

Abstract

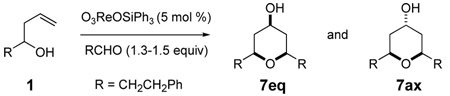
The rhenium(VII) complex O3ReOSiPh3 are particularly effective catalyst for Prins cyclizations using aromatic and α,β-unsaturated aldehydes. The reaction conditions are mild and the highly substituted 4-hydroxy tetrahydropyran products are formed stereoselectively. Rhenium(VII) complexes appear to spontaneously form esters with alcohols and to directly activate electron rich alcohols for solvolysis. Re2O7 and perrhenic acid were equally effective in catalyzing these cyclizations.
Osborn reported that the Re(VII) complex O3ReOSiPh31 was a highly effective catalyst for allylic alcohol isomerizations.2 Grubbs studied Osborn's reaction and found that O3ReOSiPh3 isomerizes allylic alcohols stereospecifically.3 Both Osborn and Grubbs invoked a concerted [3,3] rearrangement of allylic perrhenate esters to explain this very facile and selective isomerization (Figure 1).2,3 An alternative mechanism was invoked to explain some of the peculiarities of the reaction. Osborn proposed an ionic mechanism to explain the kinetics for formation of Z-alkenes,2b and Grubbs invoked a similar mechanism to explain the loss of optical purity with certain electron-rich allylic alcohol substrates (Figure 1).3 The proposed ionic mechanism attracted our attention because it suggested that Re(VII) complexes might activate and solvolyze electron rich alcohols. We report herein a Prins cyclization initiated by Re(VII) catalysts4 based on this concept that is both very mild and shows unusual selectivity.
Figure 1.

Grubbs-Osborn mechanism for stereoselective isomerization of allylic alcohols and the alternative ionic mechanism that leads to loss of selectivity are presented
Prins cyclization reactions between homoallylic alcohols and aldehydes are generally promoted by strong Bronsted or Lewis acids.5 If halide counterions are present, they are incorporated at the 4-position of the newly formed tetrahydropyran (THP) ring.5 Oxygen atoms can be captured at the 4-position of the THP ring,6 although most acids lead to esters that must be hydrolyzed to form 4-hydroxy THP products.7 Most Prins cyclizations use super-stoichiometric acid to promote the reaction because the acid counterion is trapped in the product, thus consuming the acid.5 Re(VII) complex O3ReOSiPh3 is proposed to catalyze the Prins cyclization as illustrated in Scheme 1. Activation is achieved by forming perrhenate ester 4 from hemiacetal 3. Solvolysis of ester 4 generates the intermediate 5 as a contact ion pair. Cyclization and trapping leads to perrhenate ester 6. Ester exchange with Ph3SiOH would regenerate the catalyst. The proposed Re(VII) initiated reaction has several potential advantages over traditional Prins cyclization reactions. It avoids the use of strong acid, and the facile transesterification of perrhenate esters with alcohols allows turnover and makes the system catalytic. Additionally, the alcohol product would be produced directly from the reaction rather than requiring a subsequent hydrolysis step. These potentially attractive features led us to explore this reaction.
Scheme 1.

Proposed mechanism for activation and Prins cyclization using O3ReOSiPh3
Table 1 presents an initial exploration of the Re(VII) catalyzed Prins cyclization. The aldehyde and homoallylic alcohol were selected with the same substituent to avoid complications from oxonia-Cope induced side-chain exchanges.8 O3ReOSiPh3 catalyzed the Prins cyclization effectively in most solvents except acetonitrile. Although the reaction was effective using 1 mol % catalyst (entry 1), 5 mol % catalyst was selected to screen conditions. Methylene chloride, chloroform and hexanes all gave yields over 60% on stirring at room temperature (entries 2, 4, and 7). The reaction times varied from several hours to several days. Vanadyl esters were also investigated as catalysts,9 but they were less efficient. For example, 30 mol % of tri-n-propyl vanadate (entry 9) gave slightly more than one mole of product per mole of catalyst after several days. Phosphomolybdic acid catalysis was evaluated for comparison (entry 10) and gave a lower yield but with higher equatorial selectivity.6g Two molybdenum oxo complexes were screened as catalysts but they were ineffective.10
Table 1.
Screening Prins Cyclization Conditions with O3ReOSiPh3 and Other Catalysts
 | ||||
|---|---|---|---|---|
| entry | solvent | time (h) | yield (%) | eq/ax |
| 1a | CH2Cl2 | 36 | 54 | 1.5:1 |
| 2 | CH2Cl2 | 3 | 63 | 1.9:1 |
| 3 | CHCl3 | 48 | 67 | 2.7:1 |
| 4 | EtOAc | 21 | 33 | 4.5:1 |
| 5 | MeNO2 | 24 | 40 | 6.0:1 |
| 6 | hexanes | 48 | 62 | 15:1 |
| 7 | toluene | 48 | 38 | 4.4:1 |
| 8 | MeCN | 48 | 0 | NA |
| 9b | CH2Cl2 | 72 | 37 | 1:2.4 |
| 10c | CH2Cl2 | 20 | 43 | >10:1 |
Reaction was carried out with 1 mol % O3ReOSiPh3.
Reaction was carried out with 30 mol % VO(OPrn)3 instead of O3ReOSiPh3 and with two equivalents of the aldehyde.
Reaction catalyzed with 5 mol % H3[P(Mo3O10)4]·×H2O.
One surprising aspect of the oxo-metal catalyzed reaction is the isolation of axial Prins product (7ax) as a significant component of the product. Prins reactions normally give predominantly equatorial 4-heteroatom products, and all previously reported 4-oxygen adducts showed high equatorial selectivity.6,7 We previously reported that the axial selectivity was a function of the lifetime and reactivity of the ion pair, with very reactive nucleophiles, such as bromide anion, leading to high axial selectivity.11 Typical oxygen nucleophiles, such as CF3CO2− or AcO·BF3− have low nucleophilicity and lead to ca. 20:1 selectivity for the equatorial product.8f The Re(VII) catalyst, while still favoring the equatorial product, shows a much lower level of selectivity. The vanadate ester example (entry 9) actually favors the axial alcohol product. These experiments suggest that perrhenate and other oxo-metal counter ions are much more nucleophilic than trifluoroacetate counterion in Prins reactions and that they may show unusual patterns of reactivity and selectivity. On a practical note, the best selectivity for the equatorial product was found using O3ReOSiPh3 in hexanes (entry 6).
Prins reactions with aromatic and aliphatic aldehydes behave differently when catalyzed by O3ReOSiPh3. Scheme 2 presents the reaction between isobutyraldehyde and alcohol 1. The expected product 8 is accompanied by side products 9 and 7 that arise from facile 2-oxonia-Cope rearrangements.8 The outcome with an electron rich aromatic aldehyde is very different. Side-chain exchange products are much less prevalent, and the expected Prins product was formed in 49% yield with excellent equatorial alcohol selectivity.12,13 We believe that the greater nucleophilicity of the perrhenate anion limits oxonia-Cope rearrangement with aromatic aldehydes.14 The excellent equatorial selectivity is consistent with previous observations, where a more stable oxocarbenium ion leads to a less reactive ion pair and better equatorial selectivity.
Scheme 2.

Prins reaction with aliphatic and aromatic aldehydes
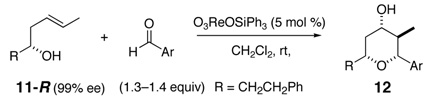
The reactivity with aromatic aldehydes was further investigated (Table 2). Substituted alcohol 11-R and its enantiomer, 11-S were used in this study.15 Both electron rich and electron poor aromatic aldehydes gave excellent yields and selectivities for the equatorial alcohol Prins product. In entries 2 and 4, the products were demonstrated to have high optical purities. Remarkably, the unprotected free phenol in entry 3 worked very well in the reaction. The very electron rich phenol in entry 7 did give some Prins product, but only very slowly. The corresponding sulfonyl-protected phenol gave an excellent yield of equatorial THP product. The aldehydes in entries 7 and 8 were previously investigated in our approach to kendomycin,13 but the Re(VII) catalyzed Prins cyclization conditions appear to be both milder and more general. Electron withdrawing substituents (entry 9, 10) lead to slightly lower yields than with electron rich aldehydes.
Table 2.
Prins Cyclization with Aromatic Aldehyde and O3ReOSiPh3
 | |||
|---|---|---|---|
| entry | aldehyde | time (h) | yield (%) |
| 1 |  |
24 | 82 |
| 2a |  |
25 | 88b |
| 3 | 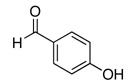 |
27 | 75 |
| 4a | 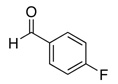 |
26 | 89b |
| 5a | 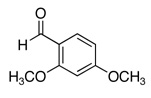 |
50 | 58 |
| 6 | 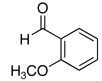 |
18 | 84 |
| 7 | 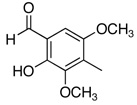 |
168 | 30d |
| 8c | 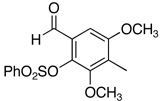 |
23 | 83 |
| 9 | 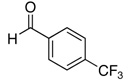 |
24 | 73 |
| 10 | 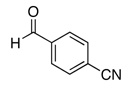 |
24 | 55 |
Enantiomeric series (11-S alcohol, 99% ee).
97% ee by HPLC analysis.
10 mol % O3ReOSiPh3.
Starting material was also recovered in 11% yield.
A common problem with aliphatic aldehydes is the generation of side-chain exchange products as shown in Scheme 2. The Re(VII) conditions appear to minimize this exchange with aromatic aldehydes. An indirect solution for selective Prins cyclization of aliphatic aldehydes might be developed using unsaturated aldehydes, followed by reduction or functionalization of the alkene.16 A few representative unsaturated aldehydes were investigated and the results are shown in Scheme 3.17 All three aldehydes worked well in the reaction, although the more complex aldehyde 17, prepared by a metathesis reaction between crotonaldehyde and the corresponding terminal alkene, was noticeably slower than the others. All of the products showed very good selectivity for the equatorial alcohol THP products. Unsaturated aldehydes will be useful for forming complex THP fragments suitable for natural product synthesis.
Scheme 3.

Prins reaction with unsaturated aldehydes
Several details of the catalytic cycle are not clear at this time, but a few control experiments have placed some limits on possible mechanisms. The Prins reaction in Table 1 in DCM returned only starting material when proton scavenger 2,6-di-tert-butyl-4-methylpyridine (2,6-DTBMP) or 4 Å sieves were added. Grubbs reported that hindered tertiary amines also halted the allylic isomerization by O3ReOSiPh3.3 Addition of water stopped the reaction.18 Added CSA had no effect on the reaction. The basic path of the reaction presumably follows the mechanism outlined in Scheme 1. One significant unanswered question is whether the activation of the hemiacetal 3 is due to perrhenate ester formation or simple acid-promoted solvolysis.
Because of ambiguities in the mechanism, we decided to test other Re(VII) catalysts, and the results are shown in Scheme 4. Solutions of 65–70% aqueous perrhenic acid18 are equally effective as O3ReOSiPh3 in the Prins cyclization of alcohol 11-R and p-anisaldehyde (19). Re2O7 is also effective for catalyzing Prins reactions. In both cases, these commercially available Re(VII) compounds lead to outcomes essentially indistinguishable from O3ReOSiPh3. Perrhenic acid is a strong acid with a pKa = −1.25,19 so an acid catalyzed activation of hemiacetal 3 (Scheme 1) is certainly possible. An indirect test for perrhenate ester formation was conducted by exploring the allylic alcohol rearrangement of allylic alcohol 21.3 Both O3ReOSiPh3 and perrhenic acid lead to efficient rearrangement to conjugated allylic alcohol 22 under similar conditions, suggesting that 65–70% aqueous perrhenic acid is capable of forming perrhenate esters under these conditions.18 Thus perrhenic acid may be active through the same mechanism (Scheme 1) proposed for O3ReOSiPh3.
Scheme 4.

Comparison of different Re(VII) catalyst
Re(VII) complexes are uniquely effective catalysts for the Prins cyclization. These catalysts will be useful for Prins cyclizations and will be of interest for the development of other catalytic processes.
Supplementary Material
Characterization data and experimental procedures for all compounds described are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
This work was supported by the National Cancer Institute (CA-081635) and by a generous gift from the Schering-Plough Research Institute. KT was supported by a fellowship from the Royal Thai Government.
References
- 1.Schoop T, Roesky HW, Noltemeyer M, Schmidt HG. Organometallics. 1993;12:571–574. [Google Scholar]
- 2.(a) Bellemin-Laponnaz S, Gisie H, Pierre Le Nye J, Osborn JA. Angew. Chem. Int. Ed. Engl. 1997;36:976–978. [Google Scholar]; (b) Bellemin-Laponnaz S, Le Ny JP, Osborn JA. Tetrahedron Lett. 2000;41:1549–1552. [Google Scholar]
- 3.(a) Morrill C, Grubbs RH. J. Am. Chem. Soc. 2005;127:2842–2843. doi: 10.1021/ja044054a. [DOI] [PubMed] [Google Scholar]; (b) Morrill C, Beutner GL, Grubbs RH. J. Org. Chem. 2006;71:7813–7825. doi: 10.1021/jo061436l. [DOI] [PubMed] [Google Scholar]
- 4.For several other related applications of high-oxidation Re catalysts in synthesis, see: Ishihara K, Furuya Y, Yamamoto H. Angew. Chem. Int. Ed. Engl. 2002;41:2983–2986. doi: 10.1002/1521-3773(20020816)41:16<2983::AID-ANIE2983>3.0.CO;2-X. Maeda Y, Nishimura T, Uemura S. Chem. Lett. 2005;34:790–791. Sherry BD, Radosevich AT, Toste FD. J. Am. Chem. Soc. 2003;125:6076–6077. doi: 10.1021/ja0343050. Sherry BD, Loy RN, Toste FD. J. Am. Chem. Soc. 2004;126:4510–4511. doi: 10.1021/ja031895t.
- 5.(a) Pastor IM, Yus M. Curr. Org. Chem. 2007;11:925–957. [Google Scholar]; (b) Snider BB. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Heathcock CH, editors. Vol. 2. New York: Pergamon Press; 1991. pp. 527–561. [Google Scholar]; (c) Adams DR, Bhatnagar SP. Synthesis. 1977:661–672. [Google Scholar]; (d) Overman LE, Pennington LD. J. Org. Chem. 2003;68:7143–7157. doi: 10.1021/jo034982c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Hanschke E. Chem. Ber. 1955;88:1053–1061. [Google Scholar]; (b) Zhang W-C, Viswanathan GS, Li C-J. Chem. Commun. 1999:291–292. [Google Scholar]; (c) Zhang W-C, Li C-J. Tetrahedron. 2000;56:2403–2411. [Google Scholar]; (d) Yadav JS, Reddy BVS, Kumar GM, Murthy CVSR. Tetrahedron Lett. 2001;42:89–91. [Google Scholar]; (e) Keh CCK, Namboodiri VV, Varma RS, Li C-J. Tetrahedron Lett. 2002;43:4993–4996. [Google Scholar]; (f) Hart DJ, Bennett CE. Org. Lett. 2003;5:1499–1502. doi: 10.1021/ol0342756. [DOI] [PubMed] [Google Scholar]; (g) Yadav JS, Reddy BVS, Kumar GGKSN, Aravind S. Synthesis. 2008:395–400. [Google Scholar]
- 7.(a) Kay IT, Williams EG. Tetrahedron Lett. 1983;24:5915–5918. [Google Scholar]; (b) Kay IT, Bartholomew D. Tetrahedron Lett. 1984;25:2035–2038. [Google Scholar]; (c) Jaber JJ, Mitsui K, Rychnovsky SD. J. Org. Chem. 2001;66:4679–4686. doi: 10.1021/jo010232w. [DOI] [PubMed] [Google Scholar]; (d) Barry CSJ, Crosby SR, Harding JR, Hughes RA, King CD, Parker GD, Willis CL. Org. Lett. 2003;5:2429–2432. doi: 10.1021/ol0346180. [DOI] [PubMed] [Google Scholar]; (e) Barry CS, Elsworth JD, Seden PT, Bushby N, Harding JR, Alder RW, Willis CL. Org. Lett. 2006;8:3319–3322. doi: 10.1021/ol0611705. [DOI] [PubMed] [Google Scholar]
- 8.(a) Lolkema LDM, Semeyn C, Ashek L, Hiemstra H, Speckamp WN. Tetrahedron. 1994;50:7129–7140. [Google Scholar]; (b) Rychnovsky SD, Marumoto S, Jaber JJ. Org. Lett. 2001;3:3815–3818. doi: 10.1021/ol0168465. [DOI] [PubMed] [Google Scholar]; (c) Alder RW, Harvey JN, Oakley MT. J. Am. Chem. Soc. 2002;124:4960–4961. doi: 10.1021/ja025902+. [DOI] [PubMed] [Google Scholar]; (d) Crosby SR, Harding JR, King CD, Parker GD, Willis CL. Org. Lett. 2002;4:577–580. doi: 10.1021/ol0102850. [DOI] [PubMed] [Google Scholar]; (e) Barry CS, Bushby N, Harding JR, Hughes RA, Parker GD, Roe R, Willis CL. Chem. Commun. 2005:3727–3729. doi: 10.1039/b504562b. [DOI] [PubMed] [Google Scholar]; (f) Jasti R, Anderson CD, Rychnovsky SD. J. Am. Chem. Soc. 2005;127:9939–9945. doi: 10.1021/ja0518326. [DOI] [PubMed] [Google Scholar]; (g) Jasti R, Rychnovsky SD. J. Am. Chem. Soc. 2006;128:13640–13648. doi: 10.1021/ja064783l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chabardes P, Kuntz E, Varagnat J. Tetrahedron. 1977;33:1775–1783. [Google Scholar]
- 10.MoO2(acac)2 Rajan OA, Chakravorty A. Inorg. Chem. 1981;20:660–664. MoO2(OSiPh3)2 Huang M, DeKock CW. Inorg. Chem. 1993;32:2287–2291.
- 11.(a) Jasti R, Vitale J, Rychnovsky SD. J. Am. Chem. Soc. 2004;126:9904–9905. doi: 10.1021/ja046972e. [DOI] [PubMed] [Google Scholar]; (b) Miles RB, Davis CE, Coates RM. J. Org. Chem. 2006;71:1493–1501. doi: 10.1021/jo052142n. [DOI] [PubMed] [Google Scholar]
- 12.This result stands in contrast to our Prins reaction approach to kendomycin, where an electron rich aromatic ring led to side chain exchange products as the major component in a TFA/DCM promoted reaction (ref. 13).
- 13.Bahnck KB, Rychnovsky SD. Chem. Commun. 2006:2388–2390. doi: 10.1039/b602937j. [DOI] [PubMed] [Google Scholar]
- 14.The aliphatic aldehyde oxonia-Cope rearrangement evident in Scheme 2 should be thermoneutral, whereas the aromatic oxonia-Cope should favor the aromatic oxocarbenium ion. In theory this effect might suppress oxonia-Cope side reactions with aromatic aldehydes, but in our experience it does not, perhaps because of a slower Prins cyclization with aromatic aldehydes. See ref. 13 for an example.
- 15.(a) Nokami J, Nomiyama K, Matsuda S, Imai N, Kataoka K. Angew. Chem. Int. Ed. 2003;42:1273–1276. doi: 10.1002/anie.200390327. [DOI] [PubMed] [Google Scholar]; (b) Nokami J, Ohga M, Nakamoto H, Matsubara T, Hussain I, Kataoka K. J. Am. Chem. Soc. 2001;123:9168–9169. doi: 10.1021/ja011257f. [DOI] [PubMed] [Google Scholar]
- 16.Chan K-P, Ling YH, Loh T-P. Chem. Commun. 2007:939–941. doi: 10.1039/b616558c. [DOI] [PubMed] [Google Scholar]
- 17.A Prins reaction between alcohol 1 and cinnamaldehyde led to the expected product in 40% yield and the side-chain exchanged product 7 in 18% yield. Further work needs to be done to make Prins reactions with terminal alkenes practical.
- 18.The "wet" reaction was run in water-saturated DCM. At 27 °C, the concentration of water would be 0.137 M (0.186 wt %: Stephenson RM. J. Chem. Eng. Data. 1992;37:80–95. ) With a catalyst concentration of 0.005 M, the molar ratio of water/Re(VII) would be ca. 27:1. For comparison, the molar ratio of commercial 70 wt % perrhenic acid is about 6:1 water/Re(VII). Presumably the difference in the water concentration led to the difference in outcome.
- 19.Bailey N, Carrington A, Lott KAK, Symons MCR. J. Chem. Soc. 1960:290–297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization data and experimental procedures for all compounds described are included. This material is available free of charge via the Internet at http://pubs.acs.org.