

Abstract
Rearrangement of chromosomal bands 1q21–23 is one of the most frequent chromosomal aberrations observed in hematological malignancy. The genes affected by these rearrangements remain poorly characterized. Typically, 1q21–23 rearrangements arise during tumor evolution and accompany disease-specific chromosomal rearrangements such as t(14;18) (BCL2) and t(8;14) (MYC), where they are thus thought to play an important role in tumor progression. The pathogenetic basis of this 1q21–23-associated disease progression is currently unknown. In this setting, we surveyed our series of follicular lymphoma for evidence of recurring 1q21–23 breaks and identified three cases in which a t(14;18)(q32;q21) was accompanied by a novel balanced t(1;22)(q22;q11). Molecular cloning of the t(1;22) in a cell line (B593) derived from one of these cases and detailed fluorescent in situ hybridization mapping in the two remaining cases identified the FCGR2B gene, which encodes the immunoreceptor tyrosine-based inhibition motif-bearing IgG Fc receptor, FcγRIIB, as the target gene of the t(1;22)(q22;q11). We demonstrate deregulation of FCGR2B leading to hyperexpression of FcγRIIb2 as the principal consequence of the t(1;22). This is evidence that IgG Fc receptors can be targets for deregulation through chromosomal translocation in lymphoma. It suggests that dysregulation of FCGR2B may play a role in tumor progression in follicular lymphoma.
Cytogenetic analysis of B-cell non-Hodgkin's lymphoma (NHL) has revealed the presence of numerous diagnostic, balanced, chromosomal translocations (primary chromosomal rearrangements), which on molecular analysis have been found to deregulate cellular protooncogenes through their juxtaposition with regulatory elements deriving from Ig heavy and light chain gene loci (1). It is now apparent that these rearrangements arise through one of three distinct mechanisms; V gene recombination, class switch recombination (for IGH), and somatic hypermutation (2, 3). Detailed analysis of the physiological roles of a number of the genes deregulated by such translocations has revealed a preponderance of genes (transcription factors, cell cycle regulatory proteins, cell death proteins) involved in the maintenance of B-cell homeostasis (1).
Molecular cloning of frequent chromosomal breakpoints has thus proved to be a useful strategy for identification of genes important in tumorigenic processes in NHL. To date, this approach has been successful in identifying genes deregulated by disease-specific chromosomal translocations such as t(14;18) (BCL2) in follicular lymphoma (4), 3q27 rearrangement (LAZ3/BCL6) in diffuse large cell lymphoma (5–7), and t(11;14) (CCND1) in mantle cell lymphoma (8). It has, however, become clear that deregulation of these genes, although necessary for tumor initiation, may not be sufficient. For instance, in a transgenic mouse model of t(14;18), B-cells exhibit extended survival (9) but are not rendered malignant (10). Likewise, in various transgenic mouse models, it has not been possible to demonstrate a dominant role for action of cyclin D1 as an oncogene (8). This indicates a requirement for further genetic changes to obtain a fully malignant phenotype. In this respect, recurring secondary chromosomal translocations in NHL are of prognostic significance and, although poorly characterized at the present time, may constitute important events in lymphomagenesis and/or tumor progression in these tumors (11).
One of the most frequent nonrandom secondary chromosomal anomalies described in B-cell NHL is rearrangement of chromosomal bands 1q21–23 (12, 13). Abnormalities of this region are particularly frequent in follicular lymphoma (20%) (14) and in diffuse large cell lymphoma (16.6%), where they have been shown to be of poor prognosis (13). Breakpoints in 1q21 are observed in 39% of marginal zone B-cell lymphoma (15), and, in Burkitts lymphoma, the t(8;14) is frequently associated with dup(1)(q21q32) (20%) (12). It should be noted that aberrations affecting 1q21–23 are not restricted to B-cell NHL, as evidenced by frequent implication of this region in primary chromosomal translocations in hematological malignancies of myeloid origin (16).
To date, only three translocations affecting 1q21–23 have been characterized in any detail. A t(1;14) observed in a B-cell acute lymphoblastic leukemia-derived cell line deregulates BCL9 (17). AF1q has been cloned as a fusion partner of MLL in a t(1;11)(q21;q23) observed in infant acute myelomonocytic leukemia (18). Finally, a gene, JTB, has recently been cloned from the breakpoint of a jumping translocation in a case of acute myelomonocytic leukemia (19). The function of these genes and therefore their pathogenic role is currently unknown. This report describes the molecular characterization of a novel t(1;22)(q22;q11) observed in B-cell NHL. This translocation is recurrent and results in deregulation of FCGR2B that encodes the low affinity IgG Fc receptor FcγRIIB.
Materials and Methods
Cell Lines, Cell Culture, and Cytogenetic Analysis.
The B593 cell line was established by xenograft of cells derived from the lymph node biopsy of a 46-year-old patient (Co) (diagnosed with grade III follicular lymphoma) onto Nude mice. Tumors were passaged in vivo for 4 months, at which time animals were killed and tumor cells were harvested. Immunophenotypic, cytological, cytogenetic, and molecular analysis (Ig gene rearrangements) confirmed identity with the original patient tumor material. The B593 cell line was established in vitro from these tumor cells. Immunophenotypic, cytogenetic, and IGL analysis have remained stable during repeated passaging in vitro. Also, normal B lymphocytes from the same patient were established as a stable cell line in vitro through Epstein–Barr virus immortalization (B-EBV cell line). The cell line Br97 was derived according to a similar protocol from the lymph node biopsy of an 89-year-old patient diagnosed with diffuse large cell lymphoma (Ra). G. Lenoir (International Agency for Research on Cancer, Lyon, France) kindly provided the Burkitt lymphoma cell lines. Other cell lines (U937, Jurkat, CEM, and Raji) were obtained through the American Type Culture Collection. Cell lines were cultured in RPMI supplemented with 10% fetal calf serum under standard conditions. Cytogenetic analysis of cell lines and primary lymphoma specimens was performed as described (20).
Translocation Breakpoint Cloning.
A partial genomic library was prepared in λZAP from B593 EcoRI-digested DNA that had been size selected in the 2- to 3-kb range by electrophoresis. Clone pBKEE2.2 containing the breakpoint sequence was obtained by screening this library with the IGLC2/3-specific probe p607-/21–68. Sequencing was performed by using an AB1 PRISM automated sequencer and AmpliTaq polymerase, FS (Perkin–Elmer). Sequences were analyzed at the National Center for Biotechnology Information against nrGDB and expressed sequence tag databases by using the Basic Local Alignment Search Tool.
PAC986b4 Isolation, Shotgun-Subcloning, and Sequencing.
The primers 5′- GAATTCAGGGCACACTATACTC-3′ and 5′-CATACACAAAGATGCTCTGAG-3′ were used to amplify a 1q22 breakpoint-specific probe (EB1). EB1 was used to screen a human genomic gridded PAC library. PAC986b4 was one of eight positive clones isolated and mapped back to chromosome 1q22 by fluorescent in situ hybridization (FISH) on B593 and normal lymphocyte metaphases. For shotgun sequencing, PAC986b4 DNA was digested to completion with HaeIII or AluI cloned into PCR-Script (Stratagene), and ≈100 clones were sequenced per library. Sequences were analyzed as above. A previously described Southern blotting strategy was used to characterize the Fc receptor genes within this clone (21, 22).
Isolation and Characterization of Yeast Artificial Chromosome (YAC), Cosmid, and Phage Clones.
YAC clone 851A10 was isolated by PCR screening of Centre d'Etude des Polymorphisme Humain YAC library pools by using EB1 primers and FISH on normal metaphase chromosomes as described (23). Phage clone 10 was obtained by screening a normal human genomic phage library with the EB1 probe. Cosmid clones cos34 and cos2 were obtained by PCR-screening colonies from a cosmid sublibrary of PAC986b4 with primers specific for FCGR2 and FCGR3 genes, respectively. The PAC sublibrary was constructed in Supercos according to the manufacturer's protocol (Stratagene). FCGR2 primers were 5′-TGTAGTGGCCTTGATCTACTGCAG-3′ and 5′-CTCACATACCACCTCCTCCAGAATG-3′ (transmembrane and 3′ intron region). FCGR3 primers 5′-GAGAATGCTGGTTCCAATTG-3′ and 5′-ACTCACCAGTCCGCATGCCAGC-3′ were derived from the published sequence of the FCGR3B promoter region (24). The SacI probe was amplified from genomic DNA by using the same primers. The 2T3 probe was derived from sequence from the T3 end of cos2. The ψL31 probe is single copy in PAC986b4 and was identified from sequencing in φ10.
Southern and Northern Blotting.
High molecular weight DNA was isolated from cell lines or frozen lymphoma tissues, was electrophoresed, and was transferred to Hybond N+ membranes (Amersham Pharmacia). Probes were labeled with 32P-dCTP by random priming. Hybridization and washing were performed according to standard procedures. Total RNA was extracted by using Trizol reagent according to the supplier's instructions (GIBCO/BRL). RNA was electrophoresed on denaturing gels and was blotted and hybridized as above. For analysis of FcγRII expression and Southern blotting of patient material, a 500-bp cDNA fragment (EC2/TM) capable of detecting all FcγRII gene transcripts was amplified by reverse transcription (RT)–PCR using primers as described (25).
RT-PCR Analysis and Mutation Analysis.
FcγRIIA/C expression was assessed by using primer pairs described previously (25). The FcγRIIB-specific primers were 5′-CCTCACCTGGAGTTCCAGGAGGGAG-3′ and 5′-AGACAATGGAGACTAAATACG-3′ (transmembrane and intracellular portion of FcγRIIB). FcγRIIb1 and FcγRIIb2 product sizes are 541 and 484 bp, respectively. Quality of cDNA was verified by amplification a 203-bp β-actin fragment. RT-PCR products were run on 1.8% agarose gels and were visualized by ethidium bromide staining. For mutation analysis, RT-PCR products were cloned into pGEM-T (Promega). Several clones corresponding to each RT-PCR product were then sequenced in both directions.
Flow Cytometric Analysis.
Membrane expression of FcγRII was measured by direct immunofluorescence using a commercial Phycoerythrin-labeled anti-CD32 monoclonal antibody according to the manufacturer's instructions (Immunotech, Luminy, France). Analysis was performed on a Becton Dickinson FACS (fluorescence-activated cell sorter) station by using cellquest software.
Fluorescence in Situ Hybridization.
YAC, PAC and cosmid clones were biotin- or digoxigenin-labeled by random priming (Appligene-Oncor) and were hybridized as described (23). Image analysis was performed with a cooled charge-coupled device camera (Photometrics, Tucson, AZ) and smartcapture software (Vysis, Paris).
Results
Southern Blot Analysis and Cloning of the t(1;22)(q21;q11) Breakpoint Junction.
Southern blot analysis of IGL rearrangements in the B593 tumor cell line compared with the B-EBV lymphoblastoid cell line, both derived from the follicular lymphoma patient Co, identified aberrant bands suspected to contain the t(1;22)(q22;q11) der(1) breakpoint (Fig. 1 A and C).
Figure 1.

Molecular characterization of the t(1;22)(q22;11) breakpoint in follicular lymphoma patient Co. (A) Southern blot analysis of IGLC rearrangements in B593 compared with original patient material (Co) using the pJλ2 probe, which detects a polymorphic EcoRI fragment encompassing the IGLC2 and IGLC3 genes (26). Short arrows indicate rearranged bands. Co is heterozygous for the 18- and 8-kb alleles; a faint 8-kb band visible in Co (horizontal arrow) but not in B593 corresponds to detection of the 8-kb germline band in normal cells in the biopsy material. In the tumor cells, the 18-kb allele is retained whereas rearrangement of the 8-kb fragment after translocation generates an aberrant 2.2-kb band. (B) Genomic sequence surrounding the der(1) breakpoint of the t(1;22)(q22;q11). The translocation juxtaposes germline chromosome 1 sequence to a somatically hypermutated V gene. A vertical arrow indicates the breakpoint site, and the Chi-like sequences are underlined. (C) Southern blot analysis of the der(1) breakpoint in B593. The p607/21–68 covering the IGLC2/3 genes and the 1q22 breakpoint-specific probe EB1 detect identical rearranged fragments (arrows).
A 2.2-kb EcoRI rearranged band, also detected in the original patient biopsy, was cloned and sequenced (GenBank accession no. AF209720). This identified ≈250 bp of novel sequence juxtaposed to an unidentifiable rearranged and somatically mutated IGLV gene (data not shown; Fig. 1B). This V gene sequence was in turn juxtaposed to a rearranged and somatically mutated IGLJ/C3 segment (not shown). Hybridization of this 250 bp of novel sequence (probe EB1) to a monochromosomal somatic cell hybrid blot identified it as originating from chromosome 1 (not shown). Also, EB1 cohybridized rearranged bands previously detected by the IGLC2/3 probe, thereby confirming this sequence as deriving from the B593 der(1) breakpoint (Fig. 1C).
We next used EB1 to screen a normal genomic phage library and isolated a 16.5-kb insert phage (φ10) corresponding to the normal chromosome 1 counterpart in the breakpoint region (Fig. 2A). The germline 1q22 sequence spanning the breakpoint was sequenced (398 bp) (GenBank accession no. AF209721). Comparison of this sequence to the der(1) breakpoint sequence revealed the latter to be germline (Fig. 1B). This indicated that the t(1;22) might have resulted from erroneous somatic hypermutation of a rearranged IGL allele that had already undergone considerable hypermutation before translocation. Interestingly, two closely spaced Chi-like sequences were identified within 70-bp of the chromosome 1 breakpoint site (Fig. 1B). Such sequences (consensus 5′-GCTGGTGG-3′) have previously been observed at other chromosomal breakpoint sites and are known targets for homologous recombination in Escherichia coli (28, 29).
Figure 2.

Genomic organization of the 1q22 breakpoint region. (A) Map of the B593 t(1;22) breakpoint region (70-kb) with respect to the low affinity IgG Fc receptor locus. Vertical lines indicate restriction enzyme sites K Kpnl and X Xhol. A solid vertical arrow represents the position of the cloned B593 breakpoint. Thin horizontal bars underneath the restriction map indicate relative positions of cosmid and phage clones, and thick horizontal bars indicate the position of genomic probes used for breakpoint cloning and cosmid mapping (described in Methods). AA693721 is a probe derived from an EST clone of the same name. The schematic representation of the low affinity Fc receptor locus is derived from previously published maps of this region (21, 27). Horizontal arrows represent the transcriptional orientation of Fc receptor genes: IIA (FCGR2A), IIIA (FCGR3A, IIB (FCGR2B), IIIB (FCGR3B), IIC (FCGR2C) (21, 27). (B) Partial metaphase showing hybridization of PAC986b4 (green signals) onto normal lymphocyte high-resolution metaphase chromosomes. PAC986b4 hybridization signals are observed in proximal 1q22.
The t(1;22)(q22;q11) Interrupts the Low Affinity IgG Fc Receptor Locus and Deregulates FCGR2B.
An initial search for 1q22-transcribed sequences in the 16.5-kb insert phage clone (φ10) proved negative. We next screened a genomic PAC library with EB1 and identified eight PAC clones. Characterization of these clones by FISH on B593 metaphases identified one (PAC986b4) that appeared to maximally overlap the 1q22 breakpoint on the centromeric side (not shown). PAC986b4 was shotgun-cloned and partially sequenced. A number of overlapping expressed sequence tags (AA693721, AA746047, and N36002) corresponding to the 3′ region of a novel gene of unknown function and sequences homologous to genes encoding the low affinity IgG Fc receptors FcγRII (CD32) and FcγRIII (CD16) were identified. Three FcγRII genes (FCGR2A, -B, and -C) and two FcγRIII genes (FCGR3A and -B) have been described and physically mapped to a region of ≈200 kb in 1q22 (21, 24, 27, 30). Accordingly, FISH with PAC986b4 on normal lymphocyte prometaphase chromosomes showed signals consistently localizing to proximal 1q22 (Fig. 2B).
Further mapping of PAC986b4 and derived cosmid subclones (cos34 and cos2) [compared to published maps (21, 27)] showed the B593 1q22 breakpoint to interrupt the low affinity Fc receptor locus just upstream of the FCGR3B promoter region and 20 kb telomeric of FCGR2B (Fig. 2), thereby juxtaposing the 5′ region of the latter with the IGL 3′ enhancer element. This identified FCGR2B that encodes the immunoreceptor tyrosine-based inhibition motif (ITIM)-bearing IgG Fc receptor FcγRIIB as the likely target gene of the t(1;22) in B593.
In keeping with this, expression of FCGR3B and of the novel gene were absent (not shown). In contrast, Northern blot analysis of FcγRII expression (-A, -B, and -C genes) in B593 with a cDNA probe spanning the first extracellular and transmembrane exons of FCGR2B (EC2/TM) revealed an at least 30-fold over-expression of two major 1.5- and 2.4-kb FcγRII transcripts, compared with that observed in the B-EBV, Br97, and Raji cell lines (Fig. 3A).
Figure 3.

FcγRII expression analysis in B593. (A) Northern blot analysis of FcγRII expression in B593 compared with cell lines of hematopoietic origin. Total RNA (20 μg) was size-fractionated on an agarose gel and was blotted and sequentially hybridized with FcγRIIB (EC2/TM) and β-actin cDNA probes. After overnight exposure, very high level expression of 1.5- and 2.4-kb transcripts was detectable in B593 compared with the other cell lines. Jurkat and CEM do not normally express FcγRII, being of T-cell origin. (B) RT-PCR assay for characterization of FcγRII transcripts in B593. A primer pair overlapping the EC2, TM, and IC exons of A/C (immunoreceptor tyrosine-based activation motif Fc receptors) (Top) and FcγRIIB transcripts (ITIM receptor) (Middle), respectively, were used. The FcγRIIB specific primer pair generates a 541-bp b1 isoform-specific transcript and a 484-bp b2 isoform-specific transcript. RNA quality was verified by amplification of a 203-bp β-actin cDNA. FcγRII expression in U937 and Raji cell lines was concordant with previously described data concerning these cell lines (25, 33). Two bands (442 and 396 bp) observed in U937 correspond to alternatively spliced forms of A and/or C transcripts. B593 and B-EBV both express a single FcγRIIA/C transcript. An aberrant FcγRIIB expression profile is observed in B593: hyperexpression of the FcγRIIb2 specific transcript [indicated by b2 (Middle)].
Multiple additional FcγRII transcripts detected in B593 but not in the other cell lines might correspond to accumulation of FcγRII splicing intermediates as a consequence of abnormally high level transcription from the FCGR2B promoter. This is probably because of its proximity to the IGL transcriptional enhancer, as previously described for the variant t(8;22) translocation that deregulates c-MYC in Burkitt's lymphoma (31).
FcγRIIA, -B, and -C receptors (expressed from the -A, -B, and -C genes) all show high homology in the extracellular and transmembrane regions, and B-cells can co-express diverse receptor isoforms (a1–a3; b1–b2; c1–c4) as a consequence of alternative splicing of FcγRIIA, -B, or -C mRNA (32, 33). To characterize FcγRII expression in B593, RT-PCR was performed by using primers that could distinguish FcγRIIA/C from FcγRIIB cDNA (Fig. 3B). A/C-specific RT-PCR detected equivalent levels of A/C message in B593 compared with the B-EBV and U937 cell lines (Fig. 3B Top). In contrast, FcγRIIB-specific RT-PCR detected high levels of the 484-bp FcγRIIb2 cDNA in B593 compared with U937, Raji, and B-EBV (Fig. 3B Middle). Expression of FcγRIIb1 appeared similar in all cell lines. These data indicated the elevated expression of FcγRII observed in B593 to specifically correspond to hyperexpression of the FcγRIIb2 receptor isoform. The high level of FcγRIIB mRNA in B593 was associated with increased levels of membrane protein expression, as detected by FACS analysis, in B593 compared with B-EBV (Fig. 4).
Figure 4.

FACS analysis of membrane FcγRII (CD32) protein expression in B593 compared with the B-EBV cell line. For both cell lines, the dotted curve indicates background fluorescence with a control antibody, and the solid black curve represents fluorescence after incubation with a CD32 (FcγRII) monoclonal antibody.
FISH Analysis of the Fc Receptor Locus in Lymphoma with 1q21–23 Breakpoints.
To assess the involvement of the FCGR2B gene in other cases of lymphoma, a total of 23 primary lymphoma cases and 3 Burkitt lymphoma (BL) cell lines with karyotypically determined 1q21–23 breakpoints were screened by FISH using a B593 breakpoint-specific CEPH YAC, 851A10. Two further cases of follicular lymphoma (Az, 650) with balanced t(1;22)(q22;q11), in addition to t(14;18)(q32;q21), and breakpoints in 851A10 were identified (not shown). It should be noted that both of these translocations had initially been cytogenetically mapped to band 1q21, which highlights the difficulty of accurate band assignment in 1q21–23. Further FISH mapping using the cosmid clones cos34 (covering FCGR2B) and cos2 (FCGR3B), both subcloned from PAC986b4, confirmed a breakpoint between FCGR2B and FCGR3B in both cases (Fig. 5 A and B).
Figure 5.
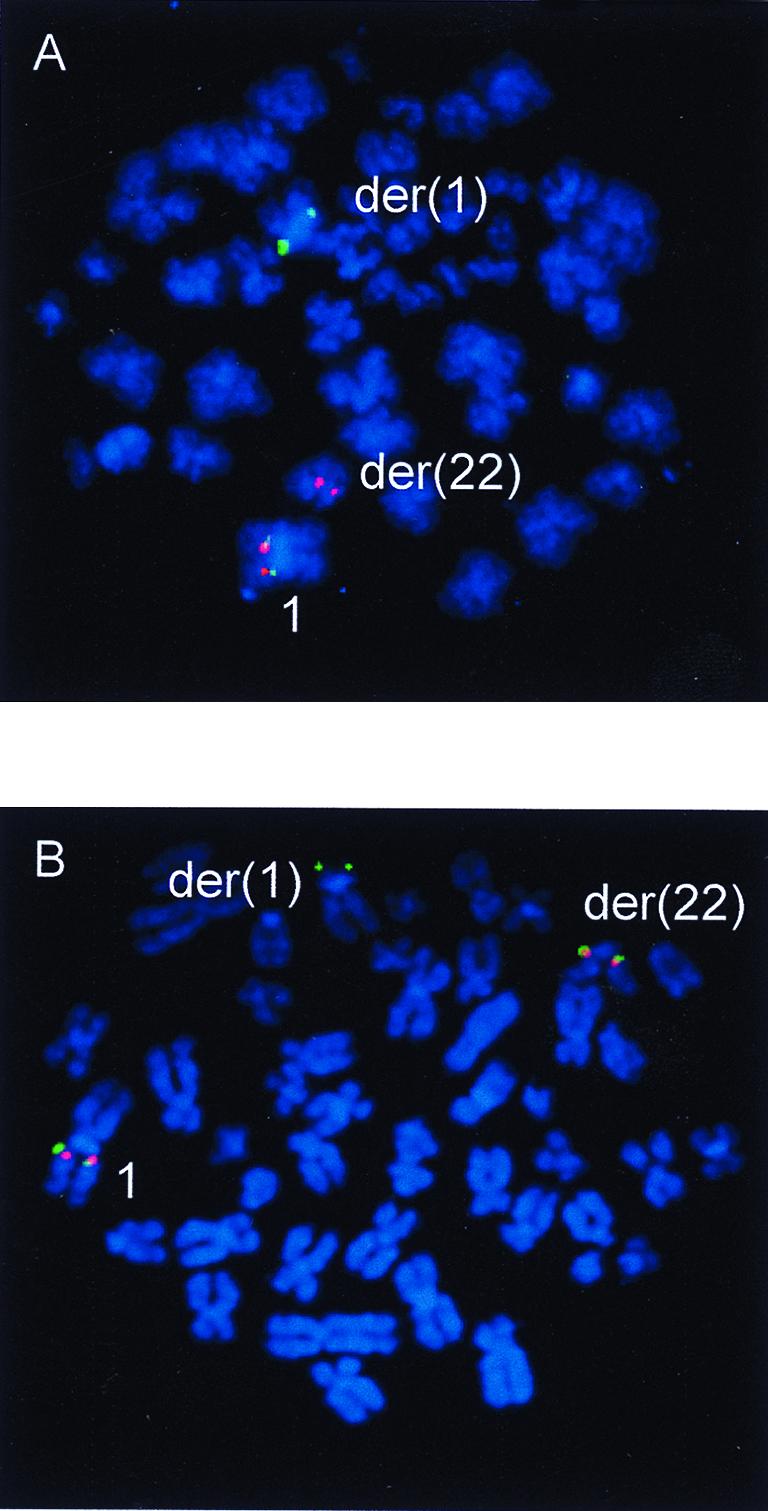
Identification of chromosomal breakpoints interrupting the Fc locus in two cases of follicular lymphoma by dual color FISH with cosmid probes cos34 (FCGR2B) (green signals) and cos2 (FCGR3B) (red signals). Chromosomes are counterstained with 4′,6-diamidino-2-phenylindole. (A) Lymphoma case Az shows hybridization of cos34 to the derivative 1 chromosome and cos2 to the der(22). (B) In case 650, a der(1) breakpoint within cos34 was detected. Cos 34 showed signals on the der(1), der(22), and normal 1 whereas cos 2 gave signals on the der(22) and normal 1 only.
Expression of FcγRIIB in Lymphoma.
RNA was available for analysis of FcγRII expression in case 650 as well as 8 primary lymphoma cases and 2 BL, with trisomy 1q encompassing the low affinity Fc locus as determined by FISH by using YAC 851A10 (data not shown). Expression was assessed by either Northern blot using the FcγRIIB EC2/TM cDNA probe (detects FcγRIIA, -B, and -C transcripts) or by FcγRIIB-specific RT-PCR and was compared with that observed in lymphoma without 1q anomalies (Fig. 6 A and B). Patients tested included seven diffuse large cell lymphoma (Sa, Te, 5501, Bar, Boy, Gn, Mo), one Burkitt lymphoma (Br), one follicular lymphoma (Ma), one splenic lymphoma with villous lymphocytes (Bo), one lymphoplasmacytoid lymphoma (Cr), one small lymphocytic lymphoma (Ba), and one marginal zone lymphoma (Mon). Case 650 showed a similar expression profile to B593: specific hyperexpression of FcγRIIb2. For all other cases, FcγRII expression appeared to vary from low to moderate, irrespective of the presence of 1q aberrations.
Figure 6.

Expression analysis of FcγRII in lymphoma. (A) Northern analysis of FcγRII expression in two Burkitt lymphoma cell lines (BL126, BL136) and four primary lymphoma samples (Sa, Te, 5501, Bar?), all with trisomy 1q compared with lymphoma case Mo (no 1q anomaly) and the T-cell line Jurkat. RNA was limited for three of these cases, so ≈10 μg instead of 20 μg was loaded for analysis (Sa, Te and Bar). The FcγRIIB probe (EC2/TM) does not distinguish between FcγRIIA, -B, or -C transcripts. (B) High level expression of the FcγRIIb2 isoform in a second case of follicular lymphoma (650) with t(1;22)(q22;q11). RT-PCR analysis of FcγRIIB expression in lymphoma case 650 compared with nine other lymphomas of diverse histological subtypes with trisomy 1q (Br, Bo, Te, Cr, and Ma) or without trisomy 1q (Br97, Ba, Boy, Gn and Mon). The RT control is shown for the FcγRIIB-specific RT-PCR only.
FCGR2B Rearrangement and Mutation Analysis.
Sequencing of cloned FcγRIIb2 cDNAs covering the entire coding region (875 bp) from B593 or 650 did not reveal mutations in the ligand-binding, transmembrane, or intracytoplasmic regions, including the ITIM motif, in either case. A single mutation was, however, detected within the signal peptide region of both cases: Ala33->Val in case 650 and Ala33->Pro in B593 (data not shown). These mutations, although recurring, did not affect the signal peptide cleavage site in either case and were therefore of uncertain significance. Finally, Southern blot analysis in B593, 650, and Az did not reveal any alterations of FCGR2B (not shown). This suggested constitutive high level expression of an intact FcγRIIB receptor to be the major consequence of the t(1;22).
Discussion
Deregulation of FCGR2B by a secondary chromosomal translocation in three cases of follicular lymphoma is strong evidence pointing to this deregulation as a possible important event in tumor evolution in this disease. The precise pathological consequences of deregulated FCGR2B and hyperexpression of a (normal) FcγRIIb2 isoform remain to be determined.
FcγRIIB is a member of the ITIM-bearing inhibitory co-receptor family that maintains immune system homeostasis through negative regulation of immune responses mediated by receptors that bear immunoreceptor tyrosine-based activation motifs such as B cell antigen receptors (BCR) (34). As an example, co-crosslinking of FcγRIIB to a BCR expressed on the same cell (through mutual binding of IgG/antigen immune complexes) down-regulates BCR-induced activatory signaling. This inhibition is mediated through FcγRIIB ITIM tyrosyl phosphorylation and subsequent recruitment of the Src homology domain-bearing inositol phosphatase SHIP (35, 36). This initiates a cascade of negative signaling responses that include reduced Ca influx (36), reduced inositol triphosphate production (IP3) (37, 38), and, in some cases, apoptosis (39, 40). Negative regulation by FcγRIIB may not be restricted to immunoreceptor tyrosine-based activation motif-bearing receptors because it has recently been shown that FcγRIIB can also inhibit c-kit-mediated cell proliferation (41).
Although FcγRIIb1 and b2 are indistinguishable in their capacities to mediate negative signaling per se, the possibility that differential splicing of FcγRIIB mRNA may be coupled to cell cycle entry during B-cell activation does exist (42). Second, FcγRIIb2, unlike FcγRIIb1, is capable of endocytosis, which is suggestive of a potential role for the former in antigen presentation (43). On the other hand, the inability of FcγRIIb1 to mediate endocytosis has been proposed as a mechanism through which co-crosslinking FcγRIIb1 (but not -b2) to the BCR can inhibit internalization of BCR-bound antigen and thereby production of MHC class II bound TCR ligands (44). Taken together, this might indicate differing functional roles for the FcγRIIb1 and -b2 isoforms in B-cells in vivo. In this respect, high-level constitutive production of FcγRIIb2 after chromosomal translocation with Ig regulatory elements as observed in this report might compromise some aspect of the regulatory function of this receptor in the control of immunoreceptor tyrosine-based activation motif-dependent or independent B-cell function.
A second pathogenic mechanism centers on emerging evidence of a possible regulatory role for FcγRIIB in B-cell development. It has been shown that anti-FcgR antibody can enhance the growth and differentiation of murine B and T lineage progenitor cells in vitro (45, 46), and FcγRIIB-deficient mice (47) (but not FcγRIIIB-deficient mice) show significantly increased B-cell compartments (45). The pathologic consequences of deregulation of FCGR2B by chromosomal translocation would therefore also depend on the timing of appearance of the translocation in a given tumor clone.
Over-expression of FcγRIIB in nonlymphoid tumor cells has been shown to significantly enhance their tumorigenic phenotype in vivo (48), and plasma concentrations of soluble FcR have been correlated with disease stage in chronic lymphocytic leukemia (49) and multiple myeloma (50). The mechanistic basis for these observations has not been investigated. It is tempting to speculate that constitutive over-expression of an IgG Fc receptor might allow tumor cells to inhibit IgG activity (avid binding of IgG antibodies) and thereby to evade (anti-tumor) antibody attack. Indeed, it is now apparent that IgG Fc binding activity is a strategy that has evolved in certain viruses to escape antibody-mediated immunity (51).
Our results showing high level expression of FcγRIIB only in cases in which FCGR2B is specifically juxtapositioned to the IGL locus and not in patients with trisomy 1q (three copies of the Fc receptor locus) is noteworthy and is suggestive of specific implication of the IGL transcriptional enhancer in deregulation of FCGR2B. This is further supported by the fact that neither FCGR2A nor FCGR3A show deregulated expression, despite being within 200 kb of the IGL enhancer after translocation. Indeed, Ig enhancer elements appear capable of modulating gene expression over large distances. For example, deregulation of CCND1 is consistently observed after t(11;14), whatever the breakpoint, up to 200 kb (52). Characterization of the IGL enhancer-mediated transcriptional activation of FCGR2B should elucidate important regulatory mechanisms controlling expression of this gene.
In summary, the specific association of a recurring t(1;22) with t(14;18) in three cases of follicular lymphoma reinforces a role for this translocation in tumor progression in this lymphoma type. However, in contrast to primary chromosomal aberrations in NHL, which are generally balanced translocations involving Ig loci, secondary chromosomal anomalies tend to be unbalanced and involve non-Ig partners (11). In this regard, the three cases described here are reminiscent of t(14;18) NHLs, which progress to high grade malignancy on acquisition of the Burkitts lymphoma-associated t(8;14). As discussed by Johannson et al., the t(8;14) in these patients, although by definition a secondary abnormality, is probably better described “as a second primary change” (12). Therefore, the possibility that the t(1;22) described here might also occur as a primary event in other lymphoma types cannot be ruled out.
Acknowledgments
We thank Marc Daëron, Saadi Khochbin, and Jean Pierre Kerckaert for valuable discussion and comments on the manuscript. We are indebted to M. C. Jacob for FACS analysis, to Danielle Marais for cytogenetic analysis, and M. Leroux for assistance with the figures. This research was supported by grants from the Association pour la Recherche sur le Cancer, La Ligue Contre le Cancer (Haute Savoie region) (Ph.D. fellowship to P.L.), Association pour la Recherche sur les Affections Malignes en Immunologie Sanguine, ESPOIR, and the European Union (Human Capital and Mobility Fellowship to M.C.).
Abbreviations
- NHL
non-Hodgkin's lymphoma
- FISH
fluorescent in situ hybridization
- ITIM
immunoreceptor tyrosine-based inhibition motif
- YAC
yeast artificial chromosome
- RT
reverse transcription
- BL
Burkitt lymphoma
- BCR
B cell antigen receptors
Footnotes
References
- 1.Rabbitts T H. Nature (London) 1994;372:143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- 2.Goossens T, Klein U, Kuppers R. Proc Natl Acad Sci USA. 1998;95:2463–2468. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hiom K, Melek M, Gellert M. Cell. 1998;94:463–470. doi: 10.1016/s0092-8674(00)81587-1. [DOI] [PubMed] [Google Scholar]
- 4.Tsujimoto Y, Cossman J, Jaffe E, Croce C M. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- 5.Kerckaert J P, Deweindt C, Tilly H, Quief S, Lecocq G, Bastard C. Nat Genet. 1993;5:66–70. doi: 10.1038/ng0993-66. [DOI] [PubMed] [Google Scholar]
- 6.Baron B W, Nucifora G, McCabe N, Espinosa R d, Le Beau M M, McKeithan T W. Proc Natl Acad Sci USA. 1993;90:5262–5266. doi: 10.1073/pnas.90.11.5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye B H, Rao P H, Chaganti R S, Dalla-Favera R. Cancer Res. 1993;53:2732–2735. [PubMed] [Google Scholar]
- 8.Callanan M, Leroux D, Magaud J P, Rimokh R. Crit Rev Oncog. 1996;7:191–203. doi: 10.1615/critrevoncog.v7.i3-4.30. [DOI] [PubMed] [Google Scholar]
- 9.McDonnell T J, Deane N, Platt F M, Nunez G, Jaeger U, McKearn J P, Korsmeyer S J. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 10.McDonnell T J, Korsmeyer S J. Nature (London) 1991;349:254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 11.Johansson B, Mertens F, Mitelman F. Genes Chromosomes Cancer. 1996;16:155–163. doi: 10.1002/(SICI)1098-2264(199607)16:3<155::AID-GCC1>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 12.Johansson B, Mertens F, Mitelman F. Blood. 1995;86:3905–3914. [PubMed] [Google Scholar]
- 13.Offit K, Wong G, Filippa D A, Tao Y, Chaganti R S. Blood. 1991;77:1508–1515. [PubMed] [Google Scholar]
- 14.Whang Peng J, Knutsen T, Jaffe E S, Steinberg S M, Raffeld M, Zhao W P, Duffey P, Condron K, Yano T, Longo D L. Blood. 1995;85:203–216. [PubMed] [Google Scholar]
- 15.Dierlamm J, Pittaluga S, Wlodarska I, Stul M, Thomas J, Boogaerts M, Michaux L, Driessen A, Mecucci C, Cassiman J J, et al. Blood. 1996;87:299–307. [PubMed] [Google Scholar]
- 16.Johansson B, Brondum-Nielsen K, Billstrom R, Schiodt I, Mitelman F. Cancer Genet Cytogenet. 1997;99:97–101. doi: 10.1016/s0165-4608(97)00198-2. [DOI] [PubMed] [Google Scholar]
- 17.Willis T G, Zalcberg I R, Coignet L J, Wlodarska I, Stul M, Jadayel D M, Bastard C, Treleaven J G, Catovsky D, Silva M L, Dyer M J. Blood. 1998;91:1873–1881. [PubMed] [Google Scholar]
- 18.Tse W, Zhu W, Chen H S, Cohen A. Blood. 1995;85:650–656. [PubMed] [Google Scholar]
- 19.Hatakeyama S, Osawa M, Omine M, Ishikawa F. Oncogene. 1999;18:2085–2090. doi: 10.1038/sj.onc.1202510. [DOI] [PubMed] [Google Scholar]
- 20.Leroux D, Le Marc'Hadour F, Gressin R, Jacob M C, Keddari E, Monteil M, Caillot P, Jalbert P, Sotto J J. Br J Haematol. 1991;77:346–353. doi: 10.1111/j.1365-2141.1991.tb08582.x. [DOI] [PubMed] [Google Scholar]
- 21.Qiu W Q, de Bruin D, Brownstein B H, Pearse R, Ravetch J V. Science. 1990;248:732–735. doi: 10.1126/science.2139735. [DOI] [PubMed] [Google Scholar]
- 22.Warmerdam P A, Nabben N M, van de Graaf S A, van de Winkel J G, Capel P J. J Biol Chem. 1993;268:7346–7349. [PubMed] [Google Scholar]
- 23.Monteil M, Callanan M, Dascalescu C, Sotto J J, Leroux D. Br J Haematol. 1996;93:656–660. doi: 10.1046/j.1365-2141.1996.d01-1675.x. [DOI] [PubMed] [Google Scholar]
- 24.Gessner J E, Grussenmeyer T, Kolanus W, Schmidt R E. J Biol Chem. 1995;270:1350–1361. doi: 10.1074/jbc.270.3.1350. [DOI] [PubMed] [Google Scholar]
- 25.Metes D, Ernst L K, Chambers W H, Sulica A, Herberman R B, Morel P A. Blood. 1998;91:2369–2380. [PubMed] [Google Scholar]
- 26.Ghanem N, Dariavach P, Bensmana M, Chibani J, Lefranc G, Lefranc M P. Exp Clin Immunogenet. 1988;5:186–195. [PubMed] [Google Scholar]
- 27.Su Y, Brooks D G, Li L, Lepercq J, Trofatter J A, Ravetch J V, Lebo R V. Proc Natl Acad Sci USA. 1993;90:10856–10860. doi: 10.1073/pnas.90.22.10856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyatt R T, Rudders R A, Zelenetz A, Delellis R A, Krontiris T G. J Exp Med. 1992;175:1575–1588. doi: 10.1084/jem.175.6.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaeger U, Purtscher B, Karth G D, Knapp S, Mannhalter C, Lechner K. Blood. 1993;81:1833–1840. [PubMed] [Google Scholar]
- 30.Peltz G A, Grundy H O, Lebo R V, Yssel H, Barsh G S, Moore K W. Proc Natl Acad Sci USA. 1989;86:1013–1017. doi: 10.1073/pnas.86.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerbitz A, Mautner J, Geltinger C, Hortnagel K, Christoph B, Asenbauer H, Klobeck G, Polack A, Bornkamm G W. Oncogene. 1999;18:1745–1753. doi: 10.1038/sj.onc.1202468. [DOI] [PubMed] [Google Scholar]
- 32.Brooks D G, Qiu W Q, Luster A D, Ravetch J V. J Exp Med. 1989;170:1369–1385. doi: 10.1084/jem.170.4.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cassel D L, Keller M A, Surrey S, Schwartz E, Schreiber A D, Rappaport E F, McKenzie S E. Mol Immunol. 1993;30:451–460. doi: 10.1016/0161-5890(93)90113-p. [DOI] [PubMed] [Google Scholar]
- 34.Daëron M. Annu Rev Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- 35.Ono M, Bolland S, Tempst P, Ravetch J V. Nature (London) 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- 36.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig M C, Ravetch J V. Nature (London) 1994;368:70–73. doi: 10.1038/368070a0. [DOI] [PubMed] [Google Scholar]
- 37.Bolland S, Pearse R N, Kurosaki T, Ravetch J V. Immunity. 1998;8:509–516. doi: 10.1016/s1074-7613(00)80555-5. [DOI] [PubMed] [Google Scholar]
- 38.Scharenberg A M, El-Hillal O, Fruman D A, Beitz L O, Li Z, Lin S, Gout I, Cantley L C, Rawlings D J, Kinet J P. EMBO J. 1998;17:1961–1972. doi: 10.1093/emboj/17.7.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch J V. Cell. 1997;90:293–301. doi: 10.1016/s0092-8674(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 40.Ashman R F, Peckham D, Stunz L L. J Immunol. 1996;157:5–11. [PubMed] [Google Scholar]
- 41.Malbec O, Fridman W H, Daëron M. J Immunol. 1999;162:4424–4429. [PubMed] [Google Scholar]
- 42.Sarmay G, Rozsnyay Z, Koncz G, Danilkovich A, Gergely J. Eur J Immunol. 1995;25:262–268. doi: 10.1002/eji.1830250143. [DOI] [PubMed] [Google Scholar]
- 43.Amigorena S, Bonnerot C, Drake J R, Choquet D, Hunziker W, Guillet J G, Webster P, Sautes C, Mellman I, Fridman W H. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- 44.Minskoff S A, Matter K, Mellman I. J Immunol. 1998;161:2079–283. [PubMed] [Google Scholar]
- 45.de Andres B, Mueller A L, Verbeek S, Sandor M, Lynch R G. Blood. 1998;92:2823–2929. [PubMed] [Google Scholar]
- 46.Sandor M, Galon J, Takacs L, Tatsumi Y, Mueller A L, Sautes C, Lynch R G. Proc Natl Acad Sci USA. 1994;91:12857–12861. doi: 10.1073/pnas.91.26.12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takai T, Ono M, Hikida M, Ohmori H, Ravetch J V. Nature (London) 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 48.Zusman T, Lisansky E, Arons E, Anavi R, Bonnerot C, Sautes C, Fridman W H, Witz I P, Ran M. Int J Cancer. 1996;68:219–227. doi: 10.1002/(SICI)1097-0215(19961009)68:2<219::AID-IJC14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 49.Astier A, Merle-Beral H, de la Salle H, Moncuit J, Cazenave J P, Fridman W H, Hanau D, Teillaud J L. Leuk Lymphoma. 1997;26:317–326. doi: 10.3109/10428199709051781. [DOI] [PubMed] [Google Scholar]
- 50.Mathiot C, Mary J Y, Tartour E, Facon T, Monconduit M, Grosbois B, Pollet J P, Michaux J L, Euller Ziegler L, Sautes C, et al. Br J Haematol. 1996;95:660–665. doi: 10.1046/j.1365-2141.1996.d01-1943.x. [DOI] [PubMed] [Google Scholar]
- 51.Lubinski J, Nagashunmugam T, Friedman H M. Semin Cell Dev Biol. 1998;9:329–337. doi: 10.1006/scdb.1998.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raynaud S D, Bekri S, Leroux D, Grosgeorge J, Klein B, Bastard C, Gaudray P, Simon M P. Genes Chromosomes Cancer. 1993;8:80–87. doi: 10.1002/gcc.2870080204. [DOI] [PubMed] [Google Scholar]