

Abstract
The death of insulin-producing β-cells is a key step in the pathogenesis of type 2 diabetes. The amyloidogenic peptide Islet Amyloid Polypeptide (IAPP, also known as amylin) has been shown to disrupt β-cell membranes leading to β-cell death. Despite the strong evidence linking IAPP to the destruction of β-cell membrane integrity and cell death, the mechanism of IAPP toxicity is poorly understood. In particular, the effect of IAPP on the bilayer structure has largely been uncharacterized. In this study, we have determined the effect of the amyloidogenic and toxic hIAPP1-37 peptide and the non-toxic and non-amyloidogenic rIAPP1-37 peptide on membranes by a combination of DSC and solid-state NMR spectroscopy. We also characterized the toxic but largely non-amyloidogenic rIAPP1-19 and hIAPP1-19 fragments. DSC shows that both amyloidogenic (hIAPP1-37) and largely non-amyloidogenic (hIAPP1-19 and rIAPP1-19) toxic versions of the peptide strongly favor the formation of negative curvature in lipid bilayers, while the non-toxic full-length rat IAPP1-37 peptide does not. This result was confirmed by solid-state NMR spectroscopy which shows that in bicelles composed of regions of high curvature and low curvature, non-toxic rIAPP1-37 binds to the regions of low curvature while toxic rIAPP1-19 binds to regions of high curvature. Similarly, solid-state NMR spectroscopy shows that the toxic rIAPP1-19 peptide significantly disrupts the lipid bilayer structure, whereas the non-toxic rIAPP1-37 does not have a significant effect. These results indicate IAPP may induce the formation of pores by the induction of excess membrane curvature and can be used to guide the design of compounds that can prevent the cell-toxicity of IAPP. This mechanism may be important to understand the toxicity of other amyloidogenic proteins. Our solid-state NMR results also demonstrate the possibility of using bicelles to measure the affinity of biomolecules for negatively or positively curved regions of the membrane, which we believe will be useful in a variety of biochemical and biophysical investigations related to the cell membrane.
INTRODUCTION
Amyloid proteins have been implicated in the etiology of numerous diseases such as type 2 diabetes,1 Alzheimer’s disease,2-4 Parkinson’s disease,5, 6 Huntington’s disease,7 and Creutzfeldt-Jakob disease.8 Investigating the mechanisms that trigger the amyloid misfolding process is critical for the development of compounds to treat these diseases. Several studies have shown that the interactions of these amyloid proteins/peptides with cell membranes catalyze the conversion from their non-toxic to toxic forms.9-17 Therefore, the affinity of amyloidogenic peptides towards the cell membrane is particularly important in the formation of toxic oligomeric intermediates that leads to the disruption of lipid bilayer structure. In this study we report an investigation of IAPP-membrane interactions that are correlated with type 2 diabetes.
Type 2 diabetes is the result of both increased resistance to insulin and decreased insulin production by β-cells.18 The relative contribution of each factor to the pathology of the disease is still uncertain, however recent research suggests that most genes predisposing individuals to type 2 diabetes are associated with the maintenance of β-cell mass and not insulin resistance,19 illustrating the key role the loss of β-cell mass plays in the early stages of the disease.20, 21 The pathological aggregation of human Islet Amyloid Polypeptide (hIAPP) has been shown to be central to this process. IAPP is a 37 residue peptide hormone that acts synergistically with insulin in glycemic control. Dense masses of IAPP composed of long cross beta-sheet amyloid fibers are found in the pancreatic islets of 90% of diabetic patients.22 Aggregates of hIAPP disrupt the integrity of the β-cell membrane, allowing entry of Ca+2 ions into the cell either through the formation of small pores or complete disruption of the membrane.23-26 Elevated intracellular calcium levels then impose oxidative stress on the β-cell as the mitochondria attempt to maintain ion balance in the cell. The ultimate result is apoptosis and β-cell death.26-29
Several characteristics of hIAPP may explain its membrane disrupting activity, but one of the most notable features of hIAPP is its tendency to distort membranes.30-32 These morphological changes in membrane structure appear to be related to the aggregation of the peptide. The linear growth of a rigid amyloid fiber attached to a flexible membrane causes a curvature strain in the membrane, which is relieved by a distortion of the membrane shape. An excessive amount of curvature strain can result in membrane fragmentation by the forcible detachment of membrane patches from the surface. 30, 33, 34 Moreover, it is likely that a distorted membrane will be more permeable to the passage of small solutes.32 Other highly curved membrane structures have also been observed after the addition of hIAPP. Highly curved striations in the membrane surface have been reported in liposomal suspensions and islet cells in the regions where the amyloid fiber contacts the membrane surface.14, 24 Curvature of the membrane causes a mechanical stress within the membrane and may be responsible for the opening of mechanosensitive calcium channels by IAPP leading to an ER stress response and β-cell death.35
Curvature plays an important role in the normal physiological functioning of biomembranes, allowing them to assume shapes that are optimal for their function. Peptides can alter the energetic cost of deforming the bilayer and, because membrane proteins are energetically coupled to the bilayer, peptide binding can also lead to conformational changes in other membrane proteins throughout the bilayer.36, 37 The creation of membrane curvature can also have a direct effect on cellular processes. For example, certain antimicrobial peptides create transient pores to kill bacteria by inducing excessive curvature in the membrane.38-41 It is possible that membrane curvature plays a similar role in the pathology of IAPP as it plays in the function of antimicrobial peptides. To investigate this possibility, we characterized IAPP induced membrane curvature in lipid bilayers and study its effects on the physicochemical properties of the bilayer. Furthermore, we have uncovered a correlation between the toxicity of IAPP and its fragments and their ability to induce negative curvature strain in lipid bilayers.
To test the effect of curvature stress in membrane damage by IAPP and to isolate the effects of non-amyloid dependent membrane damage, we have used four IAPP peptides (Figure 1) that have different propensities for the formation of amyloid fibers and for disrupting membranes. Full-length hIAPP1-37 forms amyloid fibers and displays a high toxicity to cells.1, 25, 42-46 The rat version of IAPP (rIAPP1-37) differs from hIAPP by six residues, all but one of these (the H18R substitution) are in the 20-29 region believed to be primarily responsible for the aggregation into amyloid fibers.47 The prolines within the rIAPP1-37 sequence are believed to prevent the aggregation of rIAPP1-37 into amyloid fibers. Despite possessing a similar α-helical conformation in the membrane as hIAPP1-37, rIAPP1-37 does not disrupt membranes to a significant extent and is non-toxic to cells.13, 15 The 1-19 fragment of hIAPP lacks the amyloidogenic region (residues 20-29) and does not form amyloid fibers while bound to the membrane,48 although it has a weak propensity to form amyloid fibers in solution.49 Nevertheless, despite its lack of amyloidogenicity, hIAPP1-19 has a high potential to disrupt both artificial membranes and islet cells.48, 50 Like hIAPP1-19, rIAPP1-19 is also toxic to islet cells, albeit at a reduced rate.50
Figure 1. Amino acid sequences of rat and human IAPP sequences used in this study.

The differences between the rat and human sequences are shown in red. There are disulfide bonds between residues 2 and 8 and the C-termini of the peptides are amidated like the physiologically expressed peptide.
It has been a major challenge to measure the affinity of biomolecules such as peptides or proteins for negatively or positively curved cell membrane regions. The main difficulties are in identifying a suitable model membrane that possesses both senses of curvature and a biophysical tool that can measure high-resolution structural details associated with membrane binding. In this study, we demonstrate the use of bicelles as suitable model membranes to accomplish this task. Solid-state NMR experiments on magnetically aligned 9:9:4 DMPC: DMPG: DHPC bicelles were used to determine the atomic-level disruption of lipid bilayer structures due to IAPP peptides. While 2D PDLF solid-state NMR experiments were used to determine the peptide-induced changes in the order of 13C-1H bonds in the hydrophobic core and the glycerol regions of the lipids, 31P chemical shift and 14N quadrupole coupling parameters were used to evaluate changes in the head group region. DSC experiments were used to determine the peptide-induced curvature strain on DiPoPE lipid bilayers. These results are correlated with the cell-toxicity of IAPP peptides and used to determine the mechanism of membrane disruption.
MATERIALS AND METHODS
All lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Chloroform and methanol were procured from Aldrich Chemical Inc. (Milwaukee, WI). All chemicals were used without further purification. All peptides were synthesized and purified with C-terminal amidation and a disulfide bond between residues 2 and 7 by Genscript, except hIAPP1-37 which was synthesized by SynBioSci. All peptides were >95% pure as verified by HPLC and electrospray mass spectroscopy.
Preparation of Peptide Samples
Lyophilized hIAPP1-37, hIAPP1-19, or rIAPP1-19 was dissolved in HFIP at a concentration of 10 mg/ml for one hour to break up any pre-formed aggregates present in the solution. Aliquots of the peptide stock solution were then flash-frozen in liquid nitrogen and lyophilized again for more than 16 hours at less than 1 millitorr vacuum to completely remove HFIP.51 The lyophilized pellet was then dissolved in methanol at 4 mg/ml and then the appropriate amount was added to lipids dissolved in choloroform to prepare samples for NMR or DSC experiments as given in detail below. Rat IAPP1-37, which does not aggregate, was dissolved directly in methanol at 4 mg/ml.
Preparation of Bicelle Samples
Bicelles were prepared by mixing 60 mg total of DMPC, DMPG, and DHPC (9:9:4 mole ratio, q ratio = 4.5) dissolved in chloroform with the appropriate amount of the peptide (1% rIAPP1-37, 0.5% rIAPP1-19) dissolved in methanol. The bulk solvent was removed by slowly evaporating the lipid mixture under a stream of nitrogen. Residual solvent was removed placing the sample under high vacuum overnight. The dried sample was then rehydrated by the addition of 200 μL of 10 mM pH 7.4 HEPES buffer containing 150 mM NaCl buffer to give a hydration level of 66%. The samples were then subjected to repeated heating and cooling cycles above and below the bicelle formation temperature until clear transparent solutions were formed. The sample was not subjected to freezing during the heating and cooling cycles, as previously frozen samples yielded poorly aligned bicelles.
Solid-state NMR Spectroscopy
All of the experiments were performed on a Chemagnetics/Varian Infinity 400 MHz solid-state NMR spectrometer. Each sample was equilibrated at 30° C for at least 30 minutes before starting the experiment. 31P NMR spectra were obtained using a spin-echo sequence (90°-τ-180°-τ-acquisition; τ = 125 μs) with a 90° pulse length of 5 μs under 30 kHz proton decoupling. Chemical shifts were referenced by setting the isotropic chemical shift peak of phosphoric acid to 0 ppm. 14N quadrupolar spectra were recorded using a quadrupolar echo sequence (90°-τ-90°-τ-acquisition; τ = 80 μs) without proton decoupling. Proton detected local field (PDLF) spectra were recorded as described elsewhere.52, 53 Briefly, a ramped-cross-polarization (ramp-CP) sequence with a contact time of 3 ms was used to record the 1D 13C chemical shift spectra under FLOPSY-8 proton decoupling. 2D PDLF spectra were obtained using 70 t1 increments, a 5 s recycling delay, and a 25 kHz 1H decoupling. The observed dipolar couplings were converted into order parameters using the relation:
where Dobs is the observed dipolar coupling, D0 is the dipolar coupling in the absence of motional averaging (∼21.5 kHz), θ is the angle between the membrane and the magnetic field (90°), and k is a scaling factor dependent on the homonuclear decoupling sequence employed during the t1 period (0.84 for the BLEW-8 sequence). All measurements were performed at 30°C.
Differential Scanning Calorimetry
The peptide stock solution and DiPoPE in choloroform were co-dissolved and the solution was dried under a stream of nitrogen. The peptide/lipid film was then further dried under high vacuum for several hours to remove residual solvent. Buffer (10 mM Tris/HCl, 100 mM NaCl, 1 mM EDTA, 0.002 % w/v NaN3, pH 7.4) was added to each sample to produce a 10 mg/ml lipid solution, which was briefly vortexed and then degassed without freeze thawing. The liquid crystalline (Lα) to inverted hexagonal phase (HII) transition temperature was measured with a CSC 6100 Nano II differential scanning calorimeter (Calorimetry Sciences Corp., Provo, UT). The change in Cp was recorded from 10- 60°C with a heating rate of 1°C/min.
RESULTS
DSC shows that toxic IAPP peptide fragments induce greater negative curvature strain in lipid bilayers than non-toxic rIAPP1-37
The ability of a peptide to induce curvature in a membrane can be measured by shifts in liquid crystalline (Lα) to the inverted hexagonal phase transition (HII) of DiPoPE lipids.54-58 Membranes in the inverted hexagonal phase resemble inverted (with the polar headgroup facing the center) lipid cylinders with a highly curved surface. Peptides that promote negative curvature in the membrane tend to promote this phase and accordingly decrease the phase transition temperature expected for the transition.59
Adding toxic IAPP peptides or peptide fragments to suspensions of DiPoPE reduces the lamellar liquid crystalline (Lα) to inverted hexagonal (HII) phase transition of DiPoPE (see Table 1). This reduction of the Lα to HII transition temperature indicates that these peptides induce negative curvature strain in phospholipid membranes. Human IAPP1-37 stabilized the negative curvature of the HII phase to the greatest extent, while the non-amyloidogenic rIAPP1-19 and hIAPP1-19 fragments did so to a lesser extent. Non-toxic rIAPP1-37 only negligibly stabilized the negative curvature strain of HII phase. The cooperativity of the Lα to HII transition is also greatly reduced when either rIAPP1-19 or hIAPP1-37 is added to the DiPoPE vesicle suspension (see Figure 2).
Table 1. The effect of IAPP peptides and peptide fragments on the Lα to HII transition a temperature of DiPoPE.
The Lα to HII transition temperatures of DiPoPE were determined from the a third DSC thermogram of the sample (for pure DiPoPE, this temperature was determined to be 45.5°C).
| 0.1 mol % peptide | 0.5 mol % peptide | |
|---|---|---|
| DiPoPE containing rIAPP1-37 | 43.6°C | 44.3°C |
| DiPoPE containing rIAPP1-19 | 42.3°C | 38.7°C |
| DiPoPE containing hIAPP1-37 | 39.1°C | ∼40°C |
| DiPoPE containing hIAPP1-19 | 43.3°C | 40.9°C |
Figure 2. Differential scanning calorimetry curves of the liquid crystalline (Lα) to inverted hexagonal (HII) phase transition of DiPOPE multilamellar vesicles containing IAPP.
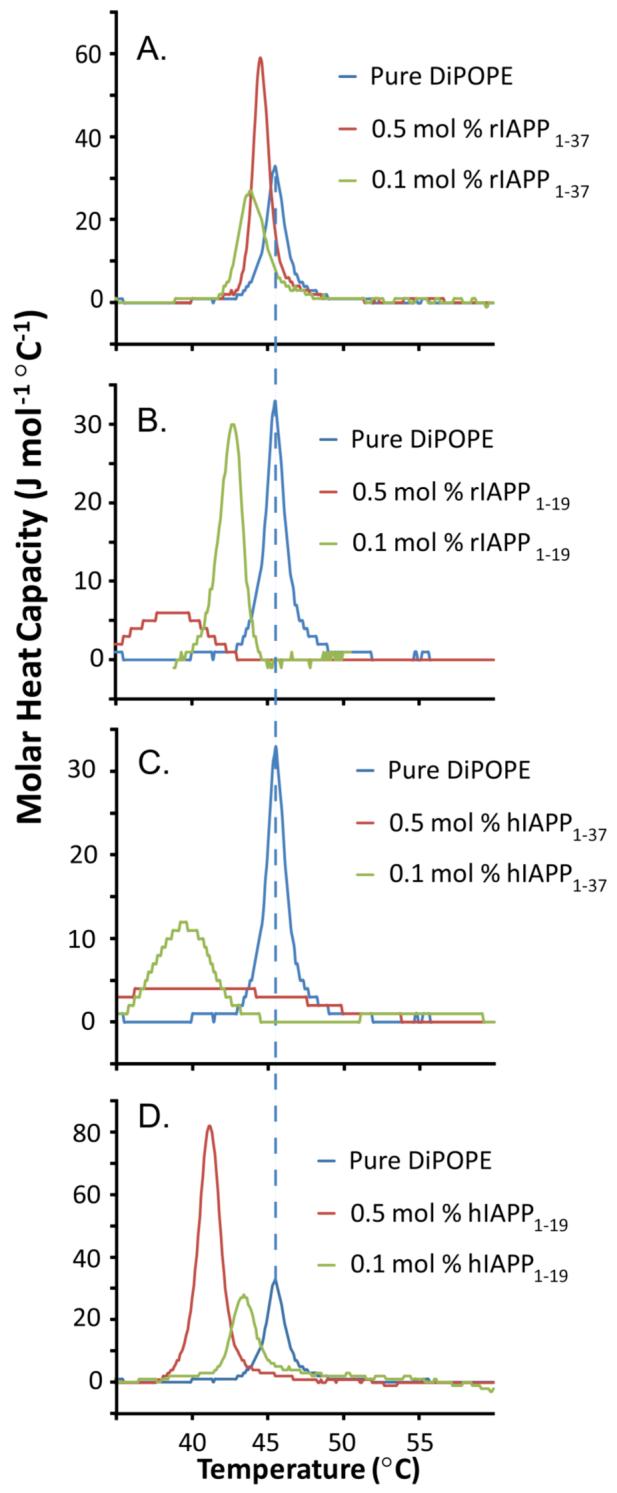
The third DSC heating scan of IAPP peptides and peptide fragments in DiPoPE is shown at the listed molar percentage of peptide. While the Lα to HII transition temperatures were reproducible, the amount of DiPoPE lipid in the DSC spectrometer cells was sensitive to sample preparation and therefore the intensities of the peaks were not completely reproducible. (A) rIAPP1-37 in DiPoPE; (B) rIAPP1-19 in DiPoPE; (C) hIAPP1-37 in DiPOPE; (D) hIAPP1-19 in DiPoPE.
NMR experiments show that IAPP peptide fragments binds to regions of high curvature in bicelles
The DSC experiments indicate toxic forms of IAPP have an affinity for curved membrane surfaces. To create membranes with flat lamellar surfaces and regions of high positive and negative curvature, we used a mixture of short (DHPC) and long (DMPC and DMPG) chain phospholipids in the form of bicelles.60-62 At the ratio of short to long chain lipids used here, bicelles form perforated lamellar bilayers that align spontaneously in the presence of an external magnetic field (see Figure 3).63, 64 The perforations are lined with DHPC molecules and are toroidally shaped with two types of curvature: negative (concave) curvature in the plane of the bilayer and positive (convex) perpendicular to the membrane surface. The sheet-like structure of the bicelles and the anisotropy of the lipids’ magnetic susceptibility tensor causes them to spontaneously align in a magnetic field. The spontaneous alignment results in sharply resolved peaks in the NMR spectrum of bicelles, facilitating high resolution studies by NMR.
Figure 3. Schematic of bicelle structure and 31P chemical shift spectra of DMPC:DMPG:DHPC bicelles containing IAPP.
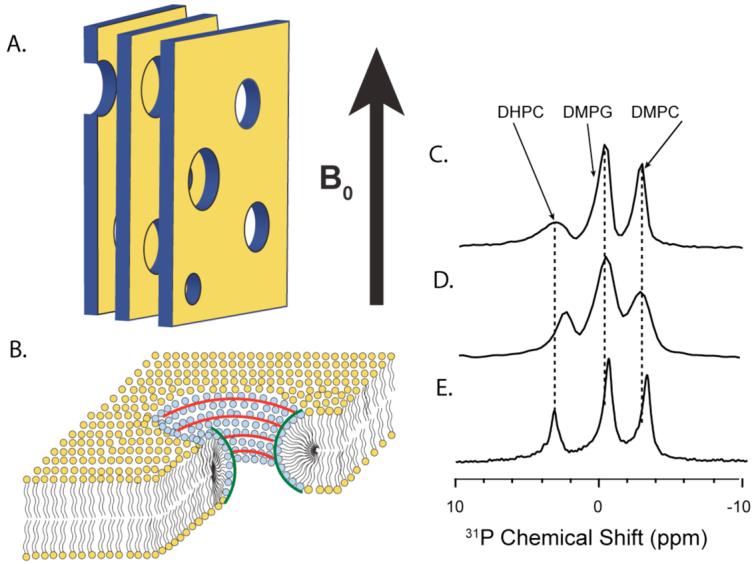
(A) A cartoon depiction of magnetically-aligned bicelles in the lamellar phase showing the parallel bicelle lamellae composed of DMPC and DMPG and the perforations composed of DHPC. The large, static magnetic field of the NMR spectrometer is indicated. (B) Zoomed in cartoon depiction of the bicelles, showing the regions of positive and negative curvature. (C) The 31P NMR spectrum of the pure bicelle sample. (D) The 31P NMR spectrum of the rIAPP1-19 bicelle sample. (E) The 31P NMR spectrum of the rIAPP1-37 bicelle sample.
Bicelles containing hIAPP1-19 formed a relatively opaque phase which did not align in the magnetic field (see Figure S1). Although the lack of alignment in hIAPP1-19 bicelles did not allow us to carry out high resolution solid-state NMR experiments, it did indicate that hIAPP1-19 has a large effect on the phase structure of the bicelle. We were also unable to prepare stable bicelles of hIAPP1-37 since hIAPP1-37 fibrillizes rapidly in a membrane environment on the time-scale of the NMR experiment. Therefore, we used bicelles mixed with rIAPP1-19 or rIAPP1-37 as model systems of toxic and non-toxic IAPP variants, respectively.50
31P NMR of bicelles shows that the rIAPP1-19 peptide fragment has a greater affinity for the highly curved perforations of bicelles, than rIAPP1-37 peptide (see Figure 3). This is evident from the -0.7 ppm shift in the 31P peak of DHPC of bicelles containing rIAPP1-19 relative to the control sample. On the other hand, the DHPC peak is unshifted for bicelles containing rIAPP1-37, indicating rIAPP1-37 has little preference for the curved perforations of the bicelles but a greater affinity than rIAPP1-19 for the long-chain phospholipids in the flat lamellae of the bicelle. The 31P peaks of DMPG and DMPC in the rIAPP1-37 bicelle both show a shift relative to the control sample (-0.5 ppm for DMPC and -0.2 ppm for DMPG), while there is no detectable shift for the corresponding bicelle sample containing rIAPP1-19. Another important observation is the absence of an isotropic peak in the 31P chemical shift spectra of bicelles, indicating that rIAPP1-37 and rIAPP1-19 did not fragment the lipid bilayer.
NMR of full-length rIAPP1-37 shows a strong interaction between the peptide and the surface of the bilayer but not between the peptide and the bilayer hydrophobic core
The 1D 13C chemical shift spectrum was used to monitor the effects of rIAPP1-37 and rIAPP1-19 on the 13C nuclei of the DMPC and DMPG lipids. The 13C nuclei of DHPC are not detectable in these spectra because the mobility of DHPC in bicelle mixtures prevents magnetization transfer from 1H nuclei to 13C nuclei during the cross-polarization period of the pulse sequence. Our previous study has shown that 13C chemical shift peaks of DHPC can be obtained by using an NOE type sensitivity enhancement or a single 90°-pulse direct excitation of 13C.62 DMPG has two glycerol moieties which produce peaks that are indistinguishable in 1D 13C NMR spectra. Changes in the 13C chemical shifts of carbons g3, g2 and β (Fig. 4C) indicate that full-length rIAPP1-37 binds near the glycerol and headgroup regions of the long-chain lipids (DMPG and DMPC). By contrast, the one-dimensional 13C spectrum of rIAPP1-19 does not show a change in the chemical shifts of the glycerol or headgroup peaks originating from long-chain lipids. This can be expected in the context of the other NMR data presented above, which indicated that rIAPP1-19 does not bind bicelles at their planar surfaces (that is the long-chain lipid region), but rather at their highly curved perforations (see Figure 3b).
Figure 4. The 13C chemical shift spectra of the headgroup and glycerol regions of DMPC:DMPG:DHPC bicelles containing IAPP.
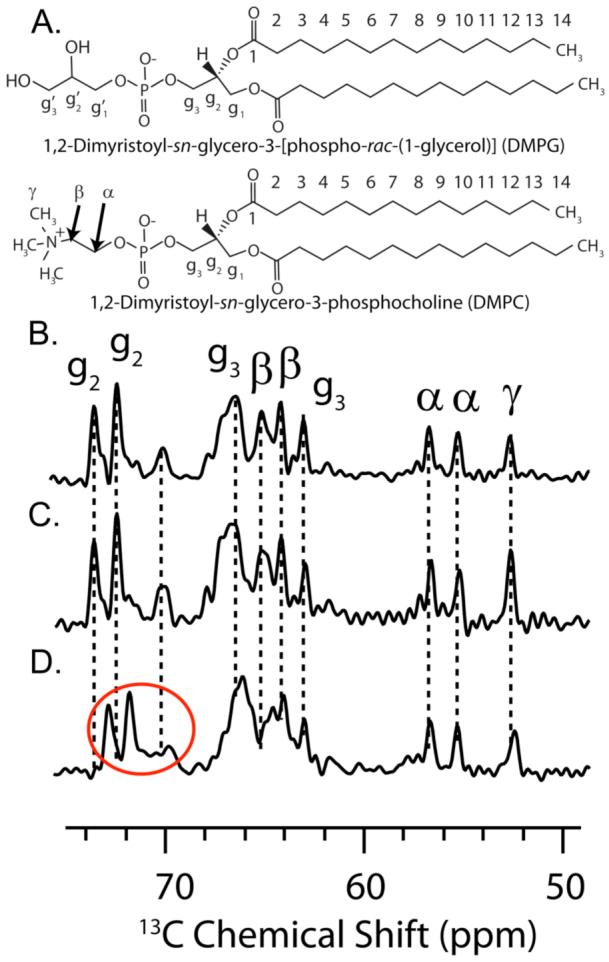
(A) Structures of the long-chain phospholipids (DMPG and DMPC) indicating the labeling convention. (B) 13C spectrum of the headgroup region of the pure bicelle sample, (C) rIAPP1-19 bicelle sample and (D) rIAPP1-37 bicelle sample. The glycerol peaks which are shifted relative to the pure bicelle peaks are circled in red.
The flexibility of lipid acyl chain tails are more drastically affected by rIAPP1-19 than by full-length rIAPP1-37
The flexibility of lipid acyl chain tails are more drastically affected by rIAPP1-19 than by full-length rIAPP1-37: although rIAPP1-19 does not bind directly to the long chains lipids in the flat lamellae, 1H-13C PDLF spectra show that rIAPP1-19 does have an effect on their flexibility. Most of the observed C-H dipolar couplings are similar to the control sample, however the very end of the acyl chain (carbons 13 and 14 of DMPC/DMPG) has two vicinal dipolar couplings associated with each carbon (Figures 5E and 6). The first vicinal dipolar coupling is very close to the dipolar coupling associated with the control sample, while the second vicinal dipolar coupling is much larger indicating a much stronger degree of order due to peptide-lipid interactions.
Figure 5. 2D PDLF spectra of rIAPP and rIAPP1-19 in DMPC:DMPG:DHPC bicelles.
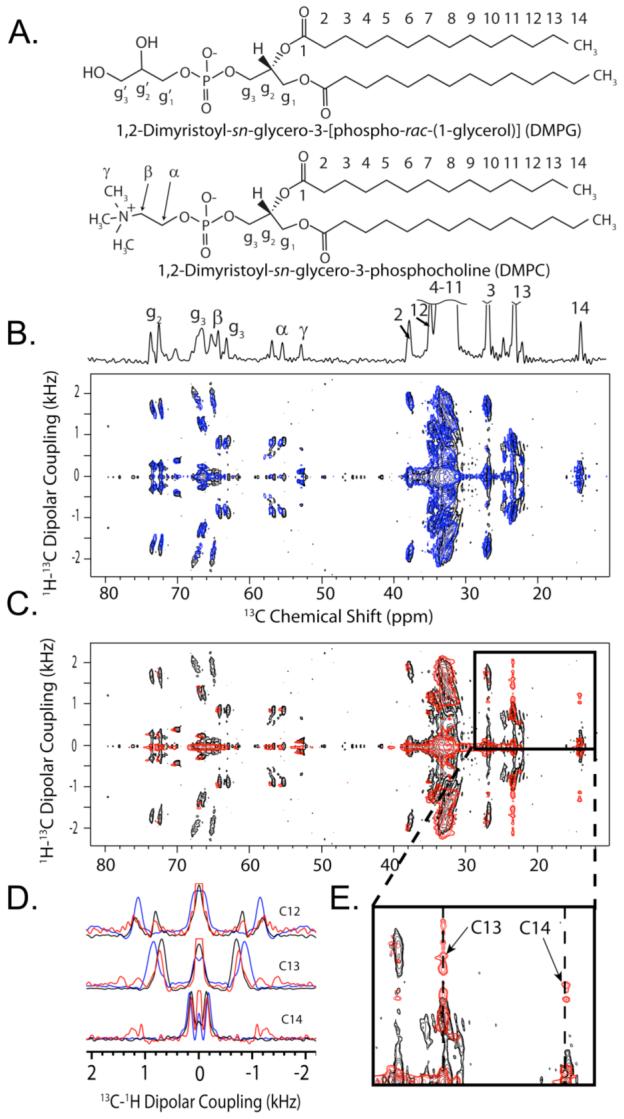
(A) The structures of the long-chain phospholipids of the bicelle samples (DMPG and DMPC) indicating the labeling convention used. Also shown is the 1D 13C chemical shift spectrum, indicating the frequency of 13C peaks in the horizontal dimension of the PDLF spectra. (B) The 2D 1H-13C PDLF spectrum of bicelles containing rIAPP1-37 (blue) superimposed upon the pure bicelle spectrum (black). (C) The 2D 1H-13C PDLF spectrum of bicelles containing rIAPP1-19 (red) superimposed upon the pure bicelle spectrum (black). (D) The 13C-1H dipolar coupling slices corresponding to carbons 12, 13, and 14 of the aliphatic fatty acid chains of DMPG and DMPC. Slices corresponding to the pure bicelle sample are shown in black; bicelles containing rIAPP1-37, blue; and bicelles containing rIAPP1-19, red. (E) A zoomed in area of the PDLF spectrum showing the significant change in the dipolar couplings associated with carbons 13 and 14 of DMPG and DMPC induced by rIAPP1-19.
Figure 6. The order parameter plots associated with DMPC:DMPG:DHPC bicelles containing (A) rIAPP1-37 and (B) rIAPP1-19.
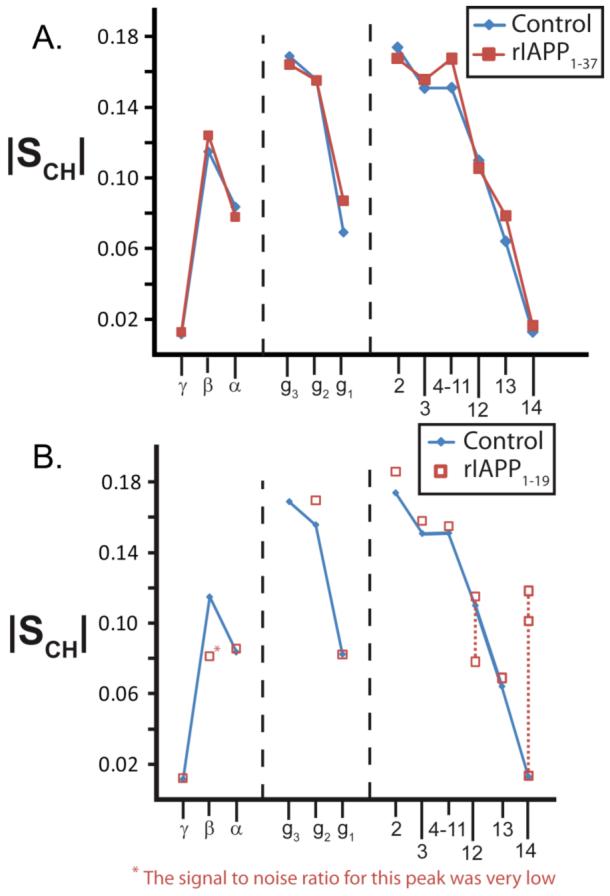
The order parameter plots are derived from their associated PDLF spectra as described in the methods section. The order parameter plot of pure bicelles is given in blue. Note that one |SCH| value for carbons 12, 13, and 14 of bicelles containing rIAPP1-19 is nearly identical to the corresponding value for the pure bicelle sample, indicating some lipids in the sample are unaffected by rIAPP1-19.
Rat IAPP1-37 has only a small effect on the flexibility of the acyl chains (Figures 5 and 6). For most of the 1H-13C bonds investigated, the effect of rIAPP1-37 on |SCH|, a measure of the rigidity of 1H-13C bonds, is not significant. Nevertheless, rIAPP1-37 does exert an effect on the flexibility of the long-chain lipid glycerol and headgroup 1H-13C bonds, as would be expected from the change in 13C chemical shifts observed in this region. Overall, the effect observed is a slight decrease in |SCH| for the 1H-13C bonds of the lipid headgroup, except for those 1H-13C bonds directly associated with the phosphate or trimethyl ammonium moieties. For this sample, the decreased flexibility in this region may be interpreted as the disruption of the inter-lipid charge-charge associations between the negatively charged phosphate group and the positively charged choline group due to interaction of rIAPP1-37 with the lipids. 14N NMR confirms this hypothesis by showing a reduced quadrupole splitting, νQ, suggesting a change in the orientation of the DMPC headgroup as a result of full-length rIAPP1-37 changing the electrostatic environment surrounding PC lipid headgroups (see Figure 7). 65-69
Figure 7. 14N NMR spectra of DMPC:DMPG:DHPC bicelles containing IAPP.
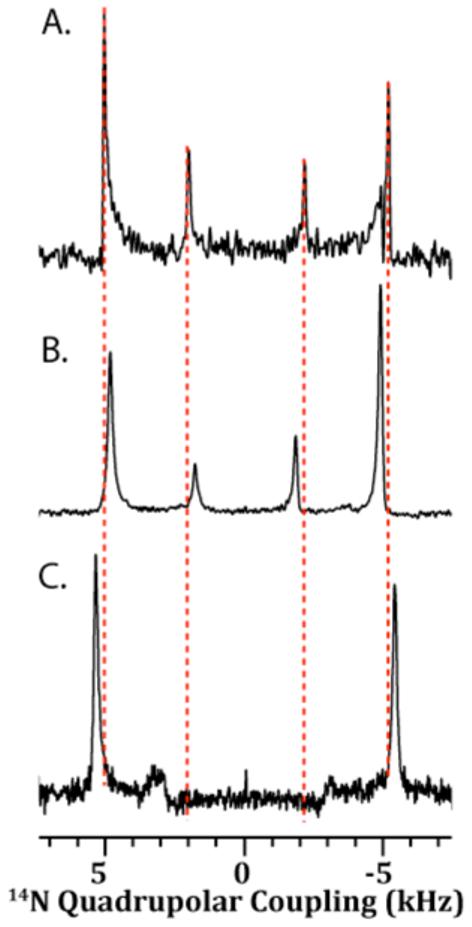
(A) The 14N quadrupolar NMR spectrum of the pure bicelle sample. The quadrupolar coupling constant (νQ) associated with DMPC is 10.2 kHz, and the νQ associated with DHPC is 4.2 kHz. DMPG does not have a 14N nucleus, and therefore there are peaks corresponding to DMPG are absent in the 14N spectrum. (B) The 14N spectrum of the rIAPP1-37 bicelle sample. The νQ associated with DMPC is 9.8 kHz and νQ associated with DHPC is 3.6 kHz. (C) The 14N NMR spectrum of the rIAPP1-19 bicelle sample. The νQ associated with DMPC is 10.8 kHz and νQ associated with DHPC is 6.4 kHz.
DISCUSSION
Misfolding of amyloid-fibril forming peptides is implicated in devastating aging-related diseases. Recent studies have shown that the ability of an amyloid peptide to bind to the cell membrane and induce disruption of lipid bilayer structure is correlated with the cytotoxicity of amyloid peptides.12, 70 Studies on model membranes have shown that the toxicity of amyloid peptides is highly dependent on the lipid composition of the membrane.14, 15, 71-73 Therefore, an understanding of amyloid peptide -lipid interactions can lead to a greater understanding of the mechanism of amyloid cytotoxicity. In this study, we have investigated the mechanism by which amylin peptides disrupt lipid bilayers using solid-state NMR and DSC experiments. We believe that the results reported in this study can be used to develop compounds to suppress the cell-toxicity of amylin, and also could be extended towards the understanding of the role of lipids on the misfolding pathway of amyloid peptides in general.
We have made the following key observations regarding the interaction of IAPP peptides with lipid membranes: (i) Both amyloidogenic (hIAPP1-37) and largely non-amyloidogenic (hIAPP1-19 and rIAPP1-19) toxic versions of the peptide strongly favor the formation of negative curvature strain on lipid bilayers. The non-toxic and non-amyloidogenic rIAPP1-37 induced only a very small amount of negative curvature. (ii) In a bicelle environment which has regions of short chain lipids with high curvature and long chain lipids with low curvature, toxic rIAPP1-19 binds to the short-chain lipids present in regions of high curvature while non-toxic rIAPP1-37 binds to the long-chain lipids present in regions of low curvature. (iii) Rat IAPP1-37 affects the chemical environment of the glycerol and phosphorus regions in the headgroup region of long-chain lipids, but does not detectably affect the chemical environment of the aliphatic carbons. (iv) In addition to binding to short chain lipids, rIAPP1-19 also significantly perturbs the motions of the aliphatic carbons of long-chain lipid’s fatty acid tails.
All of the IAPP peptides and peptide fragments studied lowered the transition temperature for the formation of the inverted hexagonal (HII) phase of DiPoPE. The inverted hexagonal phase has a high negative curvature; therefore peptides that induce negative curvature strain stabilize the HII phase and lower the transition temperature for its formation. While all the IAPP variants favored the formation of the highly curved HII phase, the negative curvature induced by non-toxic rIAPP1-37 was small in comparison to the other toxic IAPP peptides and decreased at higher concentrations. The significantly higher degree of curvature induced by toxic variants of IAPP compared to the non-toxic rIAPP1-37 suggests a correlation between the peptide’s ability to induce curvature strain and its toxicity. The ability to induce negative curvature strain is apparently not related to the presence of amyloid fibers, the fiber formation process, or a propensity for a β-sheet conformation, as the 1-19 fragments of IAPP remains stable in α-helical conformation when bound to lipid bilayers.48
The induction of negative curvature strain by toxic versions of IAPP may explain the fragmention of bilayers by hIAPP. 30-33 When hIAPP is added to planar bilayers, lipids are initially extruded out of the plain of the bilayer to form long tubular structures lined by closed bilayers with an aqueous core.30 The lipid tubules disappeared from the surface of the membrane over time as they were either sheared off the surface by mechanical stress or incorporated into amyloid fibers on the membrane surface causing severe disruption of the integrity of the membrane. The tendency of the peptide to cause curvature in the membrane may be linked to this process. The formation of lipid tubules can occur by a heterogeneous distribution of peptide with high concentrations of peptide at the tubule base, where negative curvature is highest, and low concentrations of peptide at the tubule tip.74 DSC experiments confirm that rIAPP1-19 is inhomogenously distributed throughout the membrane.50 Similar inhomogenous distributions have also been seen for hIAPP1-37, but not rIAPP1-37, in the presence of calcium.75 Notably, this process does not depend on the attachment of the membrane to a rigid amyloid fiber.
The degree and type of membrane curvature induced by a peptide is dependent on how it affects the geometry of the lipid-peptide complex, in particular the relative cross-sectional area of the lipid headgroup region near the membrane-water interface and the acyl chain region in the interior of the bilayer. In order for the membrane to remain in a flat lamellar phase, the overall shape of the peptide-lipid complex must be cylindrical with the size of the two regions matching. A cone-shaped peptide-lipid complex with a mismatch between the sizes of these two regions will induce curvature strain in the bilayer, positive curvature strain if the effective size of the head group is larger than the acyl-chain region and negative curvature strain if it is smaller. It can be seen from these considerations that binding of an amphipathic helix like IAPP at the interface between the hydrophilic headgoup region and the hydrophobic acyl chain region will induce positive curvature strain. Conversely, inserting the peptide deeply into the hydrophobic core will induce negative curvature strain.
The degree of insertion of IAPP into the membrane can be inferred by the relative perturbation of the NMR signal at each site. Rat IAPP1-37 has little effect on the order of the acyl chains and the lipid headgroup, as determined by the site-specific order parameter plot generated by the 2D PDLF experiment. This is a strong indication that the peptide binds superficially to the very top of the lipid bilayer by an electrostatic interaction with the lipid headgroup. Peptides that bind atmospherically to the membrane in this manner without penetration into the bilayer typically do not induce either positive or negative curvature strain, as demonstrated by mutations of the ENTH domain of epsin.76, 77 It could also be the case that the majority of the peptide is not bound to the membrane, however this possibility is contradicted by the 14N and 31P spectra. The 14N spectra shows a small but detectable decrease in the 14N quadrupolar splitting; which is indicative of an electrostatic interaction of rIAPP1-37 with the phosphate groups of the bilayer.
Unlike rIAPP1-37, rIAPP1-19 has a strong preference for the DHPC molecules lining the perforations of the bicelle lamellae rather than their planar regions, as shown by the strong perturbation of the 31P peak corresponding to DHPC (Figure 3D) and the negligible perturbations of the chemical shifts in the DMPC/DMPG headgroup region (Figure 4C). This preference for the perforations of the bilayer is similarly reflected in the 14N spectra which show rIAPP1-19 induces only a slight increase in the quadrupolar coupling of the long chain lipids but a relatively large increase in the quadrupolar coupling of DHPC (Figure 7B).
Rat IAPP1-19 has an unusual and significant effect on the aliphatic carbons of both DMPC and DMPG’s acyl chains in the NMR experiments. At the very end of the acyl chain (carbons 13 and 14) two vicinal dipolar couplings can be detected for each carbon. The first vicinal dipolar coupling has an order very similar to the control bicelle without peptide. The second vicinal dipolar coupling is significantly larger, indicating a region of the membrane where the terminus of the acyl chain is substantially more ordered in the presence of rIAPP1-19. This set of dipolar couplings most likely corresponds to the annulus of DMPC/DMPG lipids surrounding the bicelle perforation. An increase in order may be due to either bulky residues interfering sterically with the lipid tails of nearby long-chain lipids or interdigitation of the acyl chain termini into the adjoining leaflet due to the compression of the bilayer around the region of the perforation.78
The perforations in the bicelle possess regions of both positive curvature (parallel to the membrane normal) and negative curvature (perpendicular to the membrane normal). As such, they serve as models for the toroidal pores produced by certain antimicrobial peptides,39, 54, 55 which recent studies have noted distinct similarities to amyloidogenic proteins.30, 79-84 The toroidal pore is formed by a transient high local concentration of peptide that induces enough curvature in the membrane to form a metastable pore lined by the headgroups of phospholipids, rather than by protein as in most pore models. Although most antimicrobial peptides form toroidal pores by stabilizing positive curvature, some antimicrobials form toroidal pores by stabilizing negative curvature instead.85-87 The creation of toroidal pores as a result of the induction of negative curvature may be a novel mechanism for amyloid-induced peptide disruption. Toroidal pores have not been directly detected for IAPP, but annular structures consistent with the expected size of a toroidal pore have been observed.88, 89
Although the results presented here are for IAPP peptides, the induction of negative curvature strain may be a general mechanism of membrane disruption by amyloidogenic peptides. Solid-state NMR has shown a preferential interaction with the headgroups of PE for Aβ29-42 in POPC/POPE membranes, even at a low (10%) percentage of POPE.90 PE lipids have a high propensity for negative spontaneous curvature due to the small size of the headgroup relative to the hydrophobic core. The preferential binding of Aβ29-42 to PE lipids may reflect the similar preference of IAPP to bind to regions of high curvature shown in this study. Furthermore, Aβ1-40 has been shown to dehydrate membranes, which reduces the effective headgroup size and will likely lead to negative curvature.91 Alpha-synuclein, another neurotoxic peptide implicated in Parkinson’s disease, also has a strong preference for PE lipids over the identically charged PC lipids, suggesting lipid intrinsic curvature is an important determinant of lipid binding specificity for amyloidogenic proteins.11, 92 The preference of amyloidogenic peptides for lipids with a high negative curvature is expected to enhance membrane fusion, as membrane fusion proceeds through a high energy intermediate similar in structure to the inverted hexagonal phase.59, 93 Indeed, the amyloidogenic peptides α-synuclein, Aβ, and some prion fragments have been shown to be fusogenic.90, 94-99
CONCLUSIONS
We have reported the effects of IAPP peptides and peptide fragments on highly curved and lamellar anionic lipid systems and found that IAPP peptide fragments induce a high degree of negative curvature strain in lipid bilayers. It is therefore likely that one of the mechanisms by which IAPP peptides permeabilize lipid bilayers involves either the formation of toroidal pores or non-specific membrane disruption due to excessive negative curvature strain. The induction of a similar degree of negative curvature by 1-19 fragments of IAPP, which do not form amyloid fibers when embedded in the membrane, indicates amyloid formation is not essential for this process. Importantly, we found that non-toxic rIAPP1-37 does not induce a great degree of negative curvature strain, suggesting that toxicity may be linked to the induction of negative curvature. Since membrane curvature can be modified by ligand binding, we believe that results from this study will be useful to design compounds that can be used to prevent the interaction of IAPP with the cell membrane and therefore the cell-toxicity of IAPP. This study demonstrates that bicelles are useful for studying the affinity of membrane ligands for curved regions of the cell membrane and for investigating the role of toroidal pores in the function of membrane disruptive peptides like antimicrobial peptides, toxin peptides, and fusion peptides.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the research funds from National Institutes of Health (DK078885 to A.R.) and Michigan Diabetes Research Training Center at the University of Michigan. We acknowledge Dr. Lindsey Gottler for help with the DSC experiments. We also thank Dr. Jiadi Xu, Kazutoshi Yamamoto, and Kevin Hartman for helpful discussion.
REFERENCES
- 1.Haataja L, Gurlo T, Huang CJ, Butler PC. Endocr. Rev. 2008;29:302–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferreira ST, Vieira MNN, De Felice FG. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 3.Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Nat. Struct. Mol. Biol. 2007;14:1157–1164. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]
- 4.Chimon S, Ishii Y. J. Am. Chem. Soc. 2005;127:13472–13473. doi: 10.1021/ja054039l. [DOI] [PubMed] [Google Scholar]
- 5.Uversky VN. Curr. Prot. Pept. Sci. 2008;9:507–540. doi: 10.2174/138920308785915218. [DOI] [PubMed] [Google Scholar]
- 6.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Temussi PA, Masino L, Pastore A. EMBO J. 2003;22:355–361. doi: 10.1093/emboj/cdg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caughey B, Baron GS. Nature. 2006;443:803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 9.Khemtemourian L, Killian JA, Hoppener JW, Engel MF. Exp. Diab. Res. 2008;2008:421287. doi: 10.1155/2008/421287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayasinghe SA, Langen R. Biochim. Biophys. Acta. 2007;1768:2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 11.Jo EJ, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. J. Biol. Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 12.Beyer K. Cell Biochem. Biophys. 2007;47:285–299. doi: 10.1007/s12013-007-0014-9. [DOI] [PubMed] [Google Scholar]
- 13.Knight JD, Hebda JA, Miranker AD. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 14.Knight JD, Miranker AD. J. Mol. Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 15.Jayasinghe SA, Langen R. Biochemistry. 2005;44:12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 16.Apostolidou M, Jayasinghe SA, Langen R. J. Biol. Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson WS, Jonas A, Clayton DF, George JM. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 18.Accili D. Drug Dev. Res. 2008;69:111–115. [Google Scholar]
- 19.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 20.Kahn SE. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 21.Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR. Diabetic Med. 1998;15:290–296. doi: 10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 22.Luca S, Yau WM, Leapman R, Tycko R. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 24.Mirzabekov TA, Lin MC, Kagan BL. J. Biol. Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 25.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. J. Biol. Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 26.Mattson MP, Goodman Y. Brain Res. 1995;676:219–224. doi: 10.1016/0006-8993(95)00148-j. [DOI] [PubMed] [Google Scholar]
- 27.Mattson MP, Chan SL. Nat. Cell Biol. 2003;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- 28.Lastra G, Manrique C. Antioxid. Redox Sig. 2007;9:943–954. doi: 10.1089/ars.2007.1615. [DOI] [PubMed] [Google Scholar]
- 29.Ritzel RA, Meier JJ, Lin CY, Veldhuis JD, Butler PC. Diabetes. 2007;56:65–71. doi: 10.2337/db06-0734. [DOI] [PubMed] [Google Scholar]
- 30.Domanov YA, Kinnunen PKJ. J. Mol. Biol. 2008;376:42–54. doi: 10.1016/j.jmb.2007.11.077. [DOI] [PubMed] [Google Scholar]
- 31.Sparr E, Engel MFM, Sakharov DV, Sprong M, Jacobs J, de Kruijff B, Hoppener JWM, Killian JA. FEBS Lett. 2004;577:117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 32.Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JW. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6033–8. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brender JR, Durr UHN, Heyl D, Budarapu MB, Ramamoorthy A. Biochim. Biophys. Acta. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; Porcelli MA, Ilangovan U, Ramamoorthy A, Veglia G. Biopolymers. 2003;69:29–41. doi: 10.1002/bip.10305. [DOI] [PubMed] [Google Scholar]; Ilangovan U, Ramamoorthy A. Biopolymers. 1998;45:9–20. doi: 10.1002/(SICI)1097-0282(199801)45:1<9::AID-BIP2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 34.Green JD, Kreplak L, Goldsbury C, Blatter XL, Stolz M, Cooper GS, Seelig A, Kist-Ler J, Aebi U. J. Mol. Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 35.Casas S, Novials A, Reimann F, Gomis R, Gribble FM. Diabetologia. 2008;51:2252–2262. doi: 10.1007/s00125-008-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Epand RM, Shai YC, Segrest JP, Anantharamaiah GM. Biopolymers. 1995;37:319–338. doi: 10.1002/bip.360370504. [DOI] [PubMed] [Google Scholar]
- 37.Botelho AV, Huber T, Sakmar TP, Brown MF. Biophys. J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Domanov YA, Kinnunen PKJ. Biophys. J. 2006;91:4427–4439. doi: 10.1529/biophysj.106.091702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramamoorthy A, Thennarasu S, Lee DK, Tan AM, Maloy L. Biophys. J. 2006;91:206–216. doi: 10.1529/biophysj.105.073890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hallock KJ, Lee DK, Omnaas J, Mosberg HI, Ramamoorthy A. Biophys. J. 2002;83:1004–1013. doi: 10.1016/S0006-3495(02)75226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henzler-Wildman KA, Martinez GV, Brown MF, Ramamoorthy A. Biochemistry. 2004;43:8459–8469. doi: 10.1021/bi036284s. [DOI] [PubMed] [Google Scholar]
- 42.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 43.Konarkowska B, Aitken JF, Kistler J, Zhang SP, Cooper GJS. FEBS Journal. 2006;273:3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 44.Meier JJ, Kayed R, Lin CY, Gurlo T, Haataja L, Jayasinghe S, Langen R, Glabe CG, Butler PC. Am. J. Physiol. 2006;291:E1317–E1324. doi: 10.1152/ajpendo.00082.2006. [DOI] [PubMed] [Google Scholar]
- 45.Anguiano M, Nowak RJ, Lansbury PT. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 46.Lin CY, Gurlo T, Kayed R, Butler AE, Haataja L, Glabe CG, Butler PC. Diabetes. 2007;56:1324–1332. doi: 10.2337/db06-1579. [DOI] [PubMed] [Google Scholar]
- 47.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. J. Am. Chem. Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radovan D, Smirnovas V, Winter R. Biochemistry. 2008;47:6352–63560. doi: 10.1021/bi800503j. [DOI] [PubMed] [Google Scholar]
- 50.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. Biochemistry. 2008;47:12680–12689. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]; Nanga RPR, Brender JR, Xu JD, Veglia G, Ramamoorthy A. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Capone R, Quiroz FG, Prangkio P, Saluja I, Sauer AM, Bautista MR, Turner RS, Mayer M. Neurotoxicity Res. 2008;15:608–650. doi: 10.1007/s12640-009-9033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dvinskikh SV, Durr UHN, Yamamoto K, Ramamoorthy A. J. Am. Chem. Soc. 2007;129:794–802. doi: 10.1021/ja065536k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dvinskikh S, Durr U, Yamamoto K, Ramamoorthy A. J. Am. Chem. Soc. 2006;128:6326–6327. doi: 10.1021/ja061153a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hallock KJ, Lee DK, Ramamoorthy A. Biophys. J. 2003;84:3052–3060. doi: 10.1016/S0006-3495(03)70031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wildman KAH, Lee DK, Ramamoorthy A. Biochemistry. 2003;42:6545–6558. doi: 10.1021/bi0273563. [DOI] [PubMed] [Google Scholar]
- 56.Epand RF, Martin I, Ruysschaert JM, Epand RM. Biochem. Biophys. Res. Commun. 1994;205:1938–1943. doi: 10.1006/bbrc.1994.2897. [DOI] [PubMed] [Google Scholar]
- 57.Siegel DP, Epand RM. Biochim. Biophys. Acta. 2000;1468:87–98. doi: 10.1016/s0005-2736(00)00246-7. [DOI] [PubMed] [Google Scholar]
- 58.Epand RM, Epand RF. Biochem. Biophys. Res. Commun. 1994;202:1420–1425. doi: 10.1006/bbrc.1994.2089. [DOI] [PubMed] [Google Scholar]
- 59.Epand RM. Biochim. Biophys. Acta. 1998;1376:353–368. doi: 10.1016/s0304-4157(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 60.Sanders CR, Hare BJ, Howard KP, Prestegard JH. Prog. Nucl. Magn. Reson. Spectrosc. 1994;26:421–444. [Google Scholar]
- 61.Prosser RS, Evanics F, Kitevski JL, Al-Abdul-Wahid MS. Biochemistry. 2006;45:8453–8465. doi: 10.1021/bi060615u. [DOI] [PubMed] [Google Scholar]
- 62.Dvinskikh SV, Yamamoto K, Dürr UHN, Ramamoorthy A. J. Magn. Reson. 2008;184:228–235. doi: 10.1016/j.jmr.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gaemers S, Bax A. J. Am. Chem. Soc. 2001;123:12343–12352. doi: 10.1021/ja011967l. [DOI] [PubMed] [Google Scholar]
- 64.Harroun TA, Koslowsky M, Nieh MP, de Lannoy CF, Raghunathan VA, Katsaras J. Langmuir. 2005;21:5356–5361. doi: 10.1021/la050018t. [DOI] [PubMed] [Google Scholar]
- 65.Ramamoorthy A, Lee DK, Santos JS, Henzler-Wildman KA. J. Am. Chem. Soc. 2008;130:11023–11029. doi: 10.1021/ja802210u. [DOI] [PubMed] [Google Scholar]
- 66.Semchyschyn DJ, Macdonald PM. Magn. Reson. Chem. 2004;42:89–104. doi: 10.1002/mrc.1333. [DOI] [PubMed] [Google Scholar]
- 67.Santos JS, Lee DK, Ramamoorthy A. Magn. Reson. Chem. 2004;42:105–114. doi: 10.1002/mrc.1327. [DOI] [PubMed] [Google Scholar]
- 68.Scherer PG, Seelig J. Biochemistry. 1989;28:7720–7728. doi: 10.1021/bi00445a030. [DOI] [PubMed] [Google Scholar]
- 69.Lindstrom F, Bokvist M, Sparrman T, Grobner G. Phys. Chem. Chem. Phys. 2002;4:5524–5530. [Google Scholar]
- 70.Ulmer TS, Bax A, Cole NB, Nussbaum RL. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 71.Lau TL, Gehman JD, Wade JD, Masters CL, Barnham KJ, Separovic F. Biochim. Biophys. Acta. 2007;1768:3135–3144. doi: 10.1016/j.bbamem.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 72.Grudzielanek S, Smirnovas V, Winter R. Chem. Phys. Lipids. 2007;149:28–39. doi: 10.1016/j.chemphyslip.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 73.Wong PT, Schauerte JA, Wisser KC, Ding H, Lee EL, Steel DG, Gafni A. J. Mol. Biol. 2009;386:81–96. doi: 10.1016/j.jmb.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 74.Campelo F, McMahon HT, Kozlov MM. Biophys. J. 2008;95:2325–2339. doi: 10.1529/biophysj.108.133173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sciacca MFM, Pappalardo M, Milardi D, Grasso DM, La Rosa C. Arch. Biochem. Biophys. 2008;477:291–298. doi: 10.1016/j.abb.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 76.Ford MGJ, Mills IG, Peter BJ, Vallis Y, Praefcke GJK, Evans PR, McMahon HT. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 77.Farsad K, De Camilli P. Curr. Opin. Cell Biol. 2003;15:372–381. doi: 10.1016/s0955-0674(03)00073-5. [DOI] [PubMed] [Google Scholar]
- 78.Pabst G, Grage SL, Danner-Pongratz S, Jing W, Ulrich AS, Watts A, Lohner K, Hickel A. Biophys. J. 2008;95:5779–5788. doi: 10.1529/biophysj.108.141630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sood R, Domanov Y, Kinnunen PKJ. J. Fluoresc. 2007;17:223–234. doi: 10.1007/s10895-007-0161-9. [DOI] [PubMed] [Google Scholar]
- 80.Domanov YA, Kinnunen PKJ. Biophys. J. 2006;91:4427–4439. doi: 10.1529/biophysj.106.091702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhao H, Sood R, Jutila A, Bose S, Fimland G, Nissen-Meyer J, Kinnunen PKJ. Biochim. Biophys. Acta. 2006;1758:1461–1474. doi: 10.1016/j.bbamem.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 82.Sood R, Domanov Y, Pietiainen M, Kontinen VP, Kinnunen PKJ. Biochim. Biophys. Acta. 2008;1778:983–996. doi: 10.1016/j.bbamem.2007.11.016. [DOI] [PubMed] [Google Scholar]
- 83.Jang H, Ma B, Lal R, Nussinov R. Biophys. J. 2008;95:4631–42. doi: 10.1529/biophysj.108.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jang H, Zheng J, Lal R, Nussinov R. Trends Biochem. Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 85.Epand RF, Raguse TL, Gellman SH, Epand RM. Biochemistry. 2004;43:9527–9535. doi: 10.1021/bi049414l. [DOI] [PubMed] [Google Scholar]
- 86.Imura Y, Nishida M, Ogawa Y, Takakura Y, Matsuzaki K. Biochim. Biophys. Acta. 2007;1768:1160–1169. doi: 10.1016/j.bbamem.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 87.Timothy DA, Waring AJ, Hong M. Biochim. Biophys. Acta. 2006;1758:1285–1291. doi: 10.1016/j.bbamem.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 88.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Proc. Natl. Acad. Sci. U. S. A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vaiana SM, Ghirlando R, Yau WM, Eaton WA, Hofrichter J. Biophys. J. 2008;94:L45–L47. doi: 10.1529/biophysj.107.125146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ravault S, Soubias O, Saurel O, Thomas A, Brasseur R, Milon A. Protein Sci. 2005;14:1181–1189. doi: 10.1110/ps.041291405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuzaki K, Horikiri C. Biochemistry. 1999;38:4137–4142. doi: 10.1021/bi982345o. [DOI] [PubMed] [Google Scholar]
- 92.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Biophys. J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Epand RM. Biochim. Biophys. Acta. 2003;1614:116–121. doi: 10.1016/s0005-2736(03)00169-x. [DOI] [PubMed] [Google Scholar]
- 94.Dante S, Hauss T, Brandt A, Dencher NA. J. Mol. Biol. 2008;376:393–404. doi: 10.1016/j.jmb.2007.11.076. [DOI] [PubMed] [Google Scholar]
- 95.Dupiereux I, Zorzi W, Lins L, Brasseur R, Colson P, Heinen E, Elmoualij B. Biochem. Biophys. Res. Commun. 2005;331:894–901. doi: 10.1016/j.bbrc.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 96.Mingeot-Leclercq MP, Lins L, Bensliman M, Van Bambeke F, Van der Smissen P, Peuvot J, Schanck A, Brasseur R. Chem. Phys. Lipids. 2002;120:57–74. doi: 10.1016/s0009-3084(02)00108-1. [DOI] [PubMed] [Google Scholar]
- 97.Pillot T, Drouet B, Pincon-Raymond RL, Vandekerckhove J, Rosseneu T, Chambaz J. J. Neurochem. 2000;75:2298–2308. doi: 10.1046/j.1471-4159.2000.0752298.x. [DOI] [PubMed] [Google Scholar]
- 98.Pillot T, Drouet B, Queille S, Labeur C, Vandekerckhove J, Rosseneu M, Pincon-Raymond M, Chambaz J. J. Neurochem. 1999;73:1626–1634. doi: 10.1046/j.1471-4159.1999.0731626.x. [DOI] [PubMed] [Google Scholar]
- 99.Pillot T, Goethals M, Vanloo B, Talussot C, Brasseur R, Vandekerckhove J, Rosseneu M, Lins L. J. Biol. Chem. 1996;271:28757–28765. doi: 10.1074/jbc.271.46.28757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.