

Abstract
Although the myelin proteolipid protein gene (Plp1) is highly expressed in the central nervous system encoding the most abundant myelin protein in oligodendrocytes, it is also expressed in other tissues, including testis. Transgenic studies with mice that harbor Plp1-lacZ fusion genes suggest that Leydig cells are the source of Plp1 gene expression in testis. However, virtually nothing is known about Plp1 gene regulation in Leydig cells, which is the focus of this study. The first intron contains both positive and negative regulatory elements that are important in regulating Plp1 gene expression in oligodendrocytes. To test whether these elements are functional in Leydig cells, a battery of Plp1-lacZ fusion genes with partial deletion of Plp1 intron 1 sequence was transfected into the mouse Leydig cell line, TM3. Results presented here suggest an enhancer, which is very potent in oligodendrocytes, is only nominally active in TM3 cells. The intron also contains several negative regulatory elements that are operative in TM3 cells. Moreover a new exon (exon 1.2) was identified within the first ‘intron’ resulting in novel splice variants in TM3 cells. Western blot analysis suggests that these splice variants, along with those containing another alternatively spliced exon (exon 1.1) derived from intron 1 sequence, give rise to multiple Plp1 gene products in the mouse testis.
Keywords: Alternative splicing, Gene expression, Leydig cells, Myelin proteolipid protein, Testis, Transcriptional regulation
1. Introduction
While the myelin proteolipid protein gene (Plp1) is highly expressed in myelinating cells (oligodendrocytes) of the brain and spinal cord, it is also expressed to a lesser degree in a variety of non-neural tissues (Wight and Dobretsova, 2004, and references therein), including testis. A Plp1-lacZ transgene [PLP(+)Z], which contains mouse Plp1 genomic DNA extending from the proximal 2.4 kb of 5′-flanking DNA downstream to the first 37 bp of exon 2, is sufficient to drive lacZ reporter gene expression in a spatial manner consistent with endogenous Plp1 expression, as well as developmentally in brain (Wight et al., 1993). Yet a related transgene, PLP(−)Z, which is identical to PLP(+)Z except for the absence of Plp1 intron 1 DNA, is expressed to much lower levels in brain, where its developmental pattern is exceedingly attenuated (Li et al., 2002b). In fact expression of the PLP(−)Z transgene was so low in brain, that on initial inspection, the lines would have been characterized as non-expressing if not for a fair amount of expression in testis. Histological analysis showed that Leydig cells were the source of transgene expression in the testis (Li et al., 2002b). However, regulation of Plp1 gene expression in Leydig cells has not been elucidated until now.
Previous studies have shown that the first intron of the Plp1 gene contains several regulatory elements that are important modulators of expression in other cell types. In oligodendrocytes, there is a single positive regulatory element that resides between intron 1 positions 1,083 and 1,177, which can override repression mediated by a couple of general negative regulatory elements located elsewhere in the intron (Dobretsova and Wight, 1999). Because the positive regulatory element also displays enhancer-like qualities [i.e., orientation-independent; activity is increased with multiple copies (Dobretsova et al., 2000); can activate a heterologous promoter (Meng et al., 2005)] it was named ASE for antisilencer/enhancer. However the ASE does not function in a liver cell line (+/+ Li; ATCC, Rockville MD, USA, catalog number CRL-6467), although the general negative regulatory elements are effective in addition to a cell-type-specific negative regulatory element located near the 3′ end of intron 1 (Li et al., 2002a). In the present study we have determined the influence of these regulatory elements on Plp1 gene expression in a Leydig cell line (TM3). [The TM3 clone was derived from primary cultures of Leydig cell-enriched preparations from normal testes of 11- to 13-day-old BALB/c mice and retains many of the functions characteristic of the cell type in vivo (Mather, 1980).] As a corollary to these studies, two alternatively spliced exons were identified from what is classically defined as Plp1 intron 1 DNA. One of these, exon 1.1, has been described previously as an additional exon that is included in some of the transcripts from oligodendrocytes, neurons, and lymphoid cells (Bongarzone et al., 1999; Feng et al., 2003; Jacobs et al., 2003). However identification of the other exon, denoted here as exon 1.2, is novel. Inclusion of one or both of these exons, coupled with additional alternative splicing due to the presence of two donor splice sites in exon 3 (Nave et al., 1987), lead to a variety of splice variants in Leydig cells, which ultimately results in the expression of multiple Plp1 gene products in testis.
2. Materials and methods
2.1. Cell culture
The mouse Leydig cell line, TM3 (ATCC, catalog number CRL-1714), was grown at 37°C in a 1:1 mixture of Ham’s F-12/Dulbecco’s modified Eagle’s medium (Irvine Scientific, Santa Ana, CA, USA) supplemented with 1.2 g/L sodium bicarbonate, 15 mM HEPES, 4.5 g/L glucose, 2.5 % fetal bovine serum (Intergen, Purchase, NY, USA), 5% horse serum (HyClone, Logan, UT, USA), and maintained in an atmosphere of 5% CO2. The mouse liver cell line, +/+Li (ATCC), was grown at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) supplemented with 3.7 g/L sodium bicarbonate, 10% fetal bovine serum (Intergen), and maintained in an atmosphere of 10% CO2. The mouse immortalized oligodendrocyte cell line N20.1 (Verity et al., 1993) was grown at 34°C in a 1:1 mixture of Ham’s F-12/Dulbecco’s modified Eagle’s low glucose medium (Irvine Scientific) supplemented with 1.2 g/L sodium bicarbonate, 3.75 g/L HEPES, 5.75 g/L glucose, 100 μg/ml G-148, 10 % fetal bovine serum (Intergen), and maintained in an atmosphere of 5% CO2.
2.2. Plp1-lacZ plasmids
Generation of many of the Plp1-lacZ plasmids used in this study has been described previously. All plasmids are identical to PLP(+)Z except for deletion of some or all of Plp1 intron 1 DNA. PLP(+)Z (Wight et al., 1993) contains mouse Plp1 genomic sequences extending from the proximal 2.4 kb of 5′-flanking DNA, downstream to the first 37 bp of exon 2, which are used to drive expression of a lacZ reporter gene cassette. The PLP(−)Z construct (Wight and Dobretsova, 1997) is missing all of Plp1 intron 1 DNA, while partial deletion constructs contain only a portion of the intron and were named accordingly. For instance PLPΔ809-5807 (Dobretsova and Wight, 1999) is missing Plp1 intron 1 DNA from positions 809 to 5,807 based upon numbering the intron from positions 1 to 8,140 (Wight and Dobretsova, 1997). Plasmids PLPΔ809-5807 + F(I–V) and PLPΔ809-5807 + R(I–V) contain Plp1 intron 1 DNA positions 1,083–1,177 (I–V) inserted into the deletion junction site of PLPΔ809-5807 in the native (forward; F) or reverse (R) orientation, respectively (Dobretsova et al., 2000).
The pPUR/PLPΔ809-5807 plasmid (Dobretsova et al., 2000) was used as the foundation from which to generate pPUR/PLP(+)Z and pPUR/PLP(−)Z. The plasmid contains Plp1-lacZ sequences from PLPΔ809-5807 cloned into the pPUR vector (Clontech Laboratories, Palo Alto, CA, USA) which allows for coexpression of a puromycin-resistance selectable marker. pPUR/PLP(+)Z was produced by digestion of PLP(+)Z and pPUR/PLPΔ809-5807 with ApaI and NotI and subsequent ligation of the appropriate fragments to yield a plasmid that is essentially PLP(+)Z in background of the pPUR vector. The pPUR/PLP(−)Z plasmid was generated similarly from PLP(−)Z and pPURΔ809-5807 and is identical to pPUR/PLP(+)Z, except for the lack of Plp1 intron 1 DNA.
2.3. Transfection analysis
TM3 cells were transiently transfected with equimolar amounts of various Plp1-lacZ fusion genes (in addition to an RSV-luciferase plasmid to control for differences in transfection efficiency) using the LIPOFECTAMINE Reagent as previously described (Dobretsova and Wight, 1999). Cell lysates were prepared approximately31 h post DNA addition and reporter gene activities determined by chemiluminescent detection as previously described (Dobretsova and Wight, 1999). Results have been corrected for differences in transfection efficiency and represent the mean ± SD of βGal activity relative to that obtained for PLP(−)Z transfected cells (arbitrarily set at 100% in every experiment) from three or more independent experiments.
Stable transfections were performed similar to the methods used for transient transfection. The day prior to transfection, cells were seeded in 6-well microplates at the following densities: 2.0 × 105 (TM3), 2.4 × 105 (+/+Li), and 2.8 × 105 (N20.1) per 35-mm well. Cells were transfected with equimolar amounts of pPUR/PLP(+)Z or pPUR/PLP(−)Z linearized with AseI, which cleaves the plasmid at a single site in the vector backbone. Cells were grown in the appropriate growth medium for 48 h post-transfection and then switched to medium containing puromycin (Clontech): 4 μg/ml (TM3); 1.5 μg/ml (+/+Li); 3 μg/ml (N20.1). Puromycin-resistant clones were picked, pooled (200–300 individual colonies per pool), and expanded in culture. Lysates were prepared from a portion of each pool, and βGal activities measured as for the transient transfections. The protein concentration of lysates was determined using the BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Results are presented as βGal activity in relative light units (RLU) per microgram of total protein.
2.4. Ribonuclease protection assays (RPA)
Total RNA was isolated from a portion of each expanded pool of stably transfected cells (or untransfected cells) using the TRIZOL Reagent (Invitrogen) as recommended by the supplier. RPA analysis was performed as previously described (Li et al., 2002b) with the RPA III Kit (Ambion, Austin, TX, USA). Briefly, 25 μg of total RNA was hybridized overnight at 43.5°C with 32P-labeled riboprobes. The full-length Plp1-lacZ antisense riboprobe is 387 nt in size and includes the distal half of Plp1 exon 1 sequence along with the first 37 nt of exon 2, in addition to sequence complementary to the lacZ expression cassette (Li et al., 2002b). A 304 nt antisense riboprobe generated from the pTRI-β-actin-Mouse Template (Ambion) was also included to assess the endogenous level of β-actin mRNA, as an internal control. After hybridization, single-stranded RNA was removed using a mixture of RNase A/T1 and the remaining double-stranded RNA (protected fragments) fractionated on a 6% acrylamide-8 M urea gel. Dried gels were first analyzed on a 445 SI PhosphorImager with ImageQuantNT (Molecular Dynamics, Sunnyvale, CA, USA) and then subjected to autoradiography. Hybridization of ‘classic’ Plp1-lacZ transcripts (i.e., does not contain any Plp1 intron 1 sequence such as exon 1.1) results in protection of 335 nt of the riboprobe, while hybridization to transcripts containing alternatively spliced exon(s) derived from Plp intron 1 results in protection of a slightly smaller stretch (258 nt). Hybridization of mRNA to the β-actin riboprobe results in the protection of a 245 nt expanse.
2.5. Reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from TM3 cells (or adult mouse testis) as described earlier and treated with RQ1 RNase free DNase (1 u/μg) (Promega, Madison, WI, USA) for 1 h to eliminate residual DNA contamination. Reverse transcription (RT) was performed at 42°C for 50 min in a total volume of 20 μl using 0.5 μg of oligo(dT)12–18 primer and 200 u of Superscript II RNase H− Reverse Transcriptase (Invitrogen). The reaction was halted by heating at 70°C for 15 min followed by addition of 1.2 u of RNase H (Invitrogen) and incubation at 37°C for 20 min. One-tenth of the RT reaction was used as template for PCR with the ELONGASE Amplification System (Invitrogen), in a total volume of 50 μl. The primers used for PCR are:
Exon 1 Forward: 5′-AGAAGGAGACTGGAGAGAC-3′
Exon 1.2 Forward: 5′-TCCACAGGAAACCTAGACGAACCCAA-3′
Exon 1.2 Reverse: 5′-TCACACAGTGAGTGCTAGGTGGATG-3′
Exon 2 Reverse: 5′-CTGTACCAGTGAGAGCTTC-3′
Exon 4 Reverse: 5′-CAGAGACTGCCTATACTGG-3′
Exon 7 Reverse: 5′-TCAGAACTTGGTGCCTCGGCCCATG-3′
Initially PCR reactions were heated at 94°C for 5 min to denature the templates, followed by DNA amplification with the indicated primers according to the following conditions: Exon 1 Forward/Exon 2 Reverse – 30 sec at 94°C, 20 sec at 59°C, and 30 sec at 68°C for 35 cycles; Exon1 Forward/Exon 1.2 Reverse – 30 sec at 94°C, 30 sec at 59°C, and 50 sec at 68°C for 40 cycles; Exon 1 Forward/Exon 4 Reverse – 30 sec at 94°C, 15 sec at 59°C, and 40 sec at 68°C for 44 cycles; Exon 1.2 Forward/Exon 7 Reverse – 30 sec at 94°C, 25 sec at 60°C, and 90 sec at 68°C for 40 cycles.
RT-PCR products were separated on 1–2% agarose gels containing 0.5× TBE buffer and the bands from each reaction purified individually, using the QIAquick Gel Extraction Kit (QIAGEN Inc., Valencia, CA, USA). DNA sequences were determined directly from gel-purified fragments except in the case of RT-PCR products generated with Exon1 Forward/Exon 2 Reverse or Exon 1 Forward/Exon 1.2 Reverse primer pairs which contained fragments of overlapping sizes and thus were subcloned into the pGEM-T Vector (Promega) according to the manufacturer’s recommendations. The resulting subclones were mapped by restriction enzyme analysis and representative sized inserts sequenced. Position numbers for exon 1.1 and exon 1.2 are based on the complete sequence for Plp1 intron 1 deposited in GenBank (accession number AF003838) (Wight and Dobretsova, 1997).
2.6. Western blot analysis
A mouse at postnatal day 28 (P28) of age was anesthetized with isoflurane and exsanguinated by intracardial perfusion with PBS. Brain and testes were removed, snap frozen in liquid nitrogen, and tissue pulverized using a BioPulverizer (BioSpec Products Inc., Bartlesville, OK, USA) according to the manufacturer’s specifications. The tissue powder was resuspended in a solution of 4% SDS (250 mg tissue/5ml) and homogenized to clarity with an all-glass Dounce homogenizer. Samples were centrifuged at 16,000 × g for 5 min and the protein concentrations of the supernants (lysates) determined using the BCA Protein Assay Kit (Pierce). Western blot analysis was performed as described earlier (Dobretsova et al., 2008) with the following minor modifications. Proteins in the lysate were denatured by heating at 55°C for 5 min in gel-loading buffer prior to loading on an SDS-PAGE gel (10% polyacrylamide) and subsequently transferred to a nitrocellulose membrane. Filters were blocked with 5% non-fat dry milk in TBST (20 mM Tris, pH 7.6, 1.5 mM NaCl, 0.1% Tween-20), washed three times (15 min each) in TBST, and subsequently incubated with a mouse monoclonal antibody (plpc 1) directed against PLP carboxy-terminal residues 272–277 (Abcam Inc., Cambridge, MA, USA). The primary antibody was diluted 1/1000 in TBST containing 3% BSA and incubated with membranes overnight at 4°C. After washes and incubation with secondary antibody, immunoreactive bands were visualized with the ECL Plus Western Blotting Detection System (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).
3. Results and discussion
3.1. Transient transfection of TM3 cells with Plp1-lacZ fusion genes
To test for the presence of regulatory elements within intron 1 DNA that modulate Plp1 gene expression in Leydig cells, TM3 cells were transiently transfected with various Plp1-lacZ fusion genes. As illustrated in Fig. 1, the PLP(+)Z construct contains all of Plp1 intron 1 DNA whereas the PLP(−)Z construct is missing the entire intron. The relative level of βGal activity was much lower in cells transfected with PLP(+)Z (18%) compared to those transfected with PLP(−)Z (arbitrarily set at 100%), suggesting that the first intron contains one or more negative regulatory elements which lead to a decrease in Plp1 gene activity in TM3 cells (Fig. 1). Deletion of Plp1 intron 1 sequences from positions 809 to 5,807 (PLPΔ809-5807) further reduced the relative level of βGal activity to 7% compared to PLP(−)Z, suggesting that a positive regulatory element may have been deleted. Within this deleted sequence lies a positive regulatory element called the ASE, which enhances Plp1-lacZ gene activity in the N20.1 oligodendrocytic cell line (Dobretsova et al., 2000). To see whether the ASE is functional in TM3 cells, Plp1 intron 1 DNA encompassing positions 1083–1177 (I–V) was inserted into the deletion-junction site of PLPΔ809-5807 in either the native (“forward”; F) or reverse (R) orientation. As shown in Fig. 1, addition of the ASE to PLPΔ809-5807 was able to rescue βGal activity up to the level of that obtained with PLP(+)Z [compare PLPΔ809-5807 + F(I–V) at 25% and PLPΔ809-5807 + R(I–V) at 17% to PLP(+)Z at 18%]. Taken together these results suggest that the ASE is marginally active in TM3 cells, and that one or more negative regulatory elements residing between Plp1 intron 1 DNA positions 1–808 and/or 5,808–8,140 are also operative in TM3 cells.
Fig. 1.

Deletion-transfection analysis of Plp1-lacZ constructs in TM3 cells. Left: PLP(+)Z contains mouse Plp1 genomic sequence, extending from the proximal 2.4 kb of 5′-flanking DNA downstream to the first 37 bp of exon 2, driving expression of the lacZ reporter gene (black boxesindicate Plp1 exon 1 and exon 2 sequence while other portions of the gene are depicted by a solid line). Positions within Plp1 intron 1 DNA are numbered from 1 to 8,140 according to our previously published sequence (Wight and Dobretsova, 1997). PLPΔ809-5807 is missing Plp1 intron 1 positions 809 to 5,807, while PLP(−)Z is missing the entire intron (dashed lines indicate deleted sequence). Other constructs contain the ASE sequence (I–V) inserted at the deletion-junction site of PLPΔ809-5807 in the forward (F) or reverse (R) direction. Right: TM3 cells were transiently transfected with equimolar amounts of the indicated Plp1-lacZ construct and a fixed amount of an RSV-luciferase plasmid to correct for differences in transfection efficiency. Transfection results represent the mean ± SD values of βGal activity (n ≥ 6) relative to that obtained for PLP(−)Z, which was arbitrarily set at 100% in every experiment.
Two nested sets of Plp1-lacZ constructs with partial deletion of Plp1 intron 1 sequences were used to map the location of negative regulatory element(s) within Plp1 intron 1 which cause a decrease in expression in TM3 cells. As beforehand, levels of βGal activity in transfected TM3 cells are reported relative to that obtained with PLP(−)Z, which was arbitrarily set at 100%. Most notably, successive 5′ deletion of intron 1 sequences (Fig. 2) resulted in a dramatic increase in βGal activity when intron 1 positions 324 to 809 were deleted [compare PLPΔ809-8068 at 21% to PLPΔ324-8068 at 188%]. However further deletion of 5′ intronic sequence resulted in βGal activities close to that attained with PLP(−)Z. These results suggest that a negative regulatory element resides between Plp1 intron 1 position 324 and 809, which is consistent with earlier studies (Dobretsova and Wight, 1999; Li et al., 2002a) that functionally mapped a negative regulatory element to the same region in oligodendroglial (N20.1) and liver (+/+ Li) cells.
Fig. 2.

Transfection analysis of serial 5′ deletion constructs in TM3 cells. The Plp1-lacZ partial deletion constructs were named according to the sequence missing from Plp1 intron 1 DNA. Transfection analysis was performed as described in Fig. 1 and represent the mean ± SD values of βGal activity (n ≥ 6) relative to that obtained for PLP(−)Z, which was arbitrarily set at 100% in every experiment.
Progressive 3′ deletion of Plp intron 1 sequence beginning at position 5,807 with the second set of partial deletion constructs led to a gradual rise in βGal activity that culminated again with the PLPΔ324-8068 construct (Fig. 3). Because the level of βGal activity did not significantly differ between PLP(+)Z and PLPΔ324-7299 transfected cells (18 ± 2% and 23 ± 3%, respectively), the results from Fig. 3 suggest that additional negative regulatory element(s) resides between Plp1 intron 1 positions 7,299 and 8,068. Previously we have mapped a ‘general’ negative regulatory element to positions 7,299–7,615 that is active in both N20.1 and +/+ Li cells, and a ‘cell-type specific’ negative regulatory element (located primarily between intron 1 positions 7826–8068) which is functional in +/+ Li cells, but not in N20.1 cells (Dobretsova and Wight, 1999; Li et al., 2002a). Both of these negative regulatory elements appear to be effective in TM3 cells based upon the relative βGal activities shown in Fig. 3 [compare PLPΔ324-7299 at 23% to PLP324-7615 at 47% and PLPΔ324-7826 at 61% to PLPΔ324-8068 at 130%].
Fig. 3.
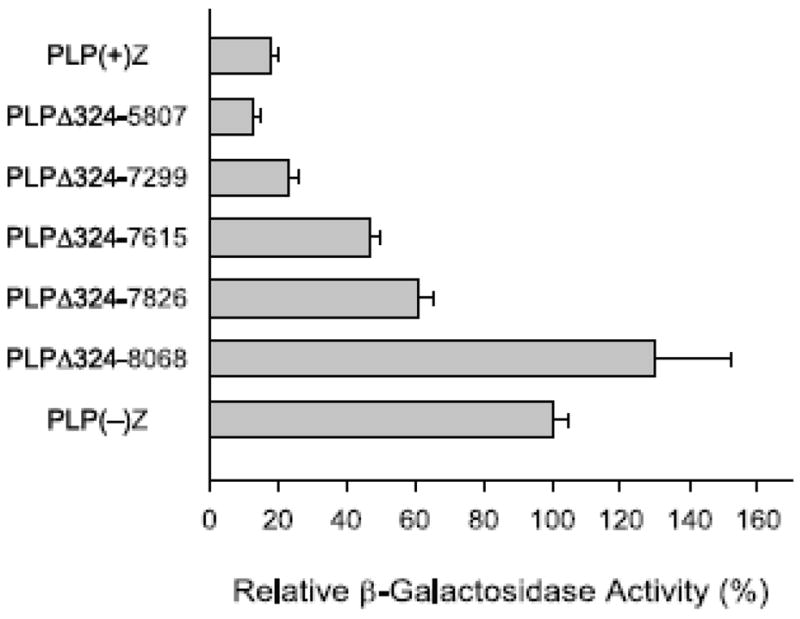
Transfection analysis of serial 3′ deletion constructs in TM3 cells. Plp1-lacZ constructs with partial deletion of Plp1 intron 1 sequence have a constant 5′ deletion endpoint at position 324 and variable 3′ deletion endpoints as indicated. Transfection analysis was performed as described in Fig. 1 and represent the mean ± SD values of βGal activity (n ≥ 6) relative to that obtained for PLP(−)Z, which was arbitrarily set at 100% in every experiment.
3.2. Analysis of cell lines stably transfected with PLP(+)Z and PLP(−)Zconstructs
To test whether the regulatory elements within the first intron are functional when chromosomally integrated, puromycin-resistant cells were selected from mouse cell lines stably transfected with either pPUR/PLP(+)Z or pPUR/PLP(−)Z. To mitigate positional effects stemming from the site of integration, 200 to 300 independent puromycin-resistant clones were pooled together. As shown in Table 1, βGal activity was higher in TM3 cells stably transfected with the ‘intronless’ construct [pPUR/PLP(−)Z] compared to the construct containing all of Plp1 intron 1 [pPUR/PLP(+)Z], similar to the trend observed with transiently transfected cells (Fig. 1). Pools of stably transfected N20.1 and +/+ Li cells were also generated with the pPUR-based constructs and analogous trends were observed for a given cell line regardless of the mode of transfection; i.e., both stably (Table 1) and transiently (Wight and Dobretsova, 1997) transfected N20.1 cells demonstrated comparable (high) levels of βGal activity between PLP(−)Z and PLP(+)Z, while βGal activity was significantly higher in +/+ Li cells stably (Table 1) or transiently (Wight and Dobretsova, 1997) transfected with PLP(−)Z, compared to PLP(+)Z.
Table 1.
β-galactosidase activity in pools of stably transfected cells*
| TM3 cells | +/+ Li cells | N20.1 cells | |
|---|---|---|---|
| pPUR/PLP(−)Z | |||
| Pool I | 30,862 | 9,831 | 77,654 |
| Pool II | 21,899 | 10,374 | 78,408 |
| Pool III | 20,673 | 10,955 | NA |
|
| |||
| pPUR/PLP(+)Z | |||
| Pool I | 5,732 | 961 | 63,364 |
| Pool II | 5,948 | 712 | 56,194 |
| Pool III | 5,856 | NA | NA |
|
| |||
| untransfected | |||
| Pool I | 75 | 143 | 249 |
| Pool II | 67 | 113 | 239 |
βGal activity (RLU) per microgram of total protein from cell lysates prepared from the indicated pools of cells
3.3. RPA analysis of Plp1-lacZ expression in stably transfected cell lines
Because the mouse Plp1 gene contains an alternatively spliced exon (designated exon 1.1; Bongarzone et al., 1999) within what is classically defined as intron 1, we decided to test whether there was a difference in alternative splice patterns between the various stably transfected cell lines. On the one hand, incorporation of exon 1.1 in mature transcripts could have had an effect on the level of βGal activity derived from PLP(+)Z transfected cells since an internal translation initiation codon must be utilized instead of the one near the end of exon 1 due to the addition of an in-frame stop codon. On the other hand, pools of puromycin-resistant cells expressing PLP(−)Z would not be affected in the same way since Plp1 intron 1 DNA (including exon 1.1) is missing from this construct. Analysis was performed by RPA using an antisense riboprobe containing sequence corresponding to Plp1 exon 1 and 2, in addition to neighboring sequence from the lacZ expression cassette. Protection of the Plp1-lacZ riboprobe (387 nt) from RNases via hybridization to ‘classic’ Plp1-lacZ transcripts would shield 335 nt from digestion, while hybridization to exon 1.1-containing transcripts would result in a smaller protected fragment (258 nt) devoid of any Plp1 exon 1 sequence. In addition, a β-actin antisense riboprobe (304 nt) was also included in the analysis as an internal control, which results in the protection of 245 nt. As shown in Fig. 4A, TM3 cells stably transfected with PLP(+)Z express a higher level of alternative splice products (i.e., Plp1-lacZ transcripts containing sequence derived from Plp1 intron 1) than for the classic transcript. Moreover, PLP(+)Z transfected +/+ Li cells demonstrated an even higher proportion of transcripts generated from alternative exon usage of Plp1 intron 1 sequence (Fig. 4B), while the reverse was seen in N20.1 transfectants, which showed higher levels of the classically spliced mRNA species (Fig. 4C). Thus the quantity of βGal activity generated in the PLP(+)Z transfected pools (Table 1) appears to be more closely associated with the level of classically spliced transcripts, rather than the entire sum of Plp1-lacZ transcripts. This could be due to a difference in the efficiency of translation initiation since transcripts that contain Plp1 exon 1.1 must use an internal AUG codon for productive translation (Bongarzone et al., 1999).
Fig. 4.

RPA analysis with pools of cells stably transfected with pPUR/PLP(+)Z or pPUR/PLP(−)Z. Total RNA was isolated from pools of puromycin-resistant cells and incubated with 32P-labeled antisense riboprobes for Plp1-lacZ (which contains Plp1 sequence only from exons 1 and 2) and β-actin. The full-length Plp1-lacZ riboprobe is 387 nt, while the full-length β-actin riboprobe is 304 nt. The fragments protected from digestion by RNases are indicated on the right side, while the position of radiolabeled markers is shown on the left side. Asterisks indicate the position of a protected fragment formed by hybridization with Plp1-lacZ transcripts that contain alternatively spliced exon(s) derived from Plp intron 1 sequence. Lane P contains the full-length (undigested) riboprobes. RNA was isolated from the indicated pools of stably transfected cells (A, TM3; B, +/+ Li; C, N20.1) or untransfected cells (lane U). Pool numbers (I, II, and III) correspond to the same pools used in Table 1.
3.4. Alternatively spliced exons derived from classical Plp1 intron 1 sequence
To determine the nature of transcripts in TM3 cells that contain alternatively spliced sequence derived from Plp1 intron 1, RT-PCR analysis was performed using an exon 1/exon 2 primer pair. In addition to the expected product, a larger fragment was generated which was longer by more than a 100 bp. Similarly sized RT-PCR products were also observed using RNA isolated from mouse tissues, including testis and liver (data not shown). RT-PCR products generated from RNA isolated from TM3 cells were subcloned and then sequenced, which revealed the existence of two alternatively spliced exons within Plp1 intron 1 DNA (Fig. 5). As expected, one of these was exon 1.1 which consists of 109 bp. The other one is a new exon, designated here as exon 1.2, which contains the 122 bp spanning Plp1 intron 1 positions 7,224–7,345, based upon numbering the intron from positions 1 to 8,140 (Wight and Dobretsova, 1997). Both exons are flanked by appropriate splice donor/acceptor sites in mouse and can be included independent of one another or together in mature transcripts (Fig. 5).
Fig. 5.

Arrangement of the Plp1 gene and sequence of alternatively spliced exons derived from what is classically defined as Plp1 intron 1. A: Arrangement of the Plp1 gene is shown at the top. Exons are illustrated by numbered boxes, which are not drawn to scale. Exons 1.1 and 1.2 (gray boxes) are not present in transcripts that encode classic gene products, PLP and DM20. [PLP is encoded by transcripts that select the distal (relative to the tsp) 5− splice site in exon 3 (mRNA contains exon 3A and 3B) and DM20 by transcripts that opt for the proximal (internal) 5− splice site (mRNA is missing exon 3B).] The start site of translation for the classic products is located near the end of exon 1 (bent arrow). RT-PCR analysis was performed with total RNA isolated from TM3 cells and the indicated primers. RT-PCR products were confirmed by DNA sequencing except for products obtained using the exon 1.2/exon 7 primer pair. RT-PCR products obtained with exon1/exon 2 and exon1/exon 1.2 primer pairs were subcloned prior to sequencing. Note that only the classic Dm20 product could be identified by direct sequencing of gel-purified RT-PCR products obtained with the exon 1/exon 4 primer pair due to a mixture of products in larger sized bands. B: Sequence for exon 1.1 (Bongarzone et al., 1999) and exon 1.2 is shown in capital letters and corresponds to Plp1 intron 1 positions 122–230 and 7,224–7,345, respectively (GenBank accession number AF003838). Lower case letters indicate surrounding intronic sequence that is 100% conserved in all splice sites. Internal ATG triplets (prospective initiation codons) are underlined in bold letters, which must be used for productive translation of transcripts that contain exon 1.1 or exon 1.2.
Exon 1.1 containing transcripts encode products that contain an additional 12 residues at the amino-terminus (counting the initiator methionine) that are restricted to the somata of oligodendrocytes and neurons in brain (Bongarzone et al., 1999). Similarly, incorporation of exon 1.2 into transcripts, by itself or in conjunction with exon 1.1, is predicted to yield products which contain two extra amino acids at the N-terminus (Met-Leu-Ala) followed by a single residue change (Gly→Ser) when compared to the classic (PLP/DM20) protein. Whether these changes affect the localization of the presumptive proteins remains to be determined. Productive translation of transcripts containing exon 1.2 (in the presence or absence of exon 1.1) requires that initiation begins at an internal AUG codon due to the presence of two in-frame stop codons just downstream of the first AUG in exon 1. It is likely that these products exist in nature since transcripts that simply contain exon 1.1 are expressed even though they possess only one in-frame stop codon downstream of the initial AUG triplet (Bongarzone et al., 1999). Indeed, a large number of transcripts in vertebrates contain AUGs upstream of the actual translation start site (Peri and Pandey, 2001).
Two classic products are generated from the Plp1 gene due to the presence of two donor sites in exon 3. PLP is encoded by transcripts that utilize the distal 5′ splice site whereas DM20 is produced if the proximal (internal) donor site is engaged (Nave et al., 1987). To see whether alternative splicing in exon 3 can occur in conjunction with the inclusion of exon 1.1 and/or exon 1.2, RT-PCR analysis was performed initially with an exon1/exon 4 primer (Fig. 5). Multiple RT-PCR products were generated using RNA isolated from TM3 cells and testis. The products were separated by gel electrophoresis, excised, and extracted for sequence analysis. DNA sequencing confirmed that the smallest product (595 bp) was generated from classic Dm20 mRNA. However direct sequencing of the other bands (which were approximately 700 bp or greater) was not successful due to a mixture of template DNA (e.g., the classic Plp1 product is 701 bp, Dm20 containing exon 1.1 is 705 bp, Dm20 containing exon 1.2 is 718 bp). To resolve this issue, RT-PCR analysis was performed with an exon 1.2/exon 7 primer pair which confirmed that exon 1.2 can be incorporated in both Plp1- and Dm20-related transcripts (Fig. 5); similar to the situation observed previously for exon 1.1 (Bongarzone et al., 1999). Thus inclusion of exon 1.1 and/or exon 1.2 in transcripts increases the likelihood of multiple Plp1 gene products in testis. Western blot analysis corroborated this supposition. In addition to PLP and DM20, protein(s) with molecular weights slightly greater than PLP were also detected in mouse testis (Fig. 6).
Fig. 6.
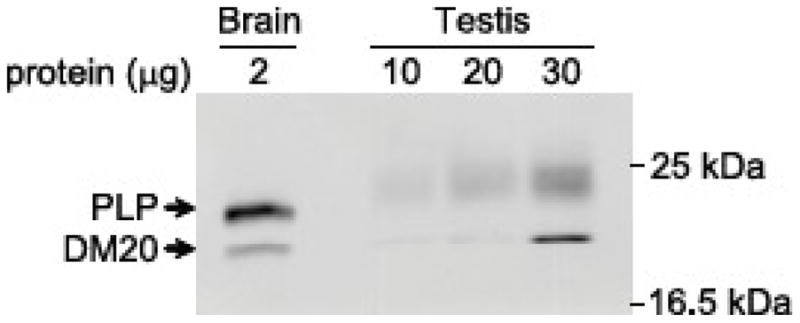
Western blot analysis of Plp1 gene products in brain and testis from a mouse at P28 of age. A mouse monoclonal antibody directed against PLP carboxy-terminal residues 272–277 was used as primary antibody, which is capable of detecting all splice isoforms.
4. Conclusions
This study demonstrates that the Plp1 gene is expressed in Leydig cells. However it is not the first “myelin-specific” gene known to be expressed in Leydig cells. Glial markers such as myelin/oligodendrocyte-specific protein, neural cell adhesion molecule, and the proteoglycan NG2 have been identified in Leydig cells and/or their progenitors (Davidoff et al., 2004). Here we report that the first intron of the Plp1 gene contains several regulatory elements that appear to affect its level of expression in Leydig (TM3) cells. For the most part, these elements function in a negative manner analogous to the situation with the mouse liver cell line, +/+ Li (Li et al., 2002a). However, a positive regulatory element (ASE) which is highly active in the N20.1 oligodendrocytic cell line, but nonfunctional in +/+ Li cells, is only marginally active in TM3 cells. Comparison of mRNA products in cell lines stably transfected with Plp1-lacZ constructs in which Plp1 intron 1 DNA is either present [PLP(+)Z] or absent [PLP(−)Z] demonstrate that the different cell lines vary in their propensity for inclusion of additional exons. The relative level of splice variants to the prototypic (classic) products commonplace in oligodendrocytes was highest in +/+ Li cells, lowest in N20.1 cells and intermediate in TM3 cells. However TM3 cells still contained slightly higher levels of splice variants compared to the classic species. Sequencing of the splice variants from TM3 cells revealed the presence of two additional (alternative) exons located within Plp1 intron 1. One of these, exon 1.1, has been described previously in neural cells (Bongarzone et al., 1999). However the other is a novel exon which we have designated exon 1.2. These non-classical exons may be included in transcripts either separately or together giving rise to multiple Plp1 gene products in testis. What role these proteins play in Leydig cells remains to be elucidated. Curiously, electron dense layered membranes termed “myelin figures” are often found adjacent to lysosomes and mitochondria in Leydig cells (Prince, 2007). Whether these membranes contain Plp1 gene products is to date, unknown.
Acknowledgments
This work was supported by grants from the National Multiple Sclerosis Society (RG 2705), the National Institutes of Health (R01 NS037821 and P30 NS047546), as well as NIH Grant Number P20 RR-16460 from the IDeA Networks of Biomedical Research Excellence (INBRE) Program of the National Center for Research Resources.
Abbreviations
- ASE
antisilencer enhancer
- βGal
β-galactosidase
- bp
base pairs
- nt
nucleotides
- PLP
proteolipid protein
- RLU
relative light units
- RSV
Rous sarcoma virus
- RT
reverse transcription
- tsp
transcription start point
- u
units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bongarzone ER, Campagnoni CW, Kampf K, Jacobs EC, Handley VW, Schonmann V, Campagnoni AT. Identification of a new exon in the myelin proteolipid protein gene encoding novel protein isoforms that are restricted to the somata of oligodendrocytes and neurons. J Neurosci. 1999;19:8349–8357. doi: 10.1523/JNEUROSCI.19-19-08349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff MS, Middendorff R, Enikolopov G, Riethmacher D, Holstein AF, Müller D. Progenitor cells of the testosterone-producing Leydig cells revealed. J Cell Biol. 2004;167:935–944. doi: 10.1083/jcb.200409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobretsova A, Johnson JA, Jones RC, Edmondson RD, Wight PA. Proteomic analysis of nuclear factors binding to an intronic enhancer in the myelin proteolipid protein gene. J Neurochem. 2008;105:1979–1995. doi: 10.1111/j.1471-4159.2008.05288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobretsova A, Kokorina NA, Wight PA. Functional characterization of a cis-acting DNA antisilencer region that modulates myelin proteolipid protein gene expression. J Neurochem. 2000;75:1368–1376. doi: 10.1046/j.1471-4159.2000.0751368.x. [DOI] [PubMed] [Google Scholar]
- Dobretsova A, Wight PA. Antisilencing: myelin proteolipid protein gene expression in oligodendrocytes is regulated via derepression. J Neurochem. 1999;72:2227–2237. doi: 10.1046/j.1471-4159.1999.0722227.x. [DOI] [PubMed] [Google Scholar]
- Feng JM, Fernandes AO, Bongarzone ER, Campagnoni CW, Kampf K, Campagnoni AT. Expression of soma-restricted proteolipid/DM20 proteins in lymphoid cells. J Neuroimmunol. 2003;144:9–15. doi: 10.1016/j.jneuroim.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Jacobs EC, Bongarzone ER, Campagnoni CW, Kampf K, Campagnoni A. Soma-restricted products of the myelin proteolipid gene are expressed primarily in neurons in the developing mouse nervous system. Dev Neurosci. 2003;25:96–104. doi: 10.1159/000072259. [DOI] [PubMed] [Google Scholar]
- Li S, Dobretsova A, Kokorina NA, Wight PA. Repression of myelin proteolipid protein gene expression is mediated through both general and cell type-specific negative regulatory elements in nonexpressing cells. J Neurochem. 2002a;82:159–171. doi: 10.1046/j.1471-4159.2002.00962.x. [DOI] [PubMed] [Google Scholar]
- Li S, Moore CL, Dobretsova A, Wight PA. Myelin proteolipid protein (Plp) intron 1 DNA is required to temporally regulate Plp gene expression in the brain. J Neurochem. 2002b;83:193–201. doi: 10.1046/j.1471-4159.2002.01142.x. [DOI] [PubMed] [Google Scholar]
- Mather JP. Establishment and characterization of two distinct mouse testicular epithelial cell lines. Biol Reprod. 1980;23:243–252. doi: 10.1095/biolreprod23.1.243. [DOI] [PubMed] [Google Scholar]
- Meng F, Zolova O, Kokorina NA, Dobretsova A, Wight PA. Characterization of an intronic enhancer that regulates myelin proteolipid protein (Plp) gene expression in oligodendrocytes. J Neurosci Res. 2005;82:346–356. doi: 10.1002/jnr.20640. [DOI] [PubMed] [Google Scholar]
- Nave KA, Lai C, Bloom FE, Milner RJ. Splice site selection in the proteolipid protein (PLP) gene transcript and primary structure of the DM-20 protein of central nervous system myelin. Proc Natl Acad Sci USA. 1987;84:5665–5669. doi: 10.1073/pnas.84.16.5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri S, Pandey A. A reassessment of the translation initiation codon in vertebrates. Trends Genet. 2001;17:685–687. doi: 10.1016/s0168-9525(01)02493-3. [DOI] [PubMed] [Google Scholar]
- Prince FP. The human Leydig cell: functional morphology and developmental history. In: Payne AH, Hardy MP, editors. The Leydig Cell in Health and Disease. Humana Press; New Jersey: 2007. pp. 71–89. [Google Scholar]
- Verity AN, Bredesen D, Vonderscher C, Handley VW, Campagnoni AT. Expression of myelin protein genes and other myelin components in an oligodendrocytic cell line conditionally immortalized with a temperature-sensitive retrovirus. J Neurochem. 1993;60:577–587. doi: 10.1111/j.1471-4159.1993.tb03188.x. [DOI] [PubMed] [Google Scholar]
- Wight PA, Dobretsova A. The first intron of the myelin proteolipid protein gene confers cell type-specific expression by a transcriptional repression mechanism in non-expressing cell types. Gene. 1997;201:111–117. doi: 10.1016/s0378-1119(97)00435-6. [DOI] [PubMed] [Google Scholar]
- Wight PA, Dobretsova A. Where, when and how much: regulation of myelin proteolipid protein gene expression. Cell Mol Life Sci. 2004;61:810–821. doi: 10.1007/s00018-003-3309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight PA, Duchala CS, Readhead C, Macklin WB. A myelin proteolipid protein-lacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J Cell Biol. 1993;123:443–454. doi: 10.1083/jcb.123.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]