Familiäre adenomatöse polyposis; pp. 303–325. [Google Scholar]
- 27.Olschwang S, Laurent-Puig P, Groden J, White R, Thomas G. Germ-line mutations in the first 14 exons of the adenomatous polyposis coli (APC) gene. Am J Hum Genet. 1993;52:273–279. [PMC free article] [PubMed] [Google Scholar]
- 28.Aretz S, Stienen D, Uhlhaas S, Pagenstecher C, Mangold E, Caspari R, Propping P, Friedl W. Large submicroscopic genomic APC deletions are a common cause of typical familial adenomatous polyposis. J Med Genet. 2005;42:185–192. doi: 10.1136/jmg.2004.022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunyan DJ, Eccles DM, Sillibourne J, Wilkins E, Thomas NS, Shea-Simonds J, Duncan PJ, Curtis CE, Robinson DO, Harvey JF, Cross NC. Dosage analysis of cancer predisposition genes by multiplex ligation-dependent probe amplification. Br J Cancer. 2004;91:1155–1159. doi: 10.1038/sj.bjc.6602121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michils G, Tejpar S, Thoelen R, van Cutsem E, Vermeesch JR, Fryns JP, Legius E, Matthijs G. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum Mutat. 2005;25:125–134. doi: 10.1002/humu.20122. [DOI] [PubMed] [Google Scholar]
- 31.Aretz S, Uhlhaas S, Goergens H, Siberg K, Vogel M, Pagenstecher C, Mangold E, Caspari R, Propping P, Friedl W. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer. 2006;119:807–814. doi: 10.1002/ijc.21905. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen M, Franken PF, Reinards TH, Weiss MM, Wagner A, van der Klift H, Kloosterman S, Houwing-Duistermaat JJ, Aalfs CM, Ausems MG, Brocker-Vriends AH, Gomez Garcia EB, Hoogerbrugge N, Menko FH, Sijmons RH, Verhoef S, Kuipers EJ, Morreau H, Breuning MH, Tops CM, Wijnen JT, Vasen HF, Fodde R, Hes FJ. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP) J Med Genet. 2005;42:e54. doi: 10.1136/jmg.2005.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 34.Montera M, Piaggio F, Marchese C, Gismondi V, Stella A, Resta N, Varesco L, Guanti G, Mareni C. A silent mutation in exon 14 of the APC gene is associated with exon skipping in a FAP family. J Med Genet. 2001;38:863–867. doi: 10.1136/jmg.38.12.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aretz S, Uhlhaas S, Sun Y, Pagenstecher C, Mangold E, Caspari R, Möslein G, Schulmann K, Propping P, Friedl W. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24:370–380. doi: 10.1002/humu.20087. [DOI] [PubMed] [Google Scholar]
- 36.Moisio AL, Jarvinen H, Peltomaki P. Genetic and clinical characterisation of familial adenomatous polyposis: a population based study. Gut. 2002;50:845–850. doi: 10.1136/gut.50.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sieber OM, Lamlum H, Crabtree MD, Rowan AJ, Barclay E, Lipton L, Hodgson S, Thomas HJ, Neale K, Phillips RK, Farrington SM, Dunlop MG, Mueller HJ, Bisgaard ML, Bulow S, Fidalgo P, Albuquerque C, Scarano MI, Bodmer W, Tomlinson IP, Heinimann K. Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or “multiple” colorectal adenomas. Proc Natl Acad Sci USA. 2002;99:2954–2958. doi: 10.1073/pnas.042699199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCart A, Latchford A, Volikos E, Rowan A, Tomlinson I, Silver A. A novel exon duplication event leading to a truncating germ-line mutation of the APC gene in a familial adenomatous polyposis family. Fam Cancer. 2006;5:205–208. doi: 10.1007/s10689-006-7471-y. [DOI] [PubMed] [Google Scholar]
- 39.Heinimann K, Thompson A, Locher A, Furlanetto T, Bader E, Wolf A, Meier R, Walter K, Bauerfeind P, Marra G, Muller H, Foernzler D, Dobbie Z. Nontruncating APC germ-line mutations and mismatch repair deficiency play a minor role in APC mutation-negative polyposis. Cancer Res. 2001;61:7616–7622. [PubMed] [Google Scholar]
- 40.Kanter-Smoler G, Fritzell K, Rohlin A, Engwall Y, Hallberg B, Bergman A, Meuller J, Gronberg H, Karlsson P, Bjork J, Nordling M. Clinical characterization and the mutation spectrum in Swedish adenomatous polyposis families. BMC Med. 2008;6:10. doi: 10.1186/1741-7015-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pedemonte S, Sciallero S, Gismondi V, Stagnaro P, Biticchi R, Haeouaine A, Bonelli L, Nicolo G, Groden J, Bruzzi P, Aste H, Varesco L. Novel germline APC variants in patients with multiple adenomas. Genes Chromosomes Cancer. 1998;22:257–267. doi: 10.1002/(sici)1098-2264(199808)22:4<257::aid-gcc1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 42.Varesco L, Gismondi V, Presciuttini S, Groden J, Spirio L, Sala P, Rossetti C, De Benedetti L, Bafico A, Heouaine A, Grammatico P, Del Porto G, White R, Bertario L, Ferrara G. Mutation in a splice-donor site of the APC gene in a family with polyposis and late age of colonic cancer death. Hum Genet. 1994;93:281–286. doi: 10.1007/BF00212023. [DOI] [PubMed] [Google Scholar]
- 43.Menéndez M, Gonzalez S, Obrador-Hevia A, Dominguez A, Pujol MJ, Valls J, Canela N, Blanco I, Torres A, Pineda-Lucena A, Moreno V, Bachs O, Capella G. Functional characterization of the novel APC N1026S variant associated with attenuated familial adenomatous polyposis. Gastroenterology. 2008;134:56–64. doi: 10.1053/j.gastro.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 44.Gavert N, Yaron Y, Naiman T, Bercovich D, Rozen P, Shomrat R, Legum C, Orr-Urtreger A. Molecular analysis of the APC gene in 71 Israeli families: 17 novel mutations. Hum Mutat. 2002;19:664. doi: 10.1002/humu.9037. [DOI] [PubMed] [Google Scholar]
- 45.Ou J, Niessen RC, Lutzen A, Sijmons RH, Kleibeuker JH, de Wind N, Rasmussen LJ, Hofstra RM. Functional analysis helps to clarify the clinical importance of unclassified variants in DNA mismatch repair genes. Hum Mutat. 2007;28:1047–1054. doi: 10.1002/humu.20580. [DOI] [PubMed] [Google Scholar]
- 46.Cotton RG, Scriver CR. Proof of “disease causing” mutation. Hum Mutat. 1998;12:1–3. doi: 10.1002/(SICI)1098-1004(1998)12:1<1::AID-HUMU1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 47.Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, White TJ, Nielsen R, Clark AG, Bustamante CD. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000083. e1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Azzopardi D, Dallosso AR, Eliason K, Hendrickson BC, Jones N, Rawstorne E, Colley J, Moskvina V, Frye C, Sampson JR, Wenstrup R, Scholl T, Cheadle JP. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 2008;68:358–363. doi: 10.1158/0008-5472.CAN-07-5733. [DOI] [PubMed] [Google Scholar]