

Abstract
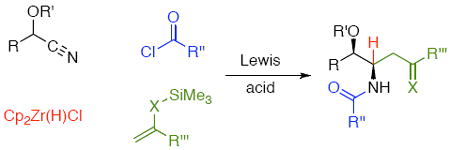
α-Branched amides are prepared by multicomponent reactions in which nitriles undergo hydrozirconation to form metalloimines that react with acyl chlorides. The resulting acylimines react with a variety of π-nucleophiles in the presence of Lewis acids to form the desired amides.
Multicomponent reactions1 have emerged as attractive processes for rapidly increasing molecular complexity and structural diversity.2 Nitriles are well-suited to be substrates in multicomponent reactions because their polarization and multiple π-bonds create numerous opportunities for sequential addition processes. We have demonstrated3 the viability of using nitriles in this capacity through the sequences that are shown in Scheme 1. Nitriles (1) react readily4 with the Schwartz reagent (Cp2Zr(H)Cl)5 to form metalloimines (2) that add into acyl chlorides6 to yield acylimines7 (3). These acylimines are useful electrophiles for the formation of a wide range of amide-containing structures.8 Specifically we have shown that tetrahydropyranyl nitrile 4 can be transformed into acyl aminal 5 through hydrozirconation, acylation, and MeOH addition,3a and nitrile 6 can be converted to tricyclic amide 7 by employing an intramolecular Friedel-Crafts reaction as the nucleophilic addition step.3b In this manuscript we show that this method can be applied to multicomponent syntheses of α-branched amides through diastereoselective bimolecular additions of π-nucleophiles into nitrile-derived acylimines. We also report that acylimines that lack branching at the α-position can be tautomerized to form E-enamides.
Scheme 1.
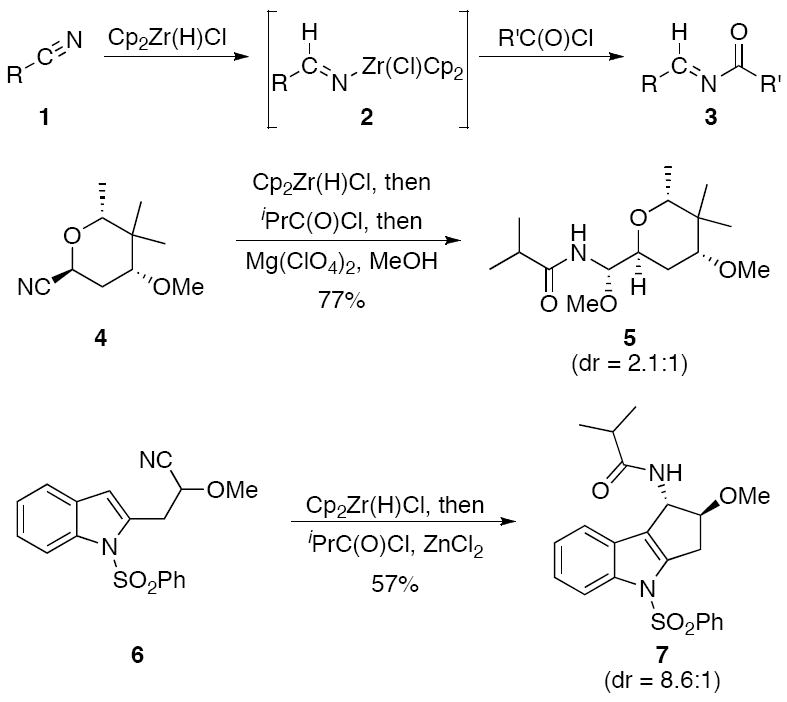
Amide formation from nitrile hydrozirconation.
We chose to investigate additions of π-nucleophiles into acylimines that are formed from cyanohydrin ether 8 (Scheme 2) in consideration of our observation3b that intramolecular carbon–carbon bond forming reactions were inefficient for substrates in which acylimine tautomerization is facile. This substrate is readily prepared from the diethyl acetal of heptanal through BiBr3-mediated ionization and TMSCN addition.9 Our initial studies focused on the use of methallyl trimethylsilane (9) as the nucleophile. Subjecting 8 to hydrozirconation and acylation with methoxyacetyl chloride followed by adding 9 did not provide the desired amide, with the acyl hemiaminal that forms from water addition during the work-up being the only isolable product. We postulated that, in contrast to alcohols and thiols, weakly reactive π-nucleophiles require acylimine activation by a Lewis acid to promote addition. Adding Sc(OTf)3 to the reaction mixture indeed resulted in addition to form amides 10 and 11 as a 1:1 mixture in 60% combined yield (see below for stereochemical determination). Changing the Lewis acid to ZnBr2 provided a 55% yield of 11 while completely suppressing the formation of 10.
Scheme 2.
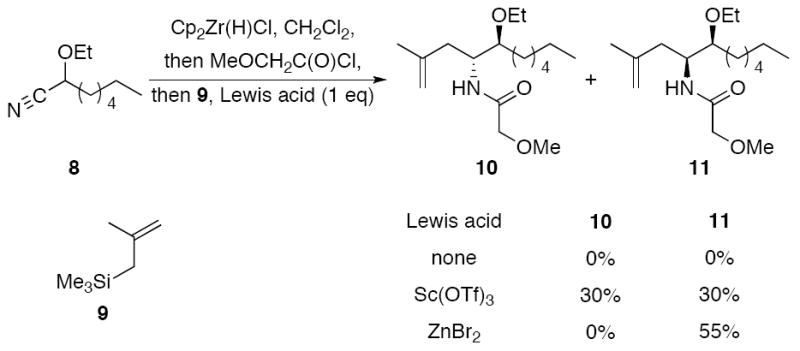
Bimolecular carbon–carbon bond formation.
We devised an independent pathway for product synthesis to assign the stereochemical relationship of the products (Scheme 3). The sequence commenced with a Masamune syn-aldol reaction10 between the (E)-dibutylboron enolate of ester 12 and heptanal to provide 13 as a single stereoisomer. Methylation of the hydroxyl group and ester cleavage formed acid 14, which was converted to carbamate 15 through a Curtius reaction under Shioiri’s conditions.11 The methyl ether was prepared rather than the ethyl ether because it could be formed under milder conditions. Cleaving the Alloc-group with Pd(PPh3)4 and Bu3SnH in the presence of methoxyacetyl chloride12 led to the formation of 16. While 16 showed several spectral features that were essentially identical to those of 10, assigning the structures unambiguously required that we prepare methoxy nitrile 17 and subject it to the multicomponent amide synthesis protocol wih catalysis by of Sc(OTf)3 and ZnBr2. In the presence of ZnBr2 we observed the formation of a single stereoisomer with spectroscopic properties that did not match those of 16, allowing us to assign this structure as syn-isomer 18. In the presence of Sc(OTf)3 we observed the formation of a 1:1 mixture of 16 and 18, thereby establishing the structural assignment. The formation of the syn-isomer through ZnBr2 catalysis is consistent with the reaction proceeding through a transition state in which the alkoxy group and the nitrogen of the acylimine chelate the Lewis acid.13
Scheme 3.
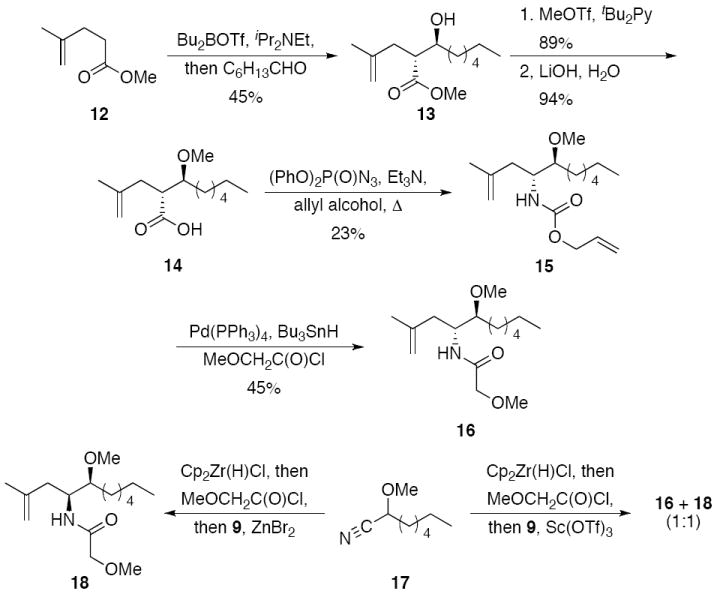
Stereochemical determination.
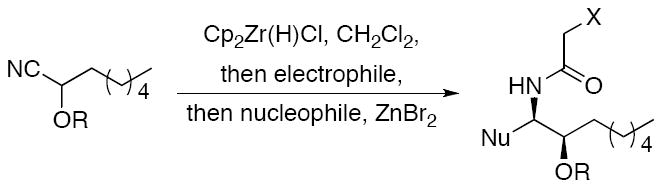
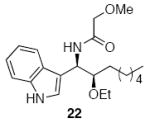
A sampling of the scope of nucleophiles and electrophiles that can be employed in this reaction is shown in Table 1. Allyl trimethylsilane provided amide 19 (entry 1), though the process was significantly less efficient than the reaction with methallyl trimethylsilane. Enolsilanes derived from acetone (entry 2) and acetophenone (entry 3) added into the acylimine smoothly to form β-amido ketones 20 and 21, respectively. Indole was a suitable nucleophile for the formation of amide 22 (entry 4), indicating that bimolecular Friedel-Crafts additions are possible with appropriately nucleophilic arenes. The reactivity trends in this series generally followed the Mayr nucleophilicity table,14 with methallyl trimethylsilane proving to be the least nucleophilic species to afford smooth reactivity. Notably allyltin reagents, though more nucleophilic than allylsilanes, did not provide the desired addition products, suggesting an incompatibility of these nucleophiles with the reaction conditions.
Table 1.
Nucleophile and electrophile variations.
 | |||||
|---|---|---|---|---|---|
| entry | nitrile | electrophile | nucleophile | product | yield (%) |
| 1 | 8 | 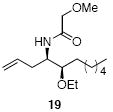 |
18 | ||
| 2 | 8 | 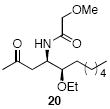 |
56 | ||
| 3b | 8 | 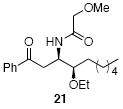 |
55 | ||
| 4 | 8 | 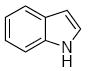 |
 |
60 | |
| 5c | 17 | 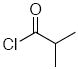 |
|
 |
47 |
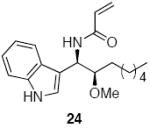
| 6d | 17 | 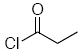 |
|
 |
42 |
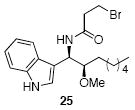
| 7e | 17 | 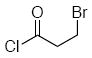 |
|
 |
46 |
Representative procedure: Cp2Zr(H)Cl (1.2 equiv) was added to a solution of the substrate in the solvent (0.1 M). The mixture stirred for 10 min at rt, then was cooled to 0 °C. The electrophile (1.2-1.5 equiv) was added and stirred for 10 min. The nucleophile (4 equiv) and the ZnBr2 (1 equiv) were added at a suitable temperature (see Supporting Information for details) and the reaction was stirred until the acyl hemiaminal was not observed by TLC.
Reaction provided a 5.2:1 ratio of diastereomeric products.
Reaction provided a 4.2:1 ratio of diastereomers.
Reaction provided a 4.9:1 ratio of diastereomers.
Reaction provided a 4.5:1 ratio of diastereomers.
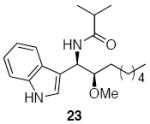
Methoxyacetyl chloride proved to be the best acylating agent that we tested with respect to product yield and diastereocontrol, but other acid chlorides can also be employed. Isobutyryl chloride (entry 5), acryloyl chloride (entry 6), and β-bromopropionyl chloride (entry 7) provided amides 23-25 when indole was utilized as the nucleophile. While the yields for these reactions were moderate the capacity to prepare useful amounts of a range of amides will prove useful for applications to library synthesis. No other structures, such as the products of inverse electron demand hetero Diels-Alder reactions,15 could be isolated from these reactions. This indicates that the moderate yields result from the competitive formation of chromatographically immobile products
When the Danishefsky diene16 (26) was used as the nucleophile (Scheme 4) the vinylogous Mannich product 27 was isolated rather than the hetero Diels-Alder product.17 This reaction appears to be quite efficient when monitored by TLC, though only low and variable yields of slightly impure product could be isolated. Subsequent studies showed that 27 is highly unstable toward acids, including silica gel, and readily undergoes fragmentation through a retro Mannich reaction. We directed our efforts toward devising a transition metal mediated cyclization to circumvent the need for Brønsted acids in an effort to convert 27 to dihydropyridone 28 without promoting fragmentation. Our recently developed protocol for adding oxygen and nitrogen nucleophiles into α,β-unsaturatedketones in the presence of Ph3PAuCl and AgSbF6 was well suited for this application.18 When we subjected partially purified 27 to these conditions we isolated 28 as a single stereoisomer in 48% yield, suggesting that this sequence can be effective for preparing dihydropyridones if superior isolation methods for the Mannich adducts can be developed.
Scheme 4.
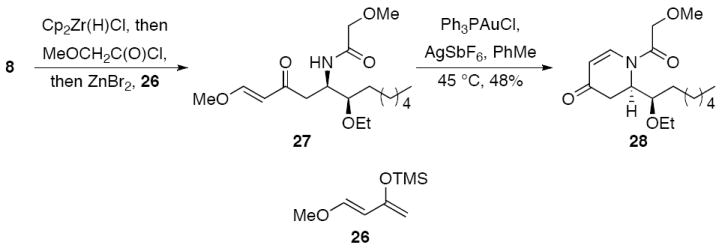
Stereoselective dihydropyridone synthesis.
Efforts to conduct these reactions with non-branched nitriles were not fruitful, in accord with prior studies,3b with dimers and higher oligomers of the acylimine intermediates being formed as the major products. We reasoned that these products were derived from the tautomerization of a portion of the acylimines to form enamides. The nucleophilic enamides can add into the residual acylimines to initiate the oligomerization reaction. In consideration of the important role of enamides in promoting the biological activity of several natural products19 and the growing number of reactions that employ enamides as nucleophiles,20 we examined the possibility of deliberately tautomerizing the acylimine intermediates from the nitrile hydrozirconation/acylation sequence. Conversions of putative acylimine intermediates into enamides have previously been reported,21 though low levels of geometrical control were observed in these processes.22
Our studies on enamide formation initially utilized non-functionalized nitrile 29 as the substrate (Scheme 5). Hydrozirconation and acylation formed the acylimine, which was then subjected to a variety of amine bases to promote tautomerization. These reactions resulted in the slow formation of complex product mixtures that contained cis- and trans-enamides along with dimerization and oligomerization products. Successful enamide formation was effected by forming the intermediate acylimine in the presence of Et3N and quickly adding BF3•OEt2. The use of THF as the solvent in this reaction was critical, since oligomerizaton occurred rapidly in CH2Cl2. Through this method 29 could be converted to trans-enamide 30 in 57% yield. Negligible amounts of cis-isomer and dimerization products were formed in this reaction. The reaction requires the presence of Et3N. BF3•OEt2 promotes substantial decomposition in the absence of Et3N. The formation of dienamides is also possible through this method. Allylic nitrile 31 was subjected to hydrozirconation, acylation, and tautomerization to form 32 in 62% yield. Conjugation enhances the tautomerization rate, with the formation of 32 being complete in less than 2 h and the formation of 30 requiring > 6 h. Thus, while tautomerization prevents unbranched nitriles from undergoing efficient addition reactions, appropriate reaction conditions have been developed for effecting a facile enamide synthesis.
Scheme 5.
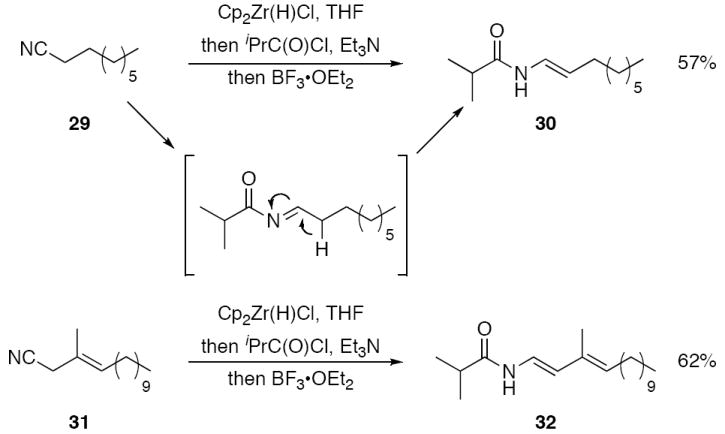
Selective enamide synthesis through tautomerization.
We have devised a general multicomponent approach to α-branched amide synthesis from nitriles through a sequence of hydrozirconation, acylation, and nucleophile addition. Allylsilanes, enolsilanes, and indole were shown to react with acylimines in the presence of a Lewis acid, provided that adjacent branching was present to inhibit decomposition through tautomerization. Reactions that employ cyanohydrin ethers as substrates proceed with good to excellent levels of chelation control when ZnBr2 is used as the Lewis acid. Acylimines that lack adjacent branching undergo selective tautomerization to form E-enamides when subjected to a mixture of Et3N and BF3•OEt2, making the protocol applicable to the synthesis of enamide-containing cytotoxins. The range of nitriles, electrophiles, and nucleophiles that are compatible with the reaction conditions indicates that the method is well suited for the preparation of structurally diverse libraries of amides from easily accessible precursors, and the capacity to control the stereochemical outcome of the process by changing the Lewis acid suggests a potential for utilizing chiral catalysts to prepare enantiomerically enriched products.
Supplementary Material
Experimental procedures and spectral data for all new compounds. Available on the World Wide Web at http://pubs.acs.org.
Acknowledgments
This work was supported by National Institutes of Health (P50-GM067082) and the Myelin Research Foundation (unrestricted research grant) for generous financial support of this work. MEG was a recipient of a Novartis graduate fellowship.
References
- 1.(a) Dömling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; (b) Ramón DJ, Yus M. Angew Chem Int Ed. 2005;44:1602. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]; (c) Bienaymé H, Hulme C, Oddon G, Schmitt P. Chem Eur J. 2000;6:3321. doi: 10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Schreiber SL. Science. 2000;287:1964. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (b) Burke MD, Schreiber SL. Angew Chem Int Ed. 2004;43:46. doi: 10.1002/anie.200300626. [DOI] [PubMed] [Google Scholar]; (c) Spring DR. Chem Soc Rev. 2005;34:472. doi: 10.1039/b312875j. [DOI] [PubMed] [Google Scholar]; (d) Tan DS. Nat Chem Biol. 2005;1:74. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]; (e) Arya P, Joseph R, Gan Z, Rakic B. Chem Biol. 2005;12:163. doi: 10.1016/j.chembiol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wan S, Green ME, Park J-H, Floreancig PE. Org Lett. 2007;9:5385. doi: 10.1021/ol702184n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiao Q, Floreancig PE. Org Lett. 2008;10:1139. doi: 10.1021/ol8000409. [DOI] [PubMed] [Google Scholar]
- 4.(a) Erker G, Frömberg W, Atwood JL, Hunter WE. Angew Chem Int Ed. 1984;23:68. [Google Scholar]; (b) Frömberg W, Erker G. J Organomet Chem. 1985;280:343. [Google Scholar]; (c) Anbhaikar NB, Herold M, Liotta DC. Heterocycles. 2004;62:217. [Google Scholar]
- 5.Cp2Zr(H)Cl is commercially available but we prefer to prepare it from Cp2ZrCl2. Hart DW, Schwartz J. J Am Chem Soc. 1974;96:8115.Buchwald SL, LaMaire SJ, Nielsen RB, Watson BT, King SM. Org Synth. 1993;71:77.
- 6.Maraval A, Igau A, Donnadieu B, Majoral JP. Eur J Org Chem. 2003:385. [Google Scholar]
- 7.For reviews of acylimines and related species, see: Petrini M, Torregiani E. Synthesis. 2007:159.Maryanoff BE, Zhang HC, Cohen JH, Turchi IJ, Maryanoff CA. Chem Rev. 2004;104:1431. doi: 10.1021/cr0306182.Speckamp WN, Moolenaar MJ. Tetrahedron. 2000;56:3817.
- 8.For other examples of nucleophilic additions into nitriles followed by acylation, see: Savarin CG, Boice GN, Murry JA, Corley E, DiMichele L, Hughes D. Org Lett. 2006;8:3903. doi: 10.1021/ol061318k.Fleming FF, Wei G, Zhang Z, Steward OW. Org Lett. 2006;8:4903. doi: 10.1021/ol0619765.
- 9.Komatsu N, Uda M, Suzuki H, Takahashi T, Domae T, Wada M. Tetrahedron Lett. 1997;38:7215. [Google Scholar]
- 10.Abiko A, Liu J, Masamune S. J Org Chem. 1996;61:2590. doi: 10.1021/jo960252b. [DOI] [PubMed] [Google Scholar]
- 11.Ninomiya K, Shioiri T, Yamada S. Tetrahedron. 1974;30:2151. [Google Scholar]
- 12.Roos E, Bernabe P, Hiemstra H, Speckamp N, Kaptein B, Boesten WHJ. J Org Chem. 1995;60:1733. [Google Scholar]
- 13.For related stereochemical discussions, see: Sugiura M, Hagio H, Hirabayashi R, Kobayashi S. J Am Chem Soc. 2001;123:12510. doi: 10.1021/ja0170448.Ella-Menye J-R, Dobbs W, Billet M, Klotz P, Mann A. Tetrahedron Lett. 2005;46:1897.
- 14.Mayr H, Kempf B, Ofial AR. Acc Chem Res. 2003;36:66. doi: 10.1021/ar020094c. [DOI] [PubMed] [Google Scholar]
- 15.(a) Scola PM, Weinreb SM. J Org Chem. 1986;51:3248. [Google Scholar]; (b) Chao W, Weinreb SM. Tetrahedron Lett. 2000;41:9199. [Google Scholar]
- Danishefsky S, Kitahara T. J Am Chem Soc. 1974;96:7807. [Google Scholar]
- 16.For examples of hetero Diels-Alder reactions between Danishefsky’s diene and aryl imines, see: Badorrey R, Cativiela C, Díaz-de-Villegas MD, Gálvez JA. Tetrahedron. 1999;55:7601.Akiyama T, Takaya J, Kagoshima H. Tetrahedron Lett. 1999;40:7831.Yuan Y, Li X, Ding K. Org Lett. 2002;4:3309. doi: 10.1021/ol0265822.Josephsohn NS, Snapper ML, Hoveyda AH. J Am Chem Soc. 2003;125:4018. doi: 10.1021/ja030033p.
- 17.Jung HH, Floreancig PE. J Org Chem. 2007;72:7359. doi: 10.1021/jo071225w. [DOI] [PubMed] [Google Scholar]
- 18.(a) Bhattacharjee A, Seguil OR, De Brabander JK. Tetrahedron Lett. 2001;42:1217. [Google Scholar]; (b) Wu Y, Liao X, Wang R, Xie X-S, De Brabander JK. J Am Chem Soc. 2002;124:3245. doi: 10.1021/ja0177713. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Kim DW, Baati R, O’Brate A, Giannakakou P. Chem Eur J. 2003;9:6177. doi: 10.1002/chem.200305230. [DOI] [PubMed] [Google Scholar]; (d) Petri AF, Sasse F, Maier ME. Eur J Org Chem. 2005:1865. [Google Scholar]
- 19.Carbery DR. Org Biomol Chem. 2008;6:3455. doi: 10.1039/b809319a. [DOI] [PubMed] [Google Scholar]
- 20.For a report of an effective tautomerization process that does not explicitly address product geometry, see: Mecozzi T, Petrini M. Synlett. 2000:73.Bayer A, Maier ME. Tetrahedron. 2004;60:6665.Shono T, Matsumura Y, Tsubata K, Sugihara Y, Yamane S-i, Kanazawa T, Aoki T. J Am Chem Soc. 1982;104:6697.
- 21.For representative examples of other recently reported methods for enamide synthesis, see: Goossen LJ, Salih KSM, Blanchot M. Angew Chem Int Ed. 2008;47:8492. doi: 10.1002/anie.200803068.Bolshan Y, Batey RA. Angew Chem Int Ed. 2008;47:2109. doi: 10.1002/anie.200704711.Pan X, Cui Q, Ma D. Org Lett. 2004;6:1809. doi: 10.1021/ol049464i.Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667. doi: 10.1021/ol035355c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for all new compounds. Available on the World Wide Web at http://pubs.acs.org.