

Abstract
One of the main challenges in high-throughput serum profiling by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is the development of proteome fractionation approaches that allow the acquisition of reproducible profiles with a maximum number of spectral features and minimum interferences from biological matrices. This study evaluates a new class of solid-phase extraction (SPE) pipette tips embedded with different chromatographic media for fractionation of model protein digests and serum samples. The materials embedded include strong anion exchange (SAX), weak cation exchange (WCX), C18, C8, C4, immobilized metal affinity chromatography (IMAC) and zirconium dioxide particles. Simple and rapid serum proteome profiling protocols based on these SPE micro tips are described and tested using a variety of MALDI matrices. We show that different types of particle-embedded SPE micro tips provide complementary information in terms of the spectral features detected for β-casein digests and control human serum samples. The effect of different sample pretreatments, such as serum dilution and ultrafiltration using molecular weight cut-off membranes, and the reproducibility observed for replicate experiments, are also evaluated. The results demonstrate the usefulness of these simple SPE tips combined with offline MALDI-TOF MS for obtaining information-rich serum profiles, resulting in a robust, versatile and reproducible open-source platform for serum biomarker discovery.
Specific serum biomarker panels are urgently needed in many areas of medicine, particularly in cancer diagnostics. Great effort is thus being directed towards the identification of molecular disease markers with high selectivity and sensitivity, with mass spectrometry (MS) being one of the tools of choice. MS-based serum biomarker discovery workflows are based on two main approaches: uni- or bi-dimensional chromatography followed by single-stage or tandem MS, which provides high peak capacity; and matrix-assisted laser desorption ionization (MALDI) MS or MS/MS without prior chromatographic separation, which provides lower coverage in terms of the maximum number of spectral features that can be detected without spectral overlap, but orders of magnitude higher sample throughput.1
Mass spectrometric serum proteome profiling by MALDI MS is widely used by medical researchers due to the frequent need for rapid analysis of large quantities of clinical samples.2–11 In this high-throughput approach, careful bioinformatic comparison of the abundance of proteins and peptides inferred from MALDI mass spectra obtained under standardized conditions enables the multivariate comparison of sample groups, with the aim of identifying proteomic patterns or ‘fingerprints’ that can be used for diagnostic purposes. Despite its unquestionable throughput, MALDI MS serum proteomic profiling can be negatively affected by the high complexity of serum samples, the presence of interfering salts, and the large dynamic range of protein concentrations, all of which can result in ionization suppression12 and/or space-charge effects in trapping instruments,13 with the concomitant inability to detect or identify low abundance biomarkers.
In this trend, one of the major challenges in serum proteome profiling is to develop optimal front end sample preparation procedures for removing salts, separating high abundance protein fractions, and selectively enriching for subsets of serum proteins and peptides (i.e. ‘subproteomes’), which can be probed in deeper detail than the original sample. A number of techniques have been developed to fractionate serum samples prior to MS analysis, including depletion columns for removal of high abundance proteins,14–17 ultrafiltration with different molecular weight cut-off membranes,18,19 solid-phase extraction (SPE) columns,20 coated magnetic beads,21 and precipitation by organic solvents.22 The biggest challenge for users of these techniques is to obtain high-enough day-to-day and batch-to-batch reproducibility to correctly evaluate biologically-significant differences within the bewildering number of mass spectral peaks detected.10,15,20
One of the most popular tools for high-throughput serum profiling in the biomedical community is surface-enhanced laser desorption/ionization (SELDI) MS, a MALDI-based technique which integrates on-target sample preparation with time-of-flight (TOF) MS peptide/protein ion detection.23 Unique to this method are functionalized MALDI targets with specific SPE or affinity-capture properties.24 Interest in this technique was sparked by early work by Liotta and coworkers which exploited the potential of SELDI-TOF MS combined with sophisticated bioinformatics algorithms for ovarian cancer diagnostics.25,26 Other research groups also applied this SELDI approach, further demonstrating the utility of applying specific surface chemistries for serum proteome partitioning, one of the most promising being strong cation-exchange functionalized targets.27 However, SELDI TOF mass spectrometers offer limited resolving power, mass accuracy and tandem MS capabilities, making biomarker identification difficult. In addition, there have been questions regarding the long-term stability of SELDI experiments. Difficulties in baseline correction, irreproducibility in sample preparation protocols, and unstable TOF mass calibration have been reported.28 More recently, in-source collision-induced dissociation has been suggested to be the origin of much of the structure uncovered by SELDI.29
As an alternative to SELDI, many groups have been investigating alternative functionalized MALDI targets that can be used across many MALDI-TOF MS platforms for on-chip sample fractionation.30,31 Another popular option is the combination of off-line serum fractionation protocols with MALDI MS, which have the advantage of decoupling sample preparation from mass analysis, thus allowing the optimization of those two steps separately.20,32 Both the on-chip and off-line methodologies share the advantage of not relying on proprietary MALDI instrumentation, and can thus be implemented in combination with a variety of high-end MALDI MS or MSn spectrometers.
In this work we evaluate the performance of off-line sample preparation for MALDI MS via particle-embedded SPE pipette micro tips. In these pipette tips, the inner plastic walls are covered with chromatographic particles with different functionalities, such as strong anion-exchange (SAX), weak cation-exchange (WCX), C18, C8, C4, ZrO2, and immobilized metal affinity chromatography (IMAC). We first present a detailed characterization of these SPE micro tips using tryptic digests of a standard protein, and then extend these studies to human serum for fractionation of low mass proteins and peptides. To the best of our knowledge, this is the first report comparing this wide variety of SPE micro tips for MALDI-TOF MS serum profiling.
Experimental
Samples and reagents
Trifluoroacetic acid (TFA), ammonium hydroxide and formic acid were obtained from Fisher Scientific (Fair Lawn, NJ, USA), acetonitrile (ACN) was from EMD Chemicals (Gibbstown, NJ, USA), nickel(II) chloride and gallium(III) nitrate hydrate were from Alfa Aesar (Ward Hill, MA, USA), healthy human serum (Stock No. S7023-50ML), ammonium citrate, β-casein, trypsin (proteomics grade), ammonium bicarbonate, α-cyano-4-hydroxycinnamic acid (CHCA), 2,5-dihydroxybenzoic acid (DHB), sinapinic acid and indoleacetic acid (IAA) were from Sigma-Aldrich (St. Louis, MO, USA). Microcon ultrafiltration 3 kDa and 50 kDa cut-off membranes were purchased from Millipore (Bedford, MA, USA). NuTip particle-embedded pipette micro tips (1-10 μL) were obtained from Glygen Corp. (Columbia, MD, USA). All aqueous solutions were prepared with nanopure water (dH2O) from a Nanopure Diamond laboratory water system (Barnstead International, Dubuque, IA, USA).
In-solution tryptic digestion of β-casein
A (2.4 μg μL−1) solution of β-casein was prepared in 6 M urea containing 50 mM ammonium bicarbonate and separated into 20 μL aliquots which were first heated for 1 h at 65°C. After cooling, 180 μL of 50 mM ammonium bicarbonate were added to each aliquot followed by addition of 48 μL of trypsin working solution (20μg mL−1) and the mixtures were incubated in a water bath for 20 h at 38°C. One aliquot was then basified by adding approximately 6 μL of 20 mM NH4Cl/NH3 aqueous buffer (pH 8.5) and the remaining aliquot was acidified using 2% TFA solution to a final pH of approximately 2.0. Purification and concentration of the acidified and basified digest aliquots was achieved by using SPE micro tips containing different chromatographic media as described below.
In silico tryptic digestion was performed with ProteinProspector.33 Variable modifications namely, methionine oxidation (Met-Ox), phosphorylation at serine, threonine, tyrosine residues (denoted as s, t, and y respectively), and carbamylations of lysine and arginine were selected. The total number of theoretically generated peptides was 225 as a result of combinations of the variable post-translational modifications. Each peptide thus generated was then characterized for its hydropathy using an online tool34 based on the Hopps-Woods scale.35 In this process, a line plot is generated for each peptide by averaging hydrophobicity values assigned to sequential amino acids in windows of five residues. If the obtained hydrophobicity line was predominantly above zero, the corresponding peptide was considered hydrophilic, and vice versa. Terms such as ‘predominantly’ or ‘partially’ hydrophilic/hydrophobic are used if a significant part of the line lies above or below zero, respectively.
Serum sample pretreatment prior to SPE
Immediately upon arrival from the vendor, the frozen serum sample was thawed, aliquoted into 1.5mL Safe-Lock Eppendorf micro test tubes and frozen at −80°C until further use. All measurements were performed on identical 500-μL aliquots of twice-thawed serum. Prior to SPE, the serum was denatured by adding 25 μL of 80% ACN to 200 μL of sample to disrupt intermolecular interactions. The mixture was then vortexed, and incubated for 30 min at ambient temperature (22–25°C). Microcon membranes (50 kDa cut-off) were rinsed three times with 0.2 mL dH2O, and used to filter 240 μL of the denatured serum mixture at 13 000 g for 25 min. A 120-μL aliquot of the filtrate was acidified by addition of 30 μL of 2% TFA solution to a final pH of ∼2.0. Similarly, a second 120 μL serum filtrate was basified to pH ∼8.5 by addition of dilute NH4Cl/NH3 aqueous buffer. The acidified and basified serum filtrate aliquots were treated with SPE micro tips as described below.
Procedure for SPE via hydrophobic (C18, C08, C4) NuTips
C18, C8 and C4 NuTips were washed with 5 × 4 μL (5 times, 4 μL each time) of 50% ACN, and equilibrated with 3 × 4 μL of 0.1% TFA solution. Then, 30 × 4μL of β-casein protein digest or acidified serum filtrate were aspirated and expelled through the tip. The tip was then washed with 2 × 4 μL of 0.1% TFA solution. Retained components were eluted with 2 μL of 50% ACN/0.1% TFA. The eluant was pipetted up and down ten times in a clean vial to ensure complete elution.
Procedure for SPE via ion-exchange NuTips
Silica strong anion-exchange (SAX)
SAX NuTips were conditioned with 5 × 4 μL of 50% ACN, and equilibrated with 3 × 4 μL of 20 mM NH4Cl/NH3 aqueous buffer (pH 8.5). Then, 30 × 4 μL of β-casein protein digest or basified serum filtrate were aspirated and expelled through the tip. The tip was then washed with 2 × 4 μL of dH2O. Retained components were eluted with 2 μL of 0.5% TFA/20% ACN, as described above for C18 tips.
Poly(aspartic acid)–silica (PolyCAT A) weak cation-exchange (WCX)
WCX NuTips were first washed with 5 × 4 μL of 50% ACN, and equilibrated with 3 × 4μL of 0.1% TFA solution. Then, 30×4 μL of acidified of β-casein protein digest or serum filtrate were sequentially aspirated and expelled through the tip. The tip was then washed with 2 × 4 μL of dH2O, and elution of the retained peptides was performed with 2 μL of 2% aqueous ammonia solution containing 20% ACN.
Procedure for SPE via IMAC NuTips
IMAC-Ga(III) and IMAC-Ni(II) NuTips were metal loaded with 10 × 4 μL of 200 mM Ga(NO3)3 and NiCl2 aqueous solutions, respectively, followed by washing with 3 × 4 μL of dH2O. Tip equilibration was performed with 3 × 4 μL of 1% acetic acid/10% ACN and with 5 × 4 μL of 0.1% acetic acid/10% ACN, respectively. Then, 30 × 4 μL of the acidified protein digest or serum filtrate were aspirated and expelled through the micro tips. Washing was done with 2 × 4 μL of dH2O, followed by elution with 2 μL of 2% aqueous ammonia solution containing 20% ACN by aspirating up and down (ten times) into a clean vial.
Procedure for SPE via ZrO2 NuTips
ZrO2 NuTips were first washed with 10 × 4 μL of 0.33% formic acid. Then, 30 × 4 μL of acidified serum filtrate or digest aliquot were aspirated/expelled, the tips were washed with 2 × 4 μL of dH2O, and the retained species were eluted with 2 μL of 2% aqueous ammonia solution containing 20% ACN.
MALDI-MS
MALDI-TOF MS analyses were performed using a Voyager DE-STR (Applied Biosystems, Framingham, MA) MALDI-TOF mass spectrometer equipped with a nitrogen laser (337 nm, 3 ns pulse width). All experiments were carried out in positive ion mode. All mass spectra were acquired in linear mode with 25 kV acceleration voltage, 92.8% grid voltage and 300 ns extraction delay. In all cases 240 laser shots were averaged for each mass spectrum. The instrument was externally calibrated prior to running any samples using angiotensin II [M+H]+ = 1046.54, ACTH (18–39) [M+H]+ = 2464.12, insulin chain β [M+H]+= 3494.65, bovine insulin [M+H]+ = 5730.61, and cytochrome C [M+H]+ = 12361.96. All spectra were baseline corrected, and smoothed prior to exporting them as ASCII files.
We performed preliminary experiments to determine a suitable MALDI matrix using four different commonly used compounds (Fig. 1S Fig. 1, Supplementary Material). Various properties of matrix molecules such as their ability to induce analyte fragmentation, ionization potentials, and proton affinity are some of the determinants of suitability of a matrix. Empirical results showed that CHCA outperformed other tested matrices in terms of observed signal-to-noise (S/N) ratio and so CHCA was chosen as the matrix for all subsequent MALDI experiments. However, more investigation may be needed to provide guidelines for selection of matrix or provide a rationale to the experimentally observed performances of these matrixes. The eluate obtained from different NuTips was mixed in a 1:1 v/v ratio with 10mg mL–1 CHCA solution in 50% ACN/0.1% TFA. A volume of 1 μL of this mixture was spotted on a stainless steel MALDI plate. The CHCA matrix solution (10 mg mL−1) used to co-crystallize with IMAC and ZrO2 eluates was prepared in 50% ACN/0.4% acetic acid/10 mM ammonium citrate.
Figure 1.
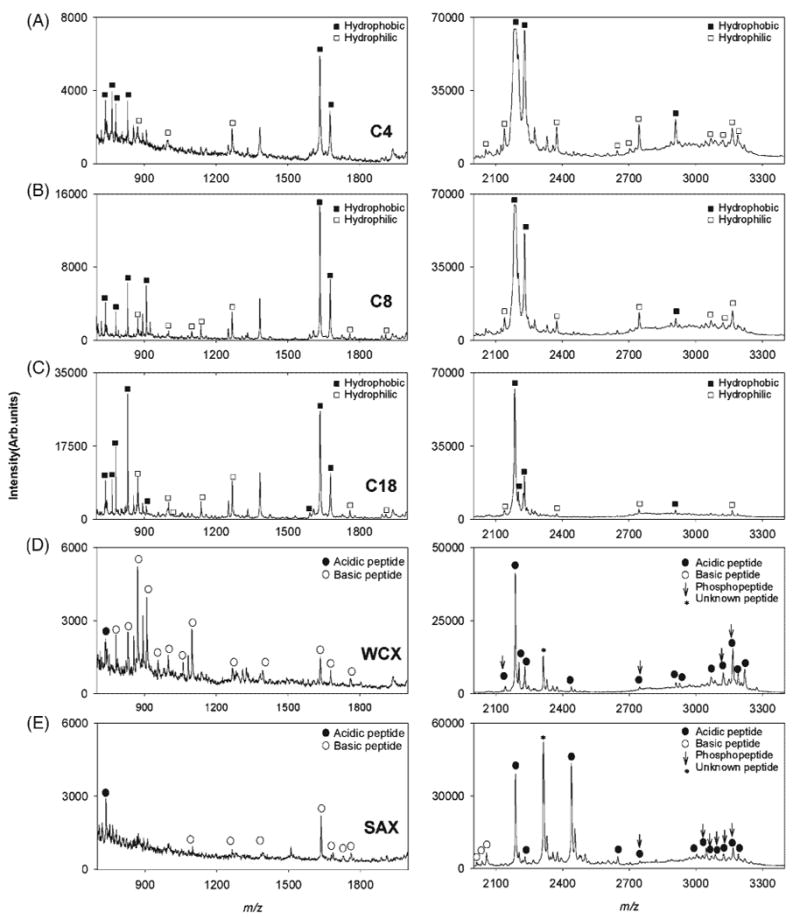
MALDI TOF mass spectra of β-casein tryptic digest treated with (A) C4, (B) C8, (C) C18, (D) WCX, and (E) SAX SPE micro tips. Left 700–2000 m/z and right 2000–3400 m/z ranges. Hydrophobic and hydrophilic peptides are denoted by filled and empty squares respectively in (A), (B) and (C). Acidic peptides (pI < 7) and basic peptides (pI > 7) are denoted by filled and empty circles respectively in (D) and (E). Phosphopeptides are marked by vertical arrows.
Results and Discussion
Selectivity study of different SPE micro tips using a model peptide mixture
A comparison of β-casein tryptic digest fractionation using particle-embedded micro tips coated with eight different materials (Fig. 1) revealed the extent to which each support selectively enriched various peptide fractions. The left panel shows the 700–2000 m/z range and the right panel the 2000–3400 m/z range. Figures 1(A)–1(C) show the MALDI spectra of digests treated with C4, C8 and C18 hydrophobic SPE micro tips, respectively. Peaks corresponding to predominantly hydrophobic or hydrophilic peptides are denoted by filled or empty squares. A detailed list of the peptides identified, along with their respective degrees of hydropathy, is given in Table 1. The most intense peaks in Figs. 1(A)–1(C) corresponded to hydrophobic peptides at m/z 780, 873, 2186, 2202, 2229 and 2909 and to predominantly hydrophobic peptides at m/z 830, 1634 and 1678. A set of less intense peaks corresponding to hydrophilic peptides was detected at m/z 1267, 907 ([M+H2O+H]+ of VKEAMAPK, 1 Met-Ox), and at m/z 2375 ([M+Na]+ of IEKFQSEEQQQTEDELQDK) with lower S/N ratio. Out of the 225 theoretical peptides generated in this tryptic digest, only 22% are hydrophobic, mostly found in the m/z <1000 range and the remainder, more hydrophilic, are predominantly present in the higher m/z range. The binding of hydrophilic peptides to hydrophobic SPE micro tips is thus explained by their higher relative abundance in the initial tryptic mixture. S/N ratios for the detected hydrophilic peptides were lower than for the hydrophobic peptides in both the higher and lower mass ranges as a result of the hydrophobic nature of SPE micro columns. Overall, inspection of the mass spectra of hydrophobic SPE eluates showed that the performance of C18 micro tips was superior in the lower m/z range, both in terms of sensitivity, reflected in the higher S/N ratios and number of peaks observed. In the higher m/z range, the MALDI mass spectrum of the C4 eluate showed more spectral features and higher intensity peaks when compared to the MALDI spectra of C8 and C18 eluates. The observed mass spectral patterns reflect the effect of the different chain lengths in each of the three materials. The shorter chain length in C4 makes it less hydrophobic and larger peptides are thus more likely to expose sufficiently-large hydrophobic regions to efficiently interact with this material. The MALDI mass spectra shown in Figs. 1(D) and 1(E) correspond to WCX and SAX eluates. Peaks corresponding to acidic or basic peptide are denoted by filled and empty circles, respectively. Theoretically, WCX materials should first retain mostly basic peptides with a net positive charge at the lower pH used for binding, and SAX material should bind acidic peptides with a net negative charge at the basic pH used. Clear differences are observed between these materials in both m/z ranges providing complementary information from the same protein digest. A detailed list of acidic and basic peptides identified in the WCX and SAX eluate mass spectra is presented in Table 2. In the low m/z range, the WCX eluate showed many peaks corresponding to eight peptides and their various modifications. One acidic peptide at m/z 742.4 (pI 6) and several basic peptides with their pI values in the 9–10.5 range were observed. The spectrum from the SAX eluate in this m/z range showed a signal corresponding to one acidic peptide, at m/z 742, and weak signals corresponding to carbamylated peptides at m/z 1099, 1267, 1634, 1678, 1683, 1726 and 1760. It has been suggested that carbamylation lowers peptide isoelectric points, providing a possible explanation for the partial retention of these peptides by the SAX micro tip.36 The acidic peptide at m/z 742 (pI 6) was seen in the mass spectra of both WCX and SAX eluates, as the net charge on this peptide shifts upon changes in the pH of the binding buffers used in either extraction and hence it will bear a net positive change in the acidic binding buffer (pH 2) used for WCX and a net negative charge in the basic binding buffer (pH 8.5) used for SAX SPE. The peak intensity and S/N ratio corresponding to this peptide were higher in the SAX eluate mass spectrum in accordance to its predominantly acidic character, whereas the carbamylated peptides at m/z 1099, 1267, 1678, 1683 and 1670, retained by the SAX and WCX micro tips, showed higher intensity peaks in the latter due to their more basic nature (pI > 7).
Table 1.
List of hydrophobic and hydrophilic peptides retained by C4, C8 and C18 SPE micro tips and corresponding signal-to-noise (S/N) ratios. Peptides detected as their water or salt adducts are marked by an asterisk. All other peptides were detected as protonated molecules. The following modifications were considered: oxidation of methionine (Met-Ox), carbamylation of lysine and arginine, and phosphorylation of serine, threonine and tyrosine (s, t, y)
| m/z | Peptide | Modifications | Hydropathy | C4 | C8 | C18 |
|---|---|---|---|---|---|---|
| 742 | GPFPIIV | Hydrophobic | 1.7 | 2.8 | 4.1 | |
| 764 | EMPFPK | 1 Met-Ox | Hydrophobic | 2.8 | 0.9 | 7.2 |
| 780 | VLPVPQK | Hydrophobic | 2.6 | 6.4 | 14.0 | |
| 830 | AVPYPQR | Predominantly Hydrophobic | 4.1 | 12.7 | 22.6 | |
| 873 | VKEAMAPK | Hydrophilic | 1.75 | 5.0 | 8.0 | |
| 910 | AVPyPQR | Hydrophobic | 1.0 | 8.9 | 11.2 | |
| 1001 | INKKIEK | 3 Carm | Hydrophilic | 2.0 | 2.6 | 7.5 |
| 1099 | HKEAMPFPK | 2 Carm | Hydrophilic | - | 3.6 | 4.0 |
| 1138 | VKEAMAPKHK | Hydrophilic | 1.6 | 9.3 | 15.6 | |
| 1267 | VKEAMAPKHK | 3 Carm | Hydrophilic | 3.3 | 13.1 | 28.5 |
| 1592 | VLPVPQKAVPYPQR | Predominantly Hydrophobic | — | — | 7.4 | |
| 1634 | VLPVPQKAVPYPQR | 1 Carm | Predominantly Hydrophobic | 16.7 | 29.7 | 46.7 |
| 1678 | VLPVPQKAVPYPQR | 2 Carm | Predominantly Hydrophobic | 8.7 | 14.9 | 9.3 |
| 1760* | EAMPKHKEMPFPK | 1 Met-Ox,2 Carm | Hydrophobic | — | — | 14.8 |
| 1910 | VKEAMAPKHKEMPFPK | 1 Carm | Hydrophilic | — | 8.8 | 6.7 |
| 2141 | FQsEEQQQtEDELQDK | Hydrophilic | 6.4 | 5.6 | 4.1 | |
| 2186 | DMPIQAFLLYQEPVLGPVR | Hydrophobic | 24.9 | 18.7 | 28.0 | |
| 2202 | DMPIQAFLLYQEPVLGPVR | 1 Met-Ox | Hydrophobic | 20.0 | 7.3 | 7.5 |
| 2229 | DMPIQAFLLYQEPVLGPVR | 1 Carm | Hydrophobic | 28.9 | 11.3 | 4.6 |
| 2375* | IEKFQSEEQQQTEDELQDK | Hydrophilic | 5.5 | 2.7 | 2.4 | |
| 2646 | ELEELNVPGEIVESLSSSEESITR | Hydrophilic | 3.0 | — | — | |
| 2703* | ELEELNVPGEIVESLSSSEESITR | Hydrophilic | 2.1 | — | — | |
| 2746* | KIEKFQsEEQQQtEDELQDK | 1 Carm | Hydrophilic | 3.6 | 2.5 | 2.5 |
| 2909 | DMPIQAFLLYQEPVLGPVRGPFPIIV | Hydrophobic | 5.0 | 1.8 | 1.7 | |
| 3044 | ELEELNVPGEIVESLSSSEESITR | 1 Carm | Hydrophilic | 3.8 | — | — |
| 3068* | ELEELNVPGEIVEsLsssEESItR | Hydrophilic | 2.0 | 1.7 | 1.0 | |
| 3168 | INKKIEKFQsEEQQQtEDELQDK | 4 Carm | Hydrophilic | 2.3 | 3.0 | 2.6 |
| 3190* | ELEELNVPGEIVESLSSSEESITRINKK | 1 Carm | Hydrophilic | 2.4 | — | — |
Table 2.
List of peptides retained by WCX, SAX, IMAC-Ga, IMAC-Ni and ZrO2 micro tips and corresponding S/N ratios. Peptides detected as their water or salt adducts are marked by an asterisk. All other peptides were detected as protonated molecules. The following modifications were considered: oxidation of methionine (Met-Ox), carbamylation of lysine and arginine (Carm), and phosphorylation of serine, threonine and tyrosine (s, y, t)
| m/z | Peptide | Modifications | pI | WCX | SAX | IMAC-Ga | IMAC-Ni | ZrO2 |
|---|---|---|---|---|---|---|---|---|
| 742 | GPFPIIV | 6 | 1.6 | 3.3 | 2.8 | 2.2 | 12.3 | |
| 780 | VLPVPQK | 10.1 | 2.3 | — | — | — | 3.3 | |
| 830 | AVPYPQR | 9.8 | 3.2 | — | 3.0 | 2.8 | 9.4 | |
| 872 | INKKIEK | 10.3 | 7.0 | — | 2.5 | — | 3.4 | |
| 910 | AVPyPQR | 9.8 | 8.3 | — | 1.6 | — | — | |
| 1056 | HKEAMPFPK | 1 Carm | 9.9 | 3.2 | — | — | — | 4.3 |
| 1001 | INKKIEK | 3 Carm | 10.3 | 2.2 | — | — | — | 4.7 |
| 1099 | HKEAMPFPK | 2 Carm | 9.9 | 5.2 | 2.0 | — | — | — |
| 1267 | VKEAMAPKHK | 3 Carm | 10.3 | 3.0 | 2.5 | 8.2 | 5.2 | 6.1 |
| 1634 | VLPVPQKAVPYPQR | 1 Carm | 10.4 | 8.6 | 10.6 | 10.3 | 12.2 | 28.7 |
| 1678 | VLPVPQKAVPYPQR | 2 Carm | 10.4 | 3.0 | — | 5.2 | 6.2 | 15.3 |
| 1683 | EAMPKHKEMPFPK | 1 Carm | 9.8 | — | 3.2 | — | — | — |
| 1726 | EAMPKHKEMPFPK | 2 Carm | 9.8 | — | 4.1 | — | — | — |
| 1728* | VLPVPQKAVPyPQR | 10.4 | — | — | — | — | 3.5 | |
| 1760* | EAMPKHKEMPFPK | 1 Met-Ox,2 Carm | 9.8 | 4.7 | 5.7 | — | — | 9.2 |
| 2013 | VKEAMPKHKEMPFPK | 1 Met-Ox,3 Carm | 10.2 | — | 3.6 | — | — | — |
| 2040 | VKEAMPKHKEMPFPK | 4 Carm | 10.2 | — | 3.0 | — | — | — |
| 2056 | VKEAMPKHKEMPFPK | 1 Met-Ox,4 Carm | 10.2 | — | 7.2 | — | — | — |
| 2061 | FQSEEQQQTEDELQDK | — | — | 8.7 | — | 4.3 | ||
| 2141 | FQsEEQQQtEDELQDK | 3.4 | 4.0 | — | 8.5 | 5.0 | 12.8 | |
| 2186 | DMPIQAFLLYQEPVLGPVR | 4.1 | 22.8 | 21.3 | 17.8 | 16.4 | 10.3 | |
| 2202 | DMPIQAFLLYQEPVLGPVR | 1 Met-Ox | 4.1 | 7.3 | 3.7 | 12.1 | 14.2 | 8.2 |
| 2229 | DMPIQAFLLYQEPVLGPVR | 1 Carm | 4.1 | 6.2 | 2.1 | 5.0 | 5.6 | 15.2 |
| 2266 | DMPIQAFLLyQEPVLGPVR | 4.1 | — | — | — | — | 3.2 | |
| 2438 | IEKFQSEEQQQTEDELQDK | 2 Carm | 3.7 | — | 18.5 | — | — | — |
| 2747* | KIEKFQsEEQQQtEDELQDK | 1 Carm | 4 | 1.7 | 2.0 | 4.2 | 3.0 | 6.5 |
| 2909 | DMPIQAFLLYQEPVLGPVRGPFPIIV | 4.4 | 2.3 | — | 1.5 | 1.6 | 2.3 | |
| 2926 | DMPIQAFLLYQEPVLGPVRGPFPIIV | 1 Met-Ox | 4.4 | 2.2 | — | 1.3 | 1.0 | — |
| 2966 | ELEELNVPGEIVEsLsssEESITR | 3.5 | — | — | — | 1.8 | 2.0 | |
| 3001 | ELEELNVPGEIVESLSSSEESITRINK | 3.9 | — | 3.1 | — | — | — | |
| 3004 | DMPIQAFLLyQEPVLGPVR | 4.4 | — | — | 1.5 | — | — | |
| 3046 | ELEELNVPGEIVESLsssEEsItR | 3.5 | — | 2.0 | 2.5 | 2.8 | 2.3 | |
| 3068* | ELEELNVPGEIVEsLsssEESItR | 3.5 | — | 2.0 | 1.7 | 2.5 | 1.5 | |
| 3087 | INKKIEKFQsEEQQQTEDELQDK | 4 Carm | 4.3 | 2.5 | 1.8 | 1.5 | 1.9 | 1.3 |
| 3124 | ELEELNVPGEIVEsLSSSEESITRINK | 1Carm | 4.3 | 1.7 | 3.2 | — | 3.5 | 1.4 |
| 3168 | INKKIEKFQsEEQQQtEDELQDK | 4 Carm | 4.3 | 5.2 | 4.0 | — | 6.5 | 2.5 |
| 3190* | ELEELNVPGEIVESLSSSEESITRINKK | 1 Carm | 4.1 | — | 3.5 | — | — | — |
| 3204 | ELEELNVPGEIVEsLsSSEESITRINK | 1Carm | 3.9 | — | — | 1.5 | 2.2 | 1.3 |
In the higher m/z range, the WCX eluate produced signals corresponding to peptides with an average pI value of 3.9 (m/z 2141, 2186, 2202 and 2229), higher than the pH to which the binding solution was adjusted (pH 2.0), making these peptides positively charged. The SAX spectrum (Fig. 1(E), right panel) showed intense peaks corresponding to mostly carbamylated peptides such as at m/z 2056 (1 Met-Ox, 4 carbamylations), 2013 (3 carbamylations), 2438 (2 carbamylations, pI 3.7) and 3168 (4 carbamylations). A comparison of WCX and SAX eluates in the higher mass range showed that peaks corresponding to acidic peptides (pI < 7) were more intense with higher S/N ratios in the mass spectrum of the SAX eluate. A few peaks corresponding to phophorylated peptides at m/z 2747 ([M+H2O+K]+, 3 carbamylations), 3046, 3068, 3087 (4 carbamylations), 3124 (1 carbamylation) and 3168 (4 carbamylations) were also detected. Retention of phosphopeptides by the SAX column is expected due to electrostatic interactions with the negatively charged phosphate groups. Only four phosphopeptides at m/z 2141, 2747, 3124 and 3168 were also retained by WCX micro tips with comparable peak intensities, as these peptides are expected to be positively charged at the acidic binding pH.
A comparative analysis of the MALDI mass spectra of eluates purified by hydrophobic and ion-exchange SPE micro tips further illustrates the binding specificity of these materials. Hydrophilic peptides of basic nature, such as INKKIEK (m/z 872) and its carbamylated form at m/z 958, HKEMPFPK (m/z 1099, 2 carbamylations and m/z 1082 with loss of ammonia) and VKEAMAPKHKEMPFPK (m/z 2056, 1 Met-Ox, 4 carbamylations) were observed in the WCX eluate mass spectra, but not in eluates from hydrophobic micro tips. Hydrophobic peptides retained by the C18, C8 and C4 SPE micro columns showed intense peaks at m/z 780, 830, 1634, 2186 and 2229, but their peak intensities were significantly lower in the WCX eluate mass spectrum with lower S/N ratios. Hydrophilic, acidic peptides (pI < 7) were seen in the mass spectrum of the SAX eluate at m/z 2062 (pI 3.4), 2438 (pI 3.7), 2395 (pI 3.7) and 2518 (pI 3.7) and were absent in the mass spectra of eluates from hydrophobic materials.
In addition to SPE and ion exchange, affinity enrichment of phosphopeptides is becoming an increasingly important tool in proteomic science and biomarker discovery, as protein phosphorylation plays a crucial role in a number of biological processes.37,38 Metals such as Ga(III), Ni(II), and Zr(IV) form complexes by coordination of the phosphate group with the zirconium electrophilic metal center. Porous ZrO2 surfaces also contain hydroxyl groups, and Lewis acid and base sites located on the Zr cation, and coordinatively unsaturated oxygen, respectively.39,40 Zirconia has recently drawn significant attention as a promising separation material due to its higher thermal stability, wider operational pH range,41 and improved selectivity towards phosphopeptides when compared to IMAC materials such as TiO2.42
Figures 2 (A)–2(C) show MALDI MS spectra obtained for eluates from the IMAC-Ni, IMAC-Ga and ZrO2 SPE micro tips, respectively. A detailed list of phosphopeptides retained by IMAC and ZrO2 materials is shown in Table 2. For completion, phosphopeptides retained by SAX and WCX microtips are also included in this table. Out of the three possible phosphopeptides in the lower m/z range, AVPyPQR (m/z 910) and VLPVPQKAVPyPQR (m/z 1671 and 1714 with 1 carbamylation), only one peak corresponding to [M+H2O+K]+ of VLPVPQKAVPyPQR was observed at m/z 1728 in the ZrO2 eluate, but was absent in the IMAC eluate spectra. For the higher m/z range, a phosphopeptide at m/z 2061 was found in the eluates of IMAC-Ga and ZrO2 but was absent in the mass spectrum of the IMAC-Ni eluate whereas a phosphopeptide at m/z 2266 was found only in the mass spectrum of the ZrO2 eluate. Phosphopeptides retained by IMAC materials and ZrO2, at m/z 2141 and 2747 were more intense in the mass spectrum of the ZrO2 eluate. Phosphopeptides at m/z 3046, 3068 and 3087 corresponding to [M+H]+, [M+Na]+ and [M+H2O+H]+of ELEELNVPGEIVEsLsssEESITR, at m/z 3124, 3167 and 3204, were of the highest S/N ratios in the IMAC-Ni eluate spectrum as compared to their respective S/N ratios in the IMAC-Ga and ZrO2 eluates. Both, IMAC and ZrO2 materials showed retention of a few basic and acidic non-phosphopeptides but their number and peak intensities were significantly higher in the spectrum of the ZrO2 eluate. These peptides are captured non-specifically due to the amphoteric character of the ZrO2 surface functional groups which results in additional ion-exchange properties. At high pH values, the predominant surface species is Zr-O− which binds positively charged species, while, at low pH values, species predominate, which are able to exchange anions. Recent studies have also demonstrated that the protein/peptide fraction retained on ZrO2 particles is a function of the pH to which the sample is preconditioned prior to SPE.39,42
Figure 2.
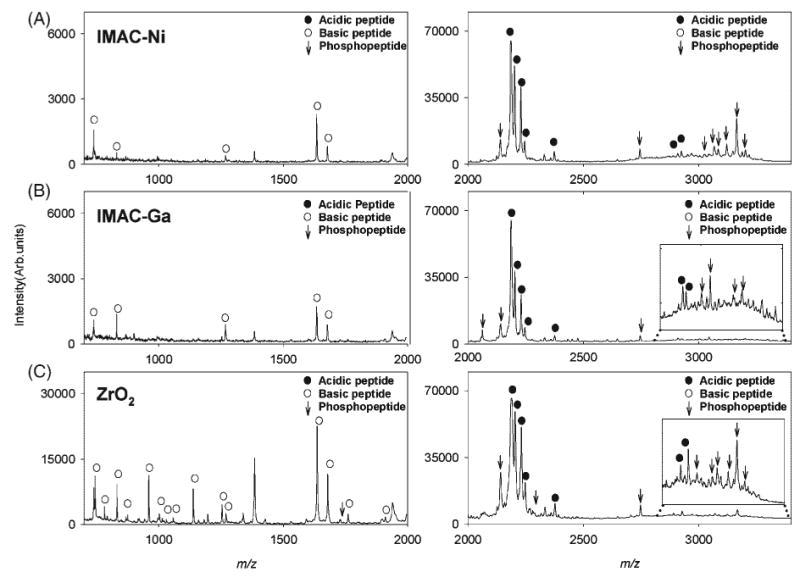
MALDI mass spectra of β-casein tryptic digest treated with (A) IMAC-Ni, (B) IMAC-Ga, and (C) ZrO2 micro tips. Left 700–2000 m/z and right 2000–3400 m/z ranges. Phosphopeptides are denoted by vertical arrows. Non-phosphopeptides with acidic and basic characters are marked by filled and empty circles, respectively.
As mentioned earlier, SAX micro tips showed non-specific retention of phosphopeptides. Therefore, the mass spectral features of SAX, IMAC materials and ZrO2 eluates were compared to investigate their phosphopeptide-binding characteristics. In the low mass range spectrum of the SAX eluate, no peaks corresponding to phosphopeptides were observed. In the higher mass range, SAX showed retention of a few phosphopeptides at m/z 2747, 3068, 3087, 3124 and 3167 that were also retained by the ZrO2 and IMAC materials, but their peak intensities were significantly lower in the of SAX eluate mass spectrum (Table 2). WCX micro tips were observed to retain four different phosphopeptides, although their peak intensities were much smaller if compared to their corresponding peaks in the SAX, ZrO2 and IMAC-Ni eluates. Acidic, carbamylated, non-phosphorylated peptides at m/z 1098, 1683 and 2439 (pI 3.5–4.0) were found in the SAX eluate mass spectrum but were absent in the spectra of IMAC and ZrO2 eluates. When extracted using IMAC and ZrO2 micro tips, these peptides exhibit net positive charges (pH of binding buffer < pI of peptide) and hence show no significant affinity for the electrophilic metal centers.
In summary, binding specificities and efficiencies of these new particle-embedded micro tips were highly dependent on their surface functionalities, isoelectric points and hydropathy of sample peptides and pH of binding buffer solution. Binding conditions, post-translational modifications and competitive and non-specific effects should all be taken into account when assessing the specificity for a given subset of species in a mixture. Following the characterization of these SPE micro tips with a model protein digest system, these were further evaluated for their applicability to human serum profiling.
Serum sample conditioning by denaturing ultrafiltration
Due to the biological complexity of human serum, ultrafiltration using molecular weight cut-off membranes facilitates the generation of mass spectrometric proteome profiles by removing high-abundance proteins.18,19 Figure 3 shows the MALDI mass spectra of human serum purified by C18 NuTips after different pre-treatments. Solid-phase extraction of the filtrate from a 50 kDa cut-off membrane by C18 micro tips yielded mass spectra with abundant features in the m/z 1000–4000 (Fig. 3(A)) and m/z 4000–10000 (Fig. 3(C)) ranges. No useful mass spectral data was obtained when the serum sample was directly analyzed by MALDI MS without ultrafiltration (Fig. 3(B), high m/z range not shown). To further explore if mass spectral information shown in Fig. 3(C) could be further improved, the 50 kDa filtrate was subsequently treated using a 3 kDa cut-off membrane (13000 g, 25 min) and the retentate was reconstituted by addition of 100 μL of 0.1% TFA solution. The analysis of the resulting sample showed improved sensitivity for molecular weight species in the 4000–10000 m/z range (Fig. 3(D)), but no new mass spectral features were observed (data not shown). As expected, no signals below 4 kDa were detected. Based on these results, we concluded that a single, 50 kDa membrane ultrafiltration step was sufficient for processing samples prior to SPE, giving the best coverage in terms of number of features detected, and with the highest sample throughput.
Figure 3.
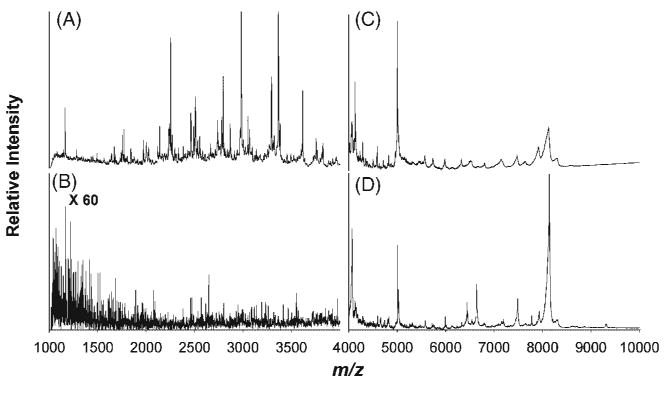
MALDI mass spectrometric profiles obtained by SPE with C18 micro tips of control human serum pre-processed by ultrafiltration using a 50 kDa membrane (A, C), without ultrafiltration (B), and with subsequent removal of low molecular weight peptides from the 50 kDa filtrate by a 3 kDa membrane (D).
Effect of serum dilution
In order to improve the filtration speed of serum samples through molecular weight cut-off filters, several dilutions were tested to optimize the experimental conditions. As shown in Fig. 4(A), a high serum concentration enhanced protein and peptide binding to the surface of C18 micro tips, resulting in optimum mass spectrometric profiles. As the serum was progressively diluted, the peak at m/z 1162.9 was observed to increase in intensity both in relative and absolute terms, while the other mass spectral features observed in Fig. 4(A) decreased, and eventually disappeared when the serum to 0.5% TFA ratio reached 1:5 (Fig. 4(C)). One possible explanation for these observations is that, as dilution increases, there is less competition for binding sites among the species present in serum, and only species with higher initial abundance are observed in the mass spectrum. The binding and ionization of these species is progressively enhanced with increasing dilution, due to the increased availability of surface binding sites, and the decrease in the number of eluted species, which reduces ionization suppression. At a certain point (Figs. 4(D) and 4(E)) the dilution effect overcomes the enhanced binding and ionization, with a concomitant decline in signal intensity. From these experiments, a ratio of 1:0.5 (v/v) of serum to 0.5% TFA aqueous solution was selected as the optimum ratio for further SPE of serum samples. This dilution was kept constant for all types of micro tips in order to compare the differences in mass spectral profiles on an equal basis.
Figure 4.
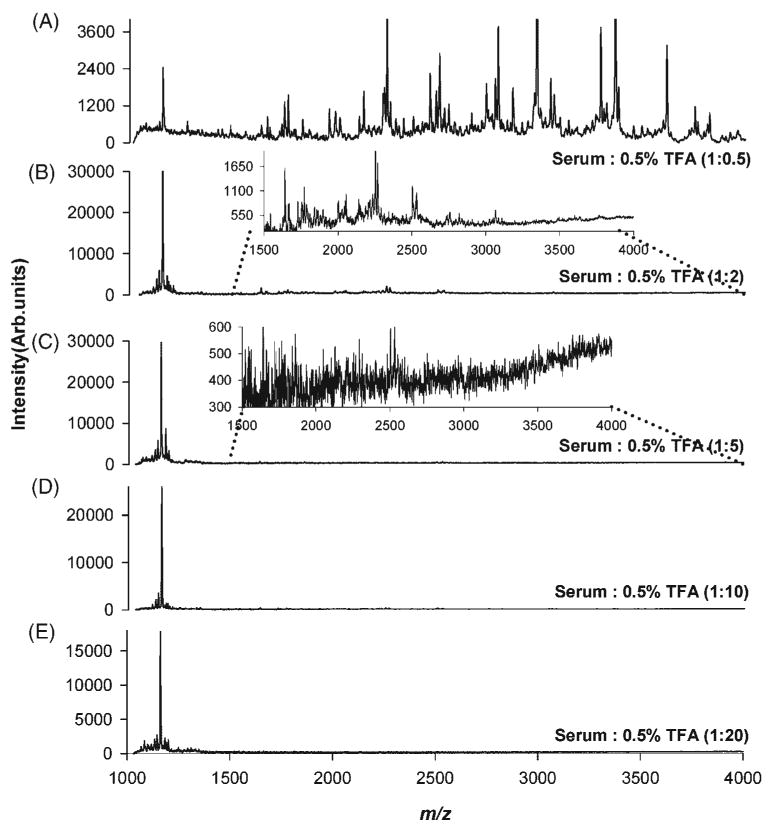
MALDI mass spectrometric profiles obtained by SPE of control human serum with C18 micro tips at different dilution ratios. Healthy individual sera samples were diluted with a 0.5% TFA solution at different serum: TFA volume ratios (v/v), (A) 1:0.5, (B) 1:2, (C) 1:5, (D) 1:10, and (E) 1:20.
Serum proteome fractionation by different materials
The MALDI MS profiles of serum samples treated with SAX, WCX, C18, C8, C4, and IMAC-Ni, IMAC-Ga, and ZrO2 micro tips are displayed in Figs. 5 and 6, respectively. Mass spectra obtained from serum extracted by C8 and C18 tips were very similar, as shown in Figs. 5(B) and 5(C) but the one extracted with C4 generated a very different spectrum (Fig. 5(A)), with peaks in a wider mass range, up to m/z 66 000. These results are in accordance with the results obtained for the pure protein digest, which showed a correlation between the length of the alkyl chain of the hydrophobic media used and the m/z range of proteins and peptides bound to it. WCX and SAX materials have oppositely charged surfaces, thus human serum SPE using these tips should lead to the generation of different MALDI mass spectra. Visual inspection of the corresponding spectra, shown in Figs. 5(D) and 5(E), revealed some apparent similarities, but closer examination and comparison of the m/z values of these profiles showed that both spectra had only very few peaks in common.
Figure 5.
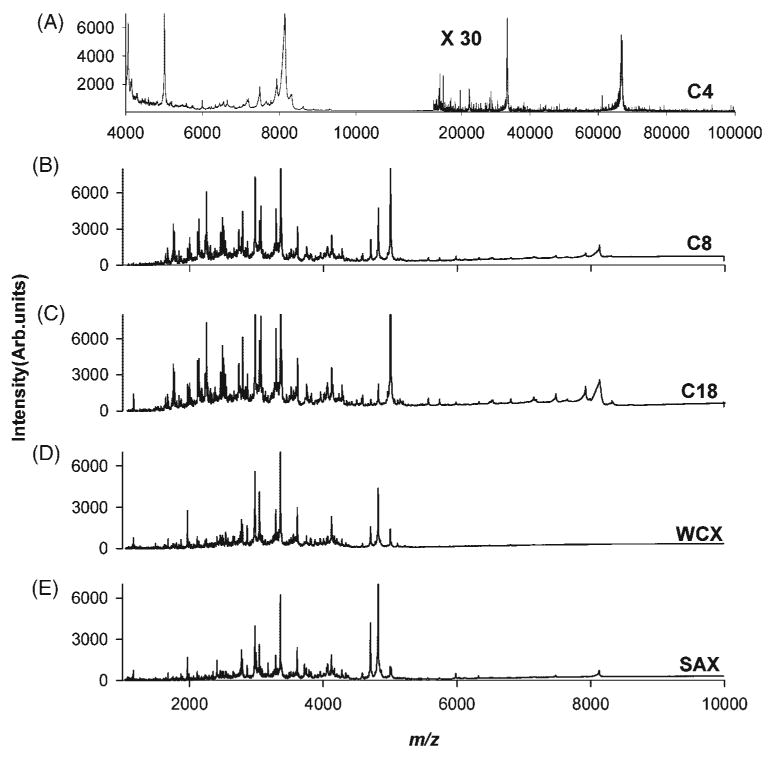
MALDI mass spectrometric profiles obtained by SPE of control human serum with (A) C4, (B) C8, (C) C18, (D) WCX, and (E) SAX micro tips.
Figure 6.
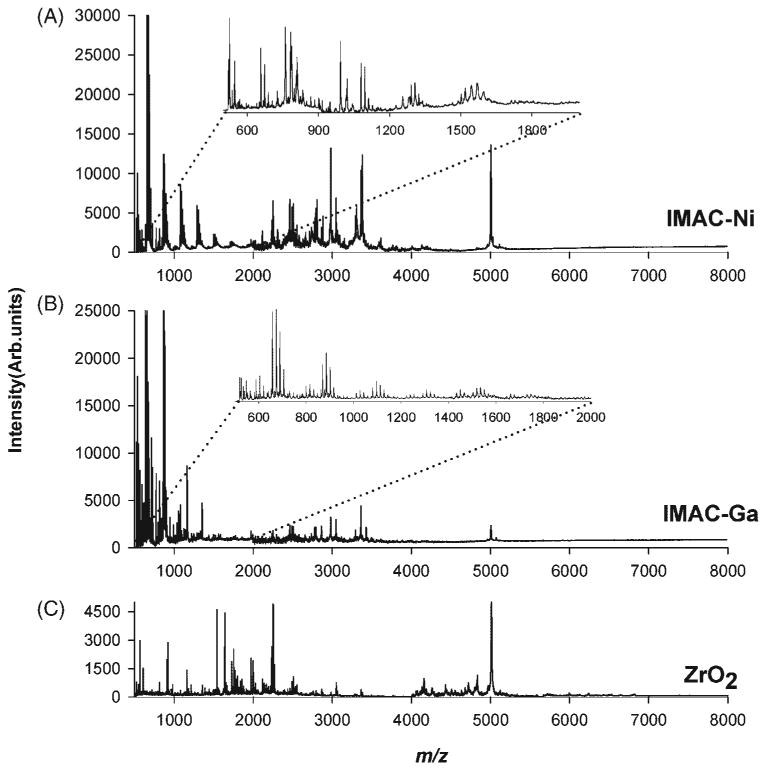
MALDI mass spectrometric profiles obtained by SPE of control human serum with (A) IMAC-Ni (II), (B) IMAC-Ga(III), and (C) ZrO2 micro tips.
Figures 6(A)–6(C) show the MALDI mass spectra of human serum extracted by IMAC-Ni, IMAC-Ga and ZrO2 micro tips, with the inset spectra showing a detail of the peaks observed in 500–2000 m/z range. The extraction efficiency of IMAC-Ga micro tips in the 2000–6000 m/z range was low compared to that of IMAC-Ni. The mass spectrum obtained for a serum sample processed using porous ZrO2 micro tips clearly differed from the ones obtained with IMAC-Ni and IMAC-Ga micro tips, suggesting that the unique surface characteristics of porous ZrO2 result in a different fraction being extracted from the same serum sample.
Peak-by-peak comparison of all the mass spectral profiles showed varying degrees of overlap in the species extracted and detected from the same serum sample and a simplified representation of these common and unique species was compiled in a Venn diagram (Fig. S2, Supplementary Material). Serum fractionation using C18 micro tip produced 106 peaks, while 113 peaks were detected using C8 micro tip (Fig. S2A, Supplementary Material). A total of 86 of these peaks overlapped between C18 and C8, but only 14 peaks overlapped when C18 and C4 were compared (Fig. S2B, Supplementary Material). Extraction with SAX and WCX micro tips produced a profile with 128 and 112 peaks, respectively, for the same serum sample (Fig. S2C, Supplementary Material), with only 18 peaks in common within the m/z accuracy afforded by the linear MALDI instrument used in this study. This observation further verified that the two oppositely charged materials offer highly complementary chemistries for serum profiling. Extractions with IMAC-Ga, IMAC-Ni and ZrO2 produced 86, 89 and 150 peaks, respectively. There were 32 overlapping peaks between the spectra obtained for C18 SPE and SAX (Fig. S2D, Supplementary Material), and 26 overlapping peaks between the spectra obtained for SAX and ZrO2 materials (Fig. S2E, Supplementary Material), while 32 peaks overlapped between IMAC-Ga and IMAC-Ni (Fig. S2F, Supplementary Material). Comparison of the spectral features extracted by ZrO2 and IMAC materials showed that there were only 18 peaks in common between ZrO2 and IMAC-Ga (Fig. S2G, Supplementary Material), and 25 overlapping peaks between ZrO2 and IMAC-Ni (Fig. S2H, Supplementary Material).
These results indicate that different embedded particles with unique surface characteristics produce different MALDI mass spectra for an identical serum sample; hence, these micro tips could be used to obtain complementary proteomic information with high sample throughput. It is important to note that the overlaps of spectral features of serum fractions extracted from different SPE microtips is the consequence of intrinsic properties like hydropathy, isoelectric point of the respective unknown proteins/peptides of the serum sample.
Inter-run reproducibility of SPE micro tips
A study of the spectral reproducibility obtainable by these micro tips is of utmost importance in profiling studies where identifying potential biomarkers from a complex sample system is the main objective. These biomarkers are identified by performing significance tests between the intensity profiles of the control and test samples. The significance of these tests is strongly affected by the variability arising from the fractionation process. An intra-run reproducibility study was thus performed using C18 and WCX micro tips in order to assess the maximum degree of variability that would be observed amongst serum profiles obtained from a single serum sample within a given experiment. This variability sets the floor of minimum biological differences that can be detected between sample groups. Eight replicate MALDI mass spectra were obtained from the same serum sample, fractionated using eight different C18 and WCX micro tips in a single laboratory session (Figs. S3 and S4, Supplementary Material). Two 2-μL eluates were obtained from each SPE micro tip and spotted separately with equal volumes of CHCA matrix. Thus there were a total of 16 spots each for the eight C18 and eight WCX micro tips used. For all C18 and WCX fractions, mass spectra acquired in the higher (m/z 5000–8000) mass ranges were appended to those acquired in the lower (m/z 1000–5000) mass range, and normalized with respect to the base peak. The asterisks denote the peaks used for coefficient of variance (CV) calculations of both m/z and peak intensity. These peaks were selected randomly over a wide mass range, disregarding peak intensities and S/N ratios. An average CV of approximately 0.04% for each m/z value was obtained in both experiments (Tables S1 and S2, Supplementary Material). The average CV of the mean intensity values was 14.9% for C18 micro tips, and 15.6% for WCX micro tips. The average %CV corresponding to spot to spot variations for both types of SPE micro tips was 3–8%. This lower intra spot %CV indicates good spot uniformity, and reproducibility of the extractions for a single tip. In all cases, sample handling operations were carried out manually, and thus, it is expected that the use of a robotic station could improve these figures. Compared to the average CV of the signal intensities, the CVs at both ends of the mass spectrum were larger, possibly originating from the lower signal-to-noise ratio in these regions (Figs. S3 and S4, Supplementary Material). Overall, the ranges in the mean intensity CV were comparable or better than those observed in previous studies,18,20 indicating that the design of the particle-embedded micro tips affords sample preparation with reproducible solution flow paths, and low pressure buildup within the tip, ensuring sufficient reproducibility for MALDI MS-based profiling studies.
Conclusions
We have evaluated and characterized particle-embedded solid-phase extraction (SPE) micro tips for their efficiency in purification, concentration, desalting and selective isolation of subgroups of peptides and small proteins from human serum. The mass spectrometric analysis of a simple protein digest with these SPE micro tips demonstrated that different embedded materials generated complementary information from identical model samples. Simple handling and minimum backpressure during sample loading and elution allowed serum profiling with good reproducibility by MALDI-TOF MS. In summary, the methods presented here provide a convenient, robust and rapid approach for peptide/protein profiling, which we expect to facilitate future biomarker discovery studies from serum proteomes.
Supplementary Material
Acknowledgments
Support from a Blanchard Assistant Professorship from the School of Chemistry and Biochemistry of the Georgia Institute of Technology to FMF, and by the Ovarian Cancer Institute at the School of Biology of the Georgia Institute of Technology is acknowledged. This work was also partially supported by Grant Number R01 AI045545-06 from the National Center for Allergic and Infectious Diseases (NIAID), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIAID or NIH. The authors wish to thank Christina Y. Hampton for proofreading the manuscript.
Contract/grant sponsor: Blanchard Assistant Professorship from the School of Chemistry and Biochemistry of the Georgia Institute of Technology.
Contract/grant sponsor: The Ovarian Cancer Institute at the School of Biology of the Georgia Institute of Technology.
Contract/grant sponsor: National Center for Allergic and Infectious Diseases; contract/grant number: R01 AI045545-06.
Footnotes
Supplementary Material: The supplementary electronic material for this paper is available in Wiley InterScience at: http://www.interscience.wiley.com/jpages/0951-4198/suppmat/.
References
- 1.Frahm JL, Howard BE, Heber S, Muddiman DC. J Mass Spectrom. 2006;41:281. doi: 10.1002/jms.1024. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed N, Oliva KT, Barker G, Hoffmann P, Reeve S, Smith IA, Quinn MA, Rice GE. Proteomics. 2005;5:4625. doi: 10.1002/pmic.200401321. [DOI] [PubMed] [Google Scholar]
- 3.Ekstrom S, Wallman L, Hok D, Marko-Varga G, Laurell T. J Proteome Res. 2006;5:1071. doi: 10.1021/pr050434z. [DOI] [PubMed] [Google Scholar]
- 4.Kozak KR, Su F, Whitelegge JP, Faull K, Reddy S, Farias-Eisner R. Proteomics. 2005;5:4589. doi: 10.1002/pmic.200500093. [DOI] [PubMed] [Google Scholar]
- 5.Purohit S, Podolsky R, Schatz D, Muir A, Hopkins D, Huang YH, She JX. Proteomics. 2006;6:6405. doi: 10.1002/pmic.200600420. [DOI] [PubMed] [Google Scholar]
- 6.Roche S, Tiers L, Provansal M, Piva MT, Lehmann S. Proteome Sci. 2006;4:20. doi: 10.1186/1477-5956-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen Y, Kim J, Strittmatter EF, Jacobs JM, Camp DG, 2nd, Fang R, Tolie N, Moore RJ, Smith RD. Proteomics. 2005;5:4034. doi: 10.1002/pmic.200401246. [DOI] [PubMed] [Google Scholar]
- 8.Tirumalai RS, Chan KC, Prieto DA, Issaq HJ, Conrads TP, Veenstra TD. Mol Cell Proteomics. 2003;2:1096. doi: 10.1074/mcp.M300031-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Villanueva J, Philip J, Entenberg D, Chaparro CA, Tanwar MK, Holland EC, Tempst P. Anal Chem. 2004;76:1560. doi: 10.1021/ac0352171. [DOI] [PubMed] [Google Scholar]
- 10.Villanueva J, Philip J, Chaparro CA, Li Y, Toledo-Crow R, DeNoyer L, Fleisher M, Robbins RJ, Tempst P. J Proteome Res. 2005;4:1060. doi: 10.1021/pr050034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Z, Luke BT, Izmirlian G, Umar A, Lynch PM, Phillips RK, Patterson S, Conrads TP, Veenstra TD, Greenwald P, Hawk ET, Ali IU. Cancer Res. 2004;64:2904. doi: 10.1158/0008-5472.can-03-3754. [DOI] [PubMed] [Google Scholar]
- 12.King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T. J Am Soc Mass Spectrom. 2000;11:942. doi: 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]
- 13.Ledford EB, Jr, Rempel DL, Gross ML. Anal Chem. 1984;56:2744. doi: 10.1021/ac00278a027. [DOI] [PubMed] [Google Scholar]
- 14.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. J Proteome Res. 2006;5:1792. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs JM, Adkins JN, Qian WJ, Liu T, Shen Y, Camp DG, 2nd, Smith RD. J Proteome Res. 2005;4:1073. doi: 10.1021/pr0500657. [DOI] [PubMed] [Google Scholar]
- 16.Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, Su Q, McGrath AM, Estock MA, Parmar PP, Zhao M, Huang ST, Zhou J, Wang F, Esquer-Blasco R, Anderson NL, Taylor J, Steiner S. Proteomics. 2003;3:1345. doi: 10.1002/pmic.200300449. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell BL, Yasui Y, Lampe JW, Gafken PR, Lampe PD. Proteomics. 2005;5:2238. doi: 10.1002/pmic.200401099. [DOI] [PubMed] [Google Scholar]
- 18.Orvisky E, Drake SK, Martin BM, Abdel-Hamid M, Ressom HW, Varghese RS, An Y, Saha D, Hortin GL, Loffredo CA, Goldman R. Proteomics. 2006;6:2895. doi: 10.1002/pmic.200500443. [DOI] [PubMed] [Google Scholar]
- 19.Harper RG, Workman SR, Schuetzner S, Timperman AT, Sutton JN. Electrophoresis. 2004;25:1299. doi: 10.1002/elps.200405864. [DOI] [PubMed] [Google Scholar]
- 20.Callesen AK, Mohammed S, Bunkenborg J, Kruse TA, Cold S, Mogensen O, Christensen R, Vach W, Jorgensen PE, Jensen ON. Rapid Commun Mass Spectrom. 2005;19:1578. doi: 10.1002/rcm.1960. [DOI] [PubMed] [Google Scholar]
- 21.de Noo ME, Tollenaar RA, Ozalp A, Kuppen PJ, Bladergroen MR, Eilers PH, Deelder AM. Anal Chem. 2005;77:7232. doi: 10.1021/ac050571f. [DOI] [PubMed] [Google Scholar]
- 22.Chertov O, Biragyn A, Kwak LW, Simpson JT, Boronina T, Hoang VM, Prieto DA, Conrads TP, Veenstra TD, Fisher RJ. Proteomics. 2004;4:1195. doi: 10.1002/pmic.200300677. [DOI] [PubMed] [Google Scholar]
- 23.Prahalad AK, Hickey RJ, Huang J, Hoelz DJ, Dobrolecki L, Murthy S, Winata T, Hock JM. Proteomics. 2006;6:3482. doi: 10.1002/pmic.200500929. [DOI] [PubMed] [Google Scholar]
- 24.Tang N, Tornatore P, Weinberger SR. Mass Spectrom Rev. 2004;23:34. doi: 10.1002/mas.10066. [DOI] [PubMed] [Google Scholar]
- 25.Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Lancet. 2002;359:572. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 26.Petricoin EF, Mills GB, Kohn EC, Liotta LA. Lancet. 2002;360:170. [Google Scholar]
- 27.Kozak KR, Amneus MW, Pusey SM, Su F, Luong MN, Luong SA, Reddy ST, Farias-Eisner R. Proc Natl Acad Sci USA. 2003;100:12343. doi: 10.1073/pnas.2033602100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baggerly KA, Morris JS, Coombes KR. Bioinformatics. 2004;20:777. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 29.Ekblad L, Baldetorp B, Ferno M, Olsson H, Bratt C. J Proteome Res. 2007;6:1609. doi: 10.1021/pr060633y. [DOI] [PubMed] [Google Scholar]
- 30.Muck A, Nesnerova P, Pichova I, Svatos A. Electrophoresis. 2005;26:2835. doi: 10.1002/elps.200400014. [DOI] [PubMed] [Google Scholar]
- 31.Dunn JD, Watson JT, Bruening ML. Anal Chem. 2006;78:1574. doi: 10.1021/ac0515982. [DOI] [PubMed] [Google Scholar]
- 32.Gobom J, Nordhoff E, Mirgorodskaya E, Ekman R, Roepstorff P. J Mass Spectrom. 1999;34:105. doi: 10.1002/(SICI)1096-9888(199902)34:2<105::AID-JMS768>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Available http://prospector.ucsf.edu/.
- 34.Available http://www.vivo.colostate.edu/molkit/hydropathy/index.html.
- 35.Hopp TP, Woods KR. Proc Natl Acad Sci USA. 1981;78:3824. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Righetti PG. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;841:14. doi: 10.1016/j.jchromb.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 37.Posewitz MC, Tempst P. Anal Chem. 1999;71:2883. doi: 10.1021/ac981409y. [DOI] [PubMed] [Google Scholar]
- 38.Zhou W, Merrick BA, Khaledi MG, Tomer KB. J Am Soc Mass Spectrom. 2000;11:273. doi: 10.1016/s1044-0305(00)00100-8. [DOI] [PubMed] [Google Scholar]
- 39.Nawrocki J, Rigney MP, McCormick A, Carr PW. J Chromatogr A. 1993;657:229. doi: 10.1016/0021-9673(93)80284-f. [DOI] [PubMed] [Google Scholar]
- 40.Nawrocki J, Dunlap C, McCormick A, Carr PW. J Chromatogr A. 2004;1028:1. doi: 10.1016/j.chroma.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 41.Hoth DC, Rivera JG, Colon LA. J Chromatogr A. 2005;1079:392. doi: 10.1016/j.chroma.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 42.Kweon HK, Hakansson K. Anal Chem. 2006;78:1743. doi: 10.1021/ac0522355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.