

Abstract
Background
Alcoholics are known to have more severe airway diseases of the lung, such as bronchitis. Little is known about why this phenomenon is observed. We hypothesized that alcohol may modulate Toll-like receptor 2 (TLR2), which regulates inflammation caused by gram-positive bacteria.
Methods
Airway epithelial cells (primary bronchial epithelial cells (NHBE) and 16HBE 14o-) were exposed to 0–100mM alcohol for 0–24 hours. Real time PCR was used to quantify TLR2 mRNA. Protein levels of TLR2 were determined using Western blots and fluorescence activated cell sorting (FACS) on cells exposed to 0, 50 and 100 mM alcohol. Finally, cells were ‘primed’ with alcohol, stimulated with a TLR2 agonist (peptidoglycan), and IL-8 release was measured.
Results
Alcohol, at biologically relevant concentrations (25–100 mM), caused a two to three-fold time- and concentration-dependent increase in TLR2 mRNA in NHBE and 16HBE 14o- cells. Western blots for TLR2 revealed a qualitative increase in TLR2 protein in cells exposed to 100mM alcohol. FACS showed that TLR2 was quantitatively increased on the surface of airway epithelial cells that were exposed to alcohol. Airway cells that were primed with alcohol produced nearly twice as much IL-8 in response to 40 ng of peptidoglycan than naive cells.
Conclusions
Alcohol upregulates TLR2 message and protein in the airway epithelium. This leads to exaggerated inflammation in response to environmental stimuli that would normally be well tolerated in airway epithelial cells. This may be a partial explanation of why alcoholics have more severe airway disease than non-alcoholics.
Keywords: TLR2, ethanol, pulmonary, inflammation
INTRODUCTION
Alcohol abuse is well known to have harmful effects on the lung. For example, heavy alcohol intake increases the risk of developing acute respiratory distress syndrome (Moss and Burnham, 2003), pneumonia (de Roux et al., 2006) and airway diseases such as bronchitis (Shaper, 1990). Understanding how alcohol affects the airway epithelium is particularly important, because it is the body’s first line of defense against inhaled microorganisms. Alcohol has previously been shown to impair mucocilliary clearance, an important aspect of innate immunity (Wyatt and Sisson, 2001). However, very little is known about how alcohol affects other aspects of innate immunity of the airway epithelium, such as Toll-like receptors.
Toll-like receptors are an essential component of the innate immunity of the airway epithelium. There are 10 Toll-like receptors; each recognizes a different pathogen associated molecular pattern. Toll-like receptor 2 (TLR2), which is present on the airway epithelium, recognizes peptidoglycan, a component of the gram-positive cell wall (Takeda and Akira, 2007). When peptidoglycan binds TLR2, it initiates a cascade of intracellular signaling, which includes activation of MAPK and NF-κB (Akira and Takeda, 2004). This activation ultimately leads to release of inflammatory cytokines, including interleukin 8 (IL-8). Although inflammation is necessary to remove the offending pathogen, over-exuberant inflammation can lead to unnecessary damage to the airway epithelium, making the lungs more vulnerable to infection.
To date, much of the study of how alcohol regulates Toll-like receptors has been in immune effector cells such as monocytes, macrophages and Kuffer cells (Yamashina et al., 2005). Alcohol has been shown to have varying effects on Toll-like receptors depending on the tissue studied and the method of alcohol exposure. For instance, alcohol has been shown to have no effect on TLR2 signaling in monocytes, except when co-stimulated with TLR2 and TLR4 agonists (Oak et al., 2006). In other models, alcohol was shown to downregulate TLR’s. For instance, acute alcohol gavage in mice was shown to decrease peritoneal macrophage cytokine production through downregulation of TLRs (Pruett et al., 2004). Intraperitoneal injection of alcohol followed by ex vivo stimulation by a TLR2 agonist resulted in downregulation of TLR2 in splenic macrophages (Goral and Kovacs, 2005). In contrast, in a murine model, striated muscle showed an increase in inflammatory cytokines in response to a TLR4 ligand after acute alcohol gavage (Frost et al., 2005).
Because the airway epithelium plays a very different role than immune effector cells, it is possible that TLR2 may be regulated in the airway in a very different way. For example, it would not be beneficial for the airway epithelium to overreact to small amounts of inhaled non-pathogenic bacteria that is present in our everyday environment. In contrast, if bacterial products were present in the bloodstream, it would be desirable for immune effector cells to mount an aggressive defense.
Because alcohol consumption is associated with an increase in bronchitis (Shaper, 1990), we hypothesized that alcohol would upregulate TLR2, priming the airway epithelium to produce an inflammatory response to an otherwise benign bacterial challenge. In this series of experiments, we determined that moderate concentrations of alcohol significantly upregulate TLR2 in airway epithelial cells. Importantly, we also demonstrated that this upregulation leads to increased IL-8 production and thus can have a functional impact on inflammation.
METHODS
Cell culture
Primary normal human bronchial epithelial cells (NHBE; Lonza Walkersville, MD: Lot numbers 2F1341, 2F1678, 4F1499) were grown in submerged culture using serum-free bronchial epithelial cell basal media (BEGM; Lonza), supplemented with BEGM SingleQuot supplements and growth factors (Lonza). The cells were cultured on plates coated with 1% vitrogen solution (Cohesion; Palo Alto, CA) Cells were maintained in a 37° incubator with 5% carbon dioxide. Cells were fed every 24–48 hours. Experiments were performed on cells that were passage 2–4. Morphology of the cells was carefully observed prior to experiments. Cells demonstrating a squamous morphology were not used. Cells passaged more than 4 times did not respond to alcohol stimulation.
The transformed human bronchial epithelial cell line, 16HBE 14o- 14o- (Cozens et al., 1994), was a gift from Dr. D. C. Gruenert of the Cardiovascular Research Institute of the University of California, San Francisco. The 16HBE 14o- cells were grown in Dulbecco’s Modified Eagle’s Media (DMEM,Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and Penicillin/Streptomycin. 16HBE 14o- were grown under the same conditions described for the NHBE.
Cell exposure to alcohol
Cells were exposed to alcohol by adding absolute ethanol (200 proof, Pharmco-AAPER, Shelbyville, KY) to their growth media at the concentrations and times described. Serum was not removed from the growth media when exposing the 16HBE 14o- to alcohol. This is because the cells did not tolerate removal of serum for the longer exposures (48 hours) required for Western blotting and fluorescence activated cell sorting (FACS) experiments.
RNA extraction
NHBE and 16HBE 14o- were grown to 60–70% confluency in 12-well tissue culture plates then exposed to alcohol at varying concentrations (0, 12.5, 25, 50 and 100mM) and time points (0, 2, 4, 6 and 24 hours). Cell monolayers were rinsed twice with Dubelco’s phosphate buffered saline (PBS; Sigma, St. Louis, MO), then removed from the plate using trypsin/EDTA (0.05%) and stored in RNA later (Applied Biosystems, Foster City, CA) until RNA extraction could be performed. RNA was extracted and genomic DNA was removed using the Magmax 96 kit (Applied Biosystems) according to the manufacturer’s instructions. Concentration and purity of the RNA was determined using the NanoDrop spectrophotometer (Nanodrop Technologies, Wilmington, DE). All RNA samples had an A260/A280 ratio of 1.9–2.0.
Real Time PCR
First strand cDNA was synthesized using 100ng of template RNA and the Taqman reverse transcription kit (Applied Biosystems). Reactions were prepared using the following cocktail: 1X TaqMan RT buffer, 5.5mM magnesium chloride, 500µM of each dNTP, 2.5µM random hexamers, 0.4 U/µl RNase inhibitor, and 1.25U/µl of MultiScribe reverse transcriptase. Samples were then incubated in a thermocycler at 25°C for 10min, then 48°C for 30 min, and then 95°C for 5 min. Real time PCR reactions were performed using 1X Taqman Master Mix (Applied Biosystems) and 1X human TLR2 primers and probes (Applied Biosystems) in 25 ul reactions. Ribosomal RNA was used as an endogenous control. PCR was performed using the ABI PRISM 7500 Sequence Detection System (Applied Biosystems). Reactions underwent the following thermocycling protocol: 50°C for 2 minutes, 95°C for 10 minutes, and then 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Each reaction was carried out in duplicate. For relative comparison of TLR2 to the ribosomal RNA endogenous control, we used the ΔΔCt method (Livak and Schmittgen, 2001).
Western blotting
16HBE 14o- cells were grown to approximately 50% confluency in 60mm dishes. They were exposed to 0, 50 and 100mM ETOH for 48 hours, with fresh growth media (DMEM with 10% FBS) with the same concentration of alcohol added at 24 hours. Cells were then lysed in a buffer containing 35 mM Tris-HCl (pH 7.4), 0.4 mM EGTA, 10 mM MgCl2, and 1 µg/ml each of leupeptin A, phenylmethylsulphonylfluoride, and aprotinin for 20 minutes on ice. The cells were then removed from the plate using a sterile cell lifter and were mechanically disrupted. Cellular debris was removed by centrifugation. Protein level in the supernatant was measured using spectrophotometry, and 50µg of protein was electrophoresed in 10% Tris-HCl polyacrilamide gels (Biorad, Hercules, CA). The blot was transferred to nitrocellulose at 10V for 45 minutes in a semi-dry transfer apparatus (Biorad). The blot was blocked in 5% Blotto at 4°C overnight, then probed with a 1:400 dilution of TLR2 (H-175) antibody (Santa Cruz; Santa Cruz, CA. Cat #10739) overnight. All antibody dilutions were made in 5% blotto. The blot was carefully washed in in PBS with 0.05% Tween-20 (PBS-Tween), PBS-tween and probed with a goat anti-rabbit Horse radish peroxidase (HRP) -conjugated secondary antibody (Jackson Immunoresearch; WestGrove, PA #111-035-003) at a 1:5000 dilution for 2 hours. The blot was carefully washed in PBS-Tween, developed using Supersignal West Pico Chemiluminescent substrate (Pierce; Rockford, IL) and exposed to X-ray film. Beta actin loading controls were performed using a mouse anti-human monoclonal anti-beta actin antibody (Sigma, #A5316) at a 1: 1000 dilution and a Rabbit anti-mouse HRP conjugated secondary antibody (Abcam, #ab6728) at a 1:2000 dilution.
Fluorescence activated cell sorting (FACS)
16HBE 14o- were grown to 50% confluency in 60mm dishes, then exposed to 0, 50, and 100 mM alcohol for 48 hours. The cells were then removed from the plate using trypsin and washed in PBS. For each condition, 1 million cells were incubated with mouse anti-human TLR2 (TL2.1 Abcam, Cambridge, MA: #ab-9100) at a 1:100 dilution in PBS for 2 hours, washed, and then incubated with Alexa 647 conjugated secondary antibody (Invitrogen, Carlsbad, CA) at a 1:1000 dilution in PBS for 1 hour. Cells were then carefully washed and fixed in 1% paraformaldehyde. Analysis was performed using a FACSCaliber flow cytometer using BD CellQuest software (Becton-Dickenson; San Jose, CA).
IL-8 Enzyme-Linked ImmunoSorbent Assay (ELISA)
16HBE 14o- cells were grown in 6-well plates to 30–40% confluency. Some cells were ‘primed’ by exposing them to 100mM alcohol for 48 hours in DMEM with 10% fetal bovine serum. The rest of the cells were maintained in DMEM with 10% fetal bovine serum. After 48 hours, the media was removed and the cell layers were washed twice with PBS. The cells were then put into serum free media (LHC-9/RPMI 1640 in a 50:50 ratio) and were stimulated with 40ng/ml of peptidoglycan derived from s. aureus (Sigma) for 24 hours. The supernatants were removed and stored at −20°C until ELISA could be performed. The IL-8 sandwich ELISA was performed as described by Wyatt et al. (Wyatt et al., 2007). Briefly, flat-bottomed 96-well poly styrene plates were coated with goat anti-human IL-8 antibody (R&D Systems, Minneapolis, MN) at a 1:500 dilution overnight at 4°C. The plates were washed, and undiluted culture supernatants and IL-8 standards (R&D Systems) were added to the plates and incubated at room temperature for 2 h. Plates were again washed, then incubated with rabbit anti-human IL-8 antibody (Endogen, Woburn, MA) at a 1:500 dilution for 1 hour. After washing, goat anti-rabbit IgG (Rockland, Gilbertsville, PA) was added at a 1:1,000 dilution for 1 hour at room temperature. The plates were again washed, and peroxidase substrate was added. The reaction was terminated with sulfuric acid, and plates were read at 490 nm in an automated ELISA reader (Dynex-MRX Revelation, Magellan Biosciences, Chantilly, VA).
Statistics
All experiments were performed a minimum of three times. All data are presented as means ± SEM. Statistics were performed using GraphPad/Prism 4. One-way analysis of variance (ANOVA) was used followed by Dunnett's Multiple Comparison Test. p values < 0.05 were considered statistically significant.
RESULTS
Alcohol increases TLR2 mRNA in a time- and concentration- dependent manner in airway epithelial cells
To determine the effect of alcohol on the expression of TLR2 in airway epithelial cells, NHBE were exposed to 0, 12.5, 25, 50 and 100 mM alcohol for 24 hours. We observed statistically significant 2–3 fold increases in TLR2 mRNA at 50 and 100 mM alcohol (Figure 1A). We did not investigate higher concentrations of alcohol, due to limited biological relevance. The peak increase in TLR2 mRNA was 2.3 fold (p<0.01) in cells exposed to 100 mM alcohol. We used this optimal concentration of alcohol to explore the time-dependence of this increase. We observed that this maximal increase (p<0.01) of TLR2 mRNA occurs at 24 hours exposure to 100 mM alcohol (Figure 1B). Limited experiments at longer time points did not show further increases (Data not shown).
Figure 1. Alcohol upregulates Toll-like Receptor 2 (TLR2) mRNA in a time- and concentration-dependent manner in both normal human bronchial epithelial cells (NHBE) and transformed human bronchial epithelial cells (16HBE 14o- ).

A. NHBE were exposed to 0–100 mM alcohol for 24 hours. B. NHBE were exposed to 100mM alcohol for 0–24 hours. C. 16HBE 14o- were exposed to 0–100 mM alcohol for 24 hours. D. 16HBE 14o- were exposed to 100 mM alcohol for 0–24 hours. All data are expressed as mean ± S.E.M. *P< 0.01 (n=5).
Alcohol-induced changes in TLR2 on NHBE cells occurred when the cells have only been in culture for a very short time at early passage (passage 2). Once NHBE cells were passed approximately 3 times they quickly lost their responsiveness to alcohol. Because cells cannot be used for experiments until passage 2, this seriously limits the quantity of experiments that can practically be done in NHBE. Because of this limitation, we investigated whether alcohol has similar effects on TLR2 in 16HBE 14o-, an immortalized bronchial epithelial cell line. We found nearly identical patterns of TLR2 mRNA expression between the two cell lines. Like NHBE, TLR2 was increased 3.3 fold (p<0.01) by 100 mM alcohol (Figure 1C). Again, similar to NHBE, the optimal time point was 24 hours, at which time we saw a 3.2 fold increase (p<0.01) in TLR2 mRNA compared to non-alcohol exposed cells (Figure 1D).
Alcohol increases the cell surface expression of TLR2 protein
To determine whether alcohol also induced an increase in TLR2 protein, we performed Western blots and FACS. In these experiments, 16HBE 14o- cells were exposed to 0, 50 and 100 mM alcohol for 48 hours. We saw a slight increase in TLR2 protein in cells exposed to 50 mM alcohol. However, there was a marked increase in TLR2 protein in cells exposed to 100 mM alcohol (Figure 2). The bands detected by anti-TLR2 antibody were seen at approximately 89 kD, the molecular weight of TLR2 protein. When compared to a gel loading control of beta-actin, the protein ratio of TLR2 to beta-actin was increased more than 2-fold in response to 100 mM alcohol (Figure 2B).
Figure 2. Alcohol upregulates TLR2 protein in whole cell lysates.


16HBE 14o- were exposed to 0, 50 and 100 mM alcohol for 48 hours. Cells were lysed and Western blot for TLR2 was performed. A. A representative blot from 3 separate experiments is shown. There is a marked increase in TLR2 protein in cells exposed to 100 mM alcohol. B. Ratio of the densitometry of TLR2 to Beta Actin bands in equal volumes of cell lysates loaded.
FACS analysis of alcohol-exposed 16HBE 14o- cells revealed that the TLR2 protein that is synthesized, is expressed on the cell surface (Figure 3A). Alcohol stimulated a 2.59-fold increase in MFI in cells exposed to 100 mM alcohol compared to control (Figure 3B). We also noted a striking increase in percentage of cells staining positive for TLR2, from 10% of control cells, to 29% in cells treated with 100 mM alcohol (Figure 3C).
Figure 3. Alcohol augments TLR2 protein expression and mobilization of TLR2 to the cell surface.
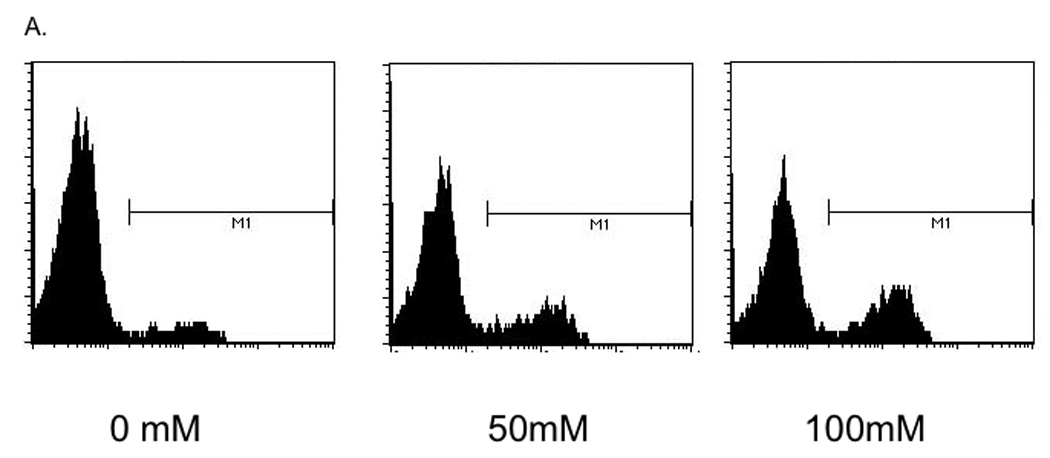

16HBE 14o- were exposed to 0, 50 and 100 mM alcohol for 48 hours and fluorescence activated cell sorting (FACS) for TLR2 was performed. A. Representative histogram showing increased TLR2 staining of cells exposed to 50 and 100 mM alcohol (n=3). B. Representative data showing increase in total Mean fluorescence intensity (MFI) with alcohol exposure (n=3). C. Representative data showing increase in percentage of cells staining positive for TLR2 with increasing alcohol concentration (n=3).
Alcohol induced up-regulation of TLR2 augments peptidoglycan-stimulated increases in interleukin-8
In order to determine whether the alcohol-induced upregulation of TLR2 was functional, we first “primed” the cells with alcohol for 48 hours followed by stimulation of the cells with a TLR2 ligand, peptidoglycan. We used a very low dose of peptidoglycan, 40ng/ml, which alone does not cause IL-8 release in airway epithelial cells. Previous studies have established that 10–100µg/ml of peptidoglycan is required to stimulate IL-8 production (Gon et al., 2004; Homma et al., 2004). We observed no increase in IL-8 in cells exposed to alcohol for 48 hours over control. Likewise, we detected no increase in IL-8 in cells stimulated with peptidoglycan alone. However, in cells that were “primed” with 100 mM alcohol for 48 hours and then stimulated with peptidoglycan, we observed a doubling of IL-8 production (Figure 4).
Figure 4. Alcohol pre-treatment augments the release of IL-8 stimulated by the TLR2 ligand, peptidoglycan, in 16HBE 14o- cells.

16HBE 14o- cells were incubated with or without alcohol for 48 hours. Cells were subsequently exposed (or not) to 40 ng/mL of peptidoglycan (PGN) for 24 hours. Supernatants were assayed for interleukin-8 (IL-8). PGN causes a significant increase in IL-8 production only in those cells that were previously exposed to alcohol. All data are expressed as mean ± S.E.M. (n=5) * P< 0.05.
DISCUSSION
In these experiments, we have demonstrated that biologically relevant concentrations of alcohol upregulate TLR2 in a concentration- and time-dependent manner in the airway epithelium. We have shown that the increase in TLR2 mRNA leads to an increase in TLR2 protein and that this increase occurs on the surface of the epithelial cells. The increase in TLR2 is likely of functional importance because alcohol pretreated cells double their IL-8 release when exposed to substimulatory levels of the TLR2 ligand, peptidoglycan. This suggests that “priming” airway cells with alcohol likely enhances airway inflammation following bacterial exposure and may drive the airway inflammation seen with acute and chronic bronchitis. We speculate that this “priming” may explain, at least in part, why alcoholics have more severe airway diseases such as bronchitis.
Alcohol can have varying effects on TLR expression and function, depending on the tissue involved and the chronicity and concentration of alcohol exposure. Our results are consistent with results in an in vivo murine model focused on the liver. In this study, mice fed a Lieber-Decarli alcohol diet were challenged with peptidoglycan injection. There was an increase in liver TLR2 mRNA and enhanced cytokine release in response to peptidoglycan in the alcohol-fed mice (Gustot et al., 2006). Our data contrasts with studies focused on immune effector cells. In one study, mice were given alcohol by gavage then challenged with zymosan (a TLR2 agonist). Alcohol-exposed mice showed less inflammatory cytokine release in peritoneal macrophages in response to the zymosan (Pruett et al., 2004). In another study, mice were administered alcohol by intra-peritoneal (ip) injection, then stimulated with zymosan. Alcohol treatment inhibited inflammatory cytokine production by zymosan (Goral and Kovacs, 2005). These data suggest that the regulation of TLR2 in the airway epithelium may be much more similar to that in the liver than that seen in monocytes and macrophages. Our observations may also be explained by differences in the length of exposure to alcohol. In Gustot’s study, mice were fed alcohol chronically, which may have enhanced TLR2 expression. In Goral and Pruett’s studies, an acute model of alcohol exposure (either ip or gavage) was used, which led to a down-regulation of TLR2. It is possible that our model approximates a chronic exposure. Animal experiments are needed to determine if and when alcohol-driven airway TLR2 upregulation occurs in vivo.
Our data clearly show that TLR2 is upregulated in airway epithelial cells by millimolar concentrations of alcohol. While such blood alcohol levels are readily observed in heavy drinkers, it is yet to be determined what effect this may have in vivo. One could argue that an upregulation of TLR2 in the airway epithelium could be a protective effect. With more TLR2 present on the airway epithelium, the airway would be able to mount a more aggressive defense against invading bacteria. However, this is not what we see clinically in alcoholics. In our cytokine experiments, we were careful to choose a dose of peptidoglycan that does not induce inflammation in normal airway epithelial cells. The fact that alcohol primed the cells to respond to a stimulus that would normally be tolerated, suggests that this inflammation is above and beyond that which would be protective and may indeed be harmful. In these experiments, we evaluated alcohol’s effect at longer durations and higher concentrations of alcohol. We did not test lower concentrations (ie. 12.5mM) at short durations (ie. 1–2 hours). It is possible that under these conditions, which may approximate acute alcohol intoxication, alcohol might have no effect or down-regulate TLR2.
The mechanism through which alcohol upregulates TLR2 is unknown. We have previously shown that alcohol causes airway epithelial cells to produce nitric oxide (Sisson, 1995). Thus, nitric oxide may be a key signaling molecule in the regulation of airway TLR2. Nitric oxide is known to stimulate soluble guanyl cyclase cyclic GMP which activates protein kinase G (PKG) in airway epithelial cells (Wyatt et al., 2003). PKG could in turn phosphorylate a substrate that leads to increased transcription of TLR2. It is also possible that alcohol is not only increasing the expression of TLR2, but is also modulating the down-stream signaling of TLR2 in airway epithelial cells. Little is known about how alcohol modulates downstream signaling of TLR2 in airway epithelial cells. However in immune effector cells, alcohol has been shown to modulate several key mediators including: IRAK-1, JNK, (Oak et al., 2006) and ERK1/2 (Goral and Kovacs, 2005). It is possible that alcohol may also modulate these mediators in a different way than occurs in immune effector cells. It will be important to define the mechanism and consequences of alcohol’s upregulation of TLR2 in the airway epithelium in vivo. Further research should define the mechanistic differences between the regulation of TLR2 in the airway epithelium and immune effector cells.
In summary, our experiments demonstrate that alcohol upregulates TLR2 in a functional way in airway epithelial cells. Alcohol dramatically increases the amount of TLR2 on the surface of airway epithelial cells. This causes the cells to double the amount of IL-8 they release in response to an amount of peptidoglycan that would normally cause no increase in inflammatory cytokines. This is a novel finding which contrasts with what has been previously shown in alcohol-exposed monocytes and macrophages. It suggests that alcohol may regulate TLR2 differently in airway epithelial cells than in immune effector cells. These findings may explain in part why alcoholics have more severe airway disease than moderate or non-drinkers.
Acknowledgments
Sources of support:
This material is the result of work supported with resources and the use of facilities at the Omaha VA Medical Center, Omaha, NE (TAW-Merit Review Award; DJR-Merit Review Award), 1F32AA016433 (KLB), 5R37AA008769 (JHS), 5R01OH008539 (DJR).
REFERENCES
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, Finkbeiner WE, Widdicombe JH, Gruenert DC. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10(1):38–47. doi: 10.1165/ajrcmb.10.1.7507342. [DOI] [PubMed] [Google Scholar]
- de Roux A, Cavalcanti M, Marcos MA, Garcia E, Ewig S, Mensa J, Torres A. Impact of alcohol abuse in the etiology and severity of community-acquired pneumonia. Chest. 2006;129(5):1219–1225. doi: 10.1378/chest.129.5.1219. [DOI] [PubMed] [Google Scholar]
- Frost RA, Nystrom G, Burrows PV, Lang CH. Temporal differences in the ability of ethanol to modulate endotoxin-induced increases in inflammatory cytokines in muscle under in vivo conditions. Alcohol Clin Exp Res. 2005;29(7):1247–1256. doi: 10.1097/01.alc.0000171935.06914.5d. [DOI] [PubMed] [Google Scholar]
- Gon Y, Asai Y, Hashimoto S, Mizumura K, Jibiki I, Machino T, Ra C, Horie T. A20 inhibits toll-like receptor 2- and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am J Respir Cell Mol Biol. 2004;31(3):330–336. doi: 10.1165/rcmb.2003-0438OC. [DOI] [PubMed] [Google Scholar]
- Goral J, Kovacs EJ. In vivo ethanol exposure down-regulates TLR2-, TLR4-, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J Immunol. 2005;174(1):456–463. doi: 10.4049/jimmunol.174.1.456. [DOI] [PubMed] [Google Scholar]
- Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, Franchimont D, Louis H, Deviere J, Le Moine O. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. 2006;43(5):989–1000. doi: 10.1002/hep.21138. [DOI] [PubMed] [Google Scholar]
- Homma T, Kato A, Hashimoto N, Batchelor J, Yoshikawa M, Imai S, Wakiguchi H, Saito H, Matsumoto K. Corticosteroid and cytokines synergistically enhance toll-like receptor 2 expression in respiratory epithelial cells. Am J Respir Cell Mol Biol. 2004;31(4):463–499. doi: 10.1165/rcmb.2004-0161OC. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med. 2003;31(4 Suppl):S207–S212. doi: 10.1097/01.CCM.0000057845.77458.25. [DOI] [PubMed] [Google Scholar]
- Oak S, Mandrekar P, Catalano D, Kodys K, Szabo G. TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol. 2006;176(12):7628–7635. doi: 10.4049/jimmunol.176.12.7628. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol. 2004;33(2):147–155. doi: 10.1016/j.alcohol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Shaper AG. Alcohol and mortality: a review of prospective studies. Br J Addict. 1990;85(7):837–847. doi: 10.1111/j.1360-0443.1990.tb03710.x. discussion 849–61. [DOI] [PubMed] [Google Scholar]
- Sisson JH. Ethanol stimulates apparent nitric oxide-dependent ciliary beat frequency in bovine airway epithelial cells. Am J Physiol. 1995;268(4 Pt 1):L596–L600. doi: 10.1152/ajplung.1995.268.4.L596. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol Chapter 14: Unit 14 12. 2007 doi: 10.1002/0471142735.im1412s77. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Forget MA, Sisson JH. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am J Pathol. 2003;163(3):1157–1166. doi: 10.1016/S0002-9440(10)63475-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Sisson JH. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;281(3):L575–L581. doi: 10.1152/ajplung.2001.281.3.L575. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Slager RE, Devasure J, Auvermann BW, Mulhern ML, Von Essen S, Mathisen T, Floreani AA, Romberger DJ. Feedlot dust stimulation of interleukin-6 and -8 requires protein kinase Cepsilon in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293(5):L1163–L1170. doi: 10.1152/ajplung.00103.2007. [DOI] [PubMed] [Google Scholar]
- Yamashina S, Takei Y, Ikejima K, Enomoto N, Kitamura T, Sato N. Ethanol-induced sensitization to endotoxin in Kupffer cells is dependent upon oxidative stress. Alcohol Clin Exp Res. 2005;29(12 Suppl):246S–250S. doi: 10.1097/01.alc.0000191128.54871.40. [DOI] [PubMed] [Google Scholar]