

Abstract
Engagement of peptide-MHC (pMHC) by the T-cell receptor induces a conformational change in CD3ε that exposes a proline-rich sequence (PRS) and recruits the cytoskeletal adaptor Nck. This event, which precedes phosphorylation of the CD3ε ITAM, has been implicated in synapse formation and T-cell function. However, there is compelling evidence that responsiveness to TCR ligation is CD3ε PRS-independent. Here we show that the CD3ε PRS is necessary for pMHC-induced phosphorylation of CD3ε and for recruitment of PKCθ to the immune synapse in differentiated CD8+ T-lymphocytes. However, whereas these two events are dispensable for functional T-cell responsiveness to high-avidity ligands, they are required for responsiveness to low-avidity ones. Thus, in at least certain T-cell clonotypes, the CD3ε PRS amplifies weak TCR signals by promoting synapse formation and CD3ε phosphorylation.
Keywords: T cells, T Cell Receptors, Signal Transduction, Antigen Presentation, Cell Activation, Transgenic/Knockout Mice
INTRODUCTION
The poor shape complementarity of the T-cell receptor (TCR)–peptide–major histocompatibility complex (pMHC) interface allows TCRs to recognize different peptide antigens in the context of a single MHC molecule. Peptide-MHC ligands induce T-cell responses that range from partial or full activation (agonists) to antagonism (antagonists) (1, 2). The stimulatory potency of peptides for a single TCR is related to a number of factors, including their affinity for the MHC restricting element, the affinity of the corresponding pMHC complexes for the cognate TCR and the dissociation rates of the interactions.
Engagement of a TCR by a full agonist pMHC ligand induces the oligomerization of TCRs and associated co-receptors into supramolecular clusters by a serial triggering mechanism (3-5). It has been shown that the number of pMHC complexes in the clusters correlate with the half-life of the TCR-pMHC interaction, such that low concentrations of agonists and low affinity ligands are inefficient at inducing cluster formation and stable signaling (4). The efficiency of TCR signaling in the clusters also depends on the ability of pMHC to ligate the TCR long enough to trigger the recruitment and activation of Lck, CD3ζ, CD3ε and ZAP-70 (6-8). This, in turn, leads to recruitment and phosphorylation of LAT and SLP-76 which couple the TCR-CD3 complex to activation of PLCγ1, Ca2+ flux, and NFAT, and recruit the adaptors Nck and ADAP, which are involved in immune synapse formation (9-11). T-cell stimulation with partial pMHC agonists results in the activation of only subsets of these different downstream signaling events (1, 12).
Recently, it has been shown that TCR ligation is accompanied by a rapid conformational change in CD3ε that exposes a Nck-binding, proline-rich sequence (PRS) (13, 14). Nck links signaling molecules with the cytoskeleton in many cell types (15, 16). In T-cells, it links the TCR–CD3 complex with molecules responsible for the cytoskeletal rearrangements underlying TCR aggregation and cluster formation at the synapse (13, 17-19). On the basis of these observations, it has been suggested that recruitment of Nck to CD3ε regulates the earliest stages of TCR signaling and contributes to synapse formation (13). Although compelling, this hypothesis has been challenged by two key observations: recruitment of Nck to CD3ε is dispensable for T-cell responsiveness to strong stimuli (20); and Nck can be recruited to the TCR via SLP-76 or LAT, independently of the CD3ε conformational change (21). Furthermore, although exposure of the CD3ε PRS precedes phosphorylation of the CD3ε ITAM, there is no evidence indicating that the latter is a consequence of the former. Accordingly, the role of the CD3ε PRS in early TCR signaling, T-cell development and function remains a contentious issue.
The work reported here was initiated to investigate the mechanisms underlying the differential responsiveness of naïve CD8+ T-cells and their antigen-differentiated progeny to a type of weak pMHC agonists (which we have previously referred to as ‘secondary’, to distinguish them from conventional –or strong/primary– agonists) that cannot induce functional responses from naïve T-cells, but exhibit strong agonistic activity on differentiated T-cells (22). We find that, in at least certain clonotypes, the CD3ε PRS plays a key role in pMHC-induced phosphorylation of the CD3ε ITAM and in recruitment of PKCθ to the immune synapse. However, whereas these two events are dispensable for T-cell responsiveness to strong (primary) pMHC ligands, they are required for responsiveness to their weaker (secondary) counterparts. Thus, the CD3ε PRS serves to amplify weak TCR signals by promoting synapse formation and CD3ε phosphorylation.
MATERIALS AND METHODS
Mice
8.3-TCR-transgenic NOD (8.3-NOD) and NOD.RAG-2−/− mice have been described (23). B6.CD3εΔP/ΔP mice were from C. Terhorst (Harvard Medical School) (24). 8.3-TCR-transgenic CD3εΔP/ΔP mice were generated by intercrossing H-2g7-homozygous 8.3-TCR-transgenic CD3εΔP/wt (B6 × NOD) N1 mice. Animal experimentation was approved by the University of Calgary Animal Care Committee.
Peptides, pMHC tetramers and antibodies
The peptides NRP-A7 (KYNKANAFL; strong/primary agonist), NAT-32 (QYNKTGYFL) and NRP-E6 (KYNKAEAFL) (weak/secondary agonists), and TUM (KYQAVTTTL; Kd-binding non-agonist control), and the corresponding pMHC tetramers were synthesized as described (22, 25). Anti-phosphotyrosine 4G10 mAb was from Upstate Biotechnology. Anti-CD8, anti-CD3ε, and anti-Vβ8 mAbs were from Becton-Dickinson/Pharmingen (San Diego, CA). Rabbit anti-Nck, phospho-c-Jun, CD3ζ, PKCθ, ZAP-70 and goat anti-CD3ε and talin were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-Erk1/2 Abs were from Cell Signaling Technology (Beverly, MA). The anti-CD3ε PRS-specific mAb APA1/1 was from Upstate Biochemicals (Lake Placid, NY). Anti-mouse-Alexa-488 and anti-rabbit-Alexa-555 were from Molecular Probes (Portland, OR).
pMHC tetramer staining
T-cells (0.5-1 × 106) were incubated with tetramer (85.5 nM) for 45 min at 25°C in 50 μl of RPMI-1640 containing 0.1% sodium azide and 2% FBS, washed, stained with 0.5 μg of anti-CD8β-FITC and analyzed with a flow cytometer.
Isolation of CD8+ T-cells and DCs and generation of CTL
Splenic 8.3-CD8+ T-cells were isolated using mAb-coated magnetic beads (Miltenyi-Biotech, Auburn, CA). Purified T-cells (>95% CD8+ and Vβ8.1+) were used as such (referred to as ‘naïve’) or upon differentiation into CTL (referred to as ‘differentiated’) by stimulation with NRP-A7 (22). Briefly, T-cells were cultured with NRP-A7-pulsed (1 μM), irradiated NOD splenocytes for 3 d, and expanded in rIL-2-containing media for 4 additional days (rIL2; Takeda Chemical Co., Osaka, Japan). Bone marrow-derived DCs, generated as described (26), were purified using anti-CD11c mAb-coated magnetic beads and matured by overnight culture in 1 μg/ml LPS. After extensive washing, DCs were incubated with peptides (1 μM) for 2-3 h at 37°C, washed and used.
Cytokine secretion
Naïve or differentiated 8.3-CD8+ T-cells (2 × 104 cells/well) were incubated in triplicate with peptide-pulsed (1 μM) mature DCs (5 × 103/well) or pMHC tetramers (5 μg/ml), for 48 h at 37°C in 5% CO2. Supernatants were assayed for IL-2 and/or IFN-γ content by ELISA (R&D systems, Cambridge, MA).
CFSE-labeling and T-cell transfer
Naïve 8.3-CD8+ T-cells were labeled with 5 μM CFSE in PBS at 37°C for 3 minutes, washed with PBS, and injected i.v. into NOD mice (107 cells). Twenty-four h later, the mice were injected in the footpads with 100 μg of peptide in PBS. Mice were killed 96 h later to investigate the presence of proliferating CD8+ T-cells in lymphoid organs.
Ca2+ flux
Naïve or differentiated CD8+ cells were loaded with Indo-1-AM at 37°C in HBSS for 30 min. After washing, the cells were incubated for 2 min at 37°C with peptide-pulsed DCs and analyzed by FACS during the next 5 min. We measured changes in the FL4:FL5 ratio with time in T cell:DC conjugates (gated according to forward and side scatter). For measurements of Ca2+ flux induced by tetramer, T-cells were loaded with Fluo-3-AM (Molecular Probes), washed and examined for 1 min, to obtain a baseline. PE-labeled pMHC tetramers were then added at 1 μg/ml and changes in the intracellular concentration of Ca2+ determined by measuring changes in Fluo-3-AM fluorescence in the tetramer+ T-cell population.
Phospho c-Jun analyses
T-cells (106) were incubated with peptide-pulsed DCs (105), fixed at different time points in 4% formaldehyde at room temperature for 10 min and stored at −20° in 90% methanol, incubated with anti-phospho c-Jun antibodies in 2% RPMI-1640 containing 0.1% saponin at RT for 1 h, followed by anti-mouse IgG-Alexa-488 and anti-CD8-PE. Samples were gated on CD8+ cells and analysed with a flow cytometer.
Immunoprecipitation, CD3ε pull-down assays and Western blotting
T-cells (6-10 × 106) were incubated with peptide-loaded DCs (at a 6:1 ratio) in 100 μl serum-free medium and incubated at 37°C for different time points. Cells were lysed by adding 100 μl of lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 1 mM PMSF, 1 mM β-glycerol phosphate, 1 mM pervanadate) containing a protease inhibitor cocktail (Roche, Laval, QC) on ice for 15 min. In some experiments, lysates were subjected to Western blot analysis with 4G10 mAb or pErk-specific Abs. In other experiments, we immunoprecipitated post-nuclear lysates with anti-CD3ε Abs. The CD3ε pull-down assay was done as described (13). Post-nuclear cell lysates were incubated with GST beads for 1 h at 4°C and then incubated with GST-Nck SH3.1 beads (Lab Vision Corp., Freemont, CA) for 2-3 h at 4°C. Beads were washed before SDS-PAGE/Western blotting. For CD3ε-Nck co-immunoprecipitation, cells were lysed in lysis buffer containing 0.5% NP40 instead of Brij96, and immunoprecipitated with a hamster anti-mouse CD3ε mAb (clone 14C11). Total lysates and immunoprecipitates were mixed with SDS sample buffer, boiled for 5 min, resolved in 10%-12% SDS-PAGE, transferred to PVDF membranes, probed with phospho-, CD3ε- or Nck-specific Abs, and developed with the SuperSignal West Pico Chemiluminescence kit (Pierce Biotech, Rockford, Il).
Cell conjugate formation
Purified CD8+ T-cells and DCs were labeled for 5 min at 37°C with the DiIC18 or DiOC18 dyes, respectively (Molecular probes), and mixed at a 1:1 ratio for 30 min at 37°C in serum-free RPMI-1640 medium. Complexes were gently pipetted up and down 5 times with a 1 ml pipettor and analyzed by flow cytometry.
Immunofluorescence microscopy
Mature DCs were loaded with peptides (1 μM) for 1-2 h at 37°C and plated for 60 min in 4-well chamber slides, washed with serum-free RPMI-1640, and incubated with T-cells (2 × 105) for 30 min at 37°C. Cells were then fixed using the Cytofix–Cytoperm kit (BD Pharmingen) for 10 min at RT, permeabilized with 0.01% Saponin (Sigma, St. Louis, MO) in PBS for 15 min, also at RT, and stained with rabbit anti-PKCθ antibodies or anti-CD3ε (APA1/1) mAb and Alexa 488-conjugated secondary Abs. The cells were examined with a Deltavision fluroscence microscope (Applied Precission, Issaquah, WA). Cells displaying focalized PKCθ or APA1/1 staining at the T-cell–DC contact site were scored as positive for synapse formation; cells making DC contacts but displaying diffuse PKCθ staining or no APA1/1 staining, were scored as negative. Synapses were scored blindly by two different investigators.
In other experiments, conjugates of differentiated T-cells with peptide-pulsed DCs or RMA-SKd cells were stained with goat anti-talin/anti-goat-Alexa-488 and rabbit anti-PKCθ/anti-rabbit-Alexa-555. Because the transgenic T-cells of the chimeras lose eGFP fluorescence upon antigen-induced differentiation (as examined under the Deltavision microscope; data not shown), all the detectable green fluorescence in these experiments emanated from the alexa-488-labeled talin/anti-talin complexes. For synapse image reconstruction, data sets were projected as a volume option using the surpass function of Imaris® software version 4.2 (Bitplane Inc., Zurich, Switzerland). Images were then cropped around the T-cell–APC contact zone and rotated in the ZX plane. Images were further processed in Adobe Photoshop to enhance the contrast.
Retroviral-Mediated Stem Cell Gene Transfer
Retroviral producer cell lines encoding wild-type and mutant (PRS-deficient CD3ε) 2A peptide-linked TCR:CD3 chains and an IRES-eGFP cassette were produced as described (20). Bone marrow-derived stem cells from 8.3-TCR-transgenic, CD3εΔP/ΔP mice were co-cultured with the retroviral producer cell lines for 48 h. The non-adherent bone marrow cells (2-4 × 106 in PBS containing 2% FBS and 20 u/ml heparin) were injected i.v. into sub-lethally irradiated (1,100 rads) hosts. Mice were killed 8-12 wk later for phenotypic and functional studies.
Statistics
Significance was assessed using two-way ANOVA, Chi-square and Mann-Whitney U tests. Differences were considered statistically significant when P ≤ 0.05.
RESULTS
Weak/secondary pMHC agonists bind TCR with low avidity
We used naïve and antigen-differentiated CD8+ T-cells derived from mice expressing a transgenic, H-2Kd-restricted TCR (8.3-TCR) (23, 27). We chose IFNg secretion as a read-out for the experiments described herein because, unlike proliferation, IL-2 secretion or CD25 upregulation, its magnitude does not change much with T-cell differentiation. For example, the proliferative activity of differentiated 8.3-CD8+ T-cells, unlike their IFNγ secretory capacity, is at least an order of magnitude lower than that of their naïve precursors, and differentiated 8.3-CD8+ T-cells, unlike their naïve counterparts, produce negligible levels of IL-2 and already express high levels of CD25 prior to re-stimulation (data not shown). Whereas NRP-A7, a strong agonist peptide, was able to elicit substantial IFNg secretion by both naïve and differentiated 8.3-CD8+ T-cells, two different weak agonists (NAT-32 and NRP-E6) could only induce significant IFNg production by differentiated 8.3-CD8+ T-cells, even at five-fold higher concentrations of peptide (Fig. 1A). As expected, both peptides also failed to elicit the proliferation of naïve 8.3-CD8+ T-cells (Fig. 1B) or the upregulation of CD25 on the T-cells' surface (data not shown). Neither TUM (a H-2Kd-binding negative control peptide) nor NRP-A4 (an antagonist) elicited IFNg secretion by either cell type (Fig. 1A). Experiments employing 8.3-CD8+ T-cells maturing in two different genetic backgrounds (8.3-NOD and 8.3-B10.H2g7) produced similar outcomes (data not shown).
Figure 1. Agonistic properties and binding avidities of strong and weak pMHC agonists.
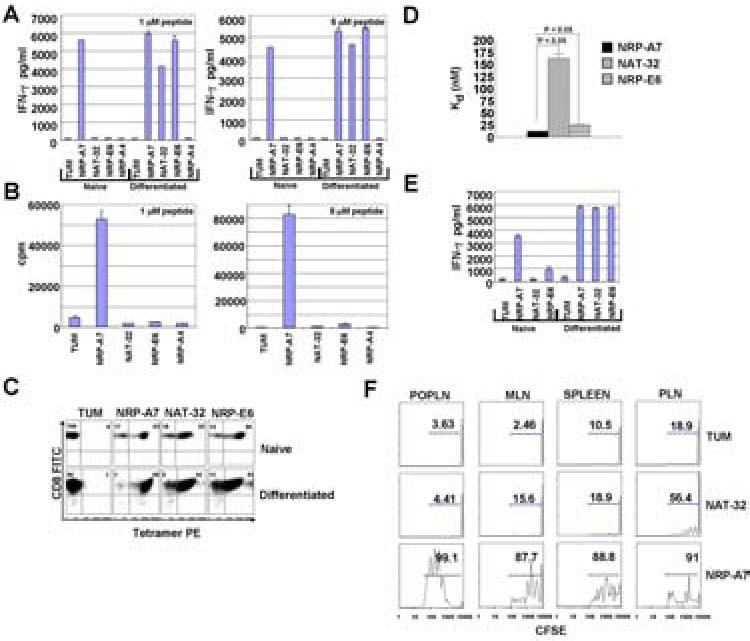
A, Secretion of IFNγ in response to peptide-pulsed (1 or 5 μM) DCs. Data are representative of 3 different experiments. B, Proliferation of naïve 8.3-CD8+ T-cells in response to peptide-pulsed (1 or 5 μM) DCs. C, Reactivity of naïve and differentiated 8.3-CD8+ T-cells to control (TUM), strong agonist (NRP-A7) and weak agonist (NAT-32 and NRP-E6) pMHC tetramers. Values correspond to % of cells binding to the corresponding tetramers. The corresponding MFI values are: 996, 174 and 617 for naïve T cells; and 1095, 297 and 231 for differentiated T-cells. Data are representative of 2-to-5 experiments (for naïve and differentiated T-cells, respectively). D, Kd values of tetramer-binding to differentiated 8.3-CD8+ T-cells. Data correspond to average ± s.e.m. of 3 experiments (12 ± 0.2, 16.3 ± 1, and 25.7 ± 1, respectively) . P values were obtained with Mann-Whitney U test. E, Secretion of IFNγ in response to pMHC tetramers (5 μg/ml) in the absence of APCs. F, Proliferation of CFSE-labeled naïve 8.3-CD8+ cells in peripheral lymphoid organs of host mice in response to 100 μg of TUM (neg. control), NRP-A7 or NAT-32 given via the footpad. Hosts were transfused with CFSE-labeled 8.3-CD8+ cells, treated with peptide 24 h later and examined for T-cell proliferation on day 3. POLN, popliteal lymph nodes; MLN, mesenteric lymph nodes; PLN, pancreatic lymph nodes. Values shown correspond to % proliferating cells within the CFSE+ gate. The data are representative of 2 experiments.
Since all three peptides stabilized Kd expression on the surface of TAP-deficient RMA-SKd cells with similar efficiency (data not shown), we reasoned that the above results might be due to differences in the 8.3-TCR binding avidity of the corresponding pMHC class I complexes. To investigate this, we compared the ability of naïve and differentiated 8.3-CD8+ T-cells to bind to NAT-32/Kd, NRP-E6/Kd and NRP-A7/Kd tetramers, and to produce IFNγ in response to tetramer challenge. Although all three tetramers stained naïve 8.3-CD8+ T-cells well (Fig. 1C), they did so with significantly different avidities (Kd values: NRP-A7/Kd << NRP-E6/Kd < NAT-32/Kd) (Fig. 1D), and elicited significantly different amounts of IFNγ from these T-cells (NRP-A7/Kd >> NRP-E6/Kd > NAT-32/Kd) (Fig. 1E). Notwithstanding these relatively large differences in binding avidity and naïve T-cell stimulatory potency, these tetramers elicited comparable amounts of IFNγ from differentiated 8.3-CD8+ T-cells (Fig. 1E).
We next asked if this inability of weak pMHC agonists to activate naïve CD8+ T-cells was also true in vivo. To this end, we examined the ability of CFSE-labeled naïve 8.3-CD8+ T-cells to proliferate in the spleen and popliteal (POPLN), mesenteric (MLN) and pancreatic lymph nodes (PLN) of NOD hosts upon adoptive transfer and peptide administration. The transfused T-cells proliferated vigorously in the lymphoid organs of NRP-A7-treated hosts but not in those of control peptide (TUM)-treated mice (Fig. 1F). As expected, NAT-32 treatment failed to elicit the proliferation of 8.3-CD8+ T-cells located in the spleen, POPLN and MLNs (Fig. 1F). Unlike TUM, however, NAT-32 triggered the proliferation of 8.3-CD8+ T-cells contained in the PLNs of these mice (Fig. 1F), presumably because these T-cells had been pre-activated in situ by their endogenous antigenic ligand (residues 206-214 of islet-specific glucose-6-phosphatase catalytic subunit-related protein). When taken together, these observations demonstrate that weak pMHC agonists cannot activate naïve CD8+ T-cells, either in vitro or in vivo, and that T-cells activated with strong pMHC agonists (i.e. NRP-A7/Kd in vitro, or endogenous IGRP206-214/Kd in vivo) somehow acquire the ability to respond to weak pMHC agonists.
Impaired distal TCR-signaling by weak agonists in naïve T-cells
Naïve 8.3-CD8+ T-cells displayed similar tyrosine phosphorylation patterns upon stimulation with strong and weak pMHC agonists (Fig. 2A). Likewise, the tyrosine phosphorylation patterns elicited by strong and weak pMHC agonists in differentiated 8.3-CD8+ T-cells, although different from those seen in their naïve precursors, were similar. To ascertain whether strong and weak agonists elicited qualitatively and/or quantitatively different TCR signals in naïve and/or differentiated CD8+ T-cells, we measured three different TCR-distal signaling events: Ca2+ signaling, which occurs within minutes of TCR ligation; Erk1/2 activation, which also occurs within minutes but can be sustained for up to 3 hours; and phospho c-Jun accumulation, which can be monitored for up to 24 hours.
Figure 2. Global assessment of TCR signaling in response to strong and weak agonists.
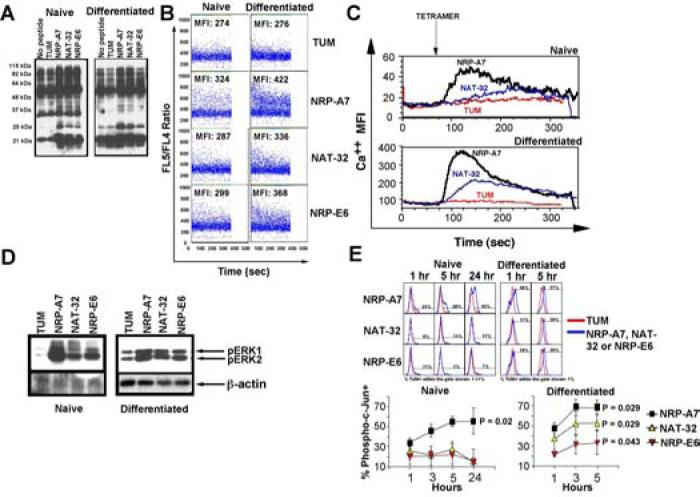
A, Overall tyrosine phosphorylation induced by pMHC tetramers. B and C, Mobilization of intracellular calcium in response to peptide-pulsed (1 μM) DCs (in B, using Indo-I-Am) or pMHC tetramers (1 μg/ml) (in C, using Fluo-3-AM). Data in C correspond to gated tetramer+ cells. D, Erk1/2 activation 10 minutes after stimulation with peptide-pulsed DCs. Western blots were probed with phospho-Erk1/2- or β-actin-specific antibodies. The densitometric values (normalized to β-actin) for NRP-A7, NAT-32 and NRP-E6 as compared to TUM were 14, 12 and 13-fold (naïve T-cells) and 2, 2 and 1.7-fold (differentiated T-cells), respectively. E, Phospho-c-Jun accumulation in response to peptide-pulsed DCs. Cells were fixed and stained with anti-CD8 and anti-phospho-c-Jun-specific antibodies after the indicated stimulation periods. Although NRP-A7-induced differentiation resulted in heightened levels of basal phospho-c-Jun as compared to the background levels seen in naïve T-cells, NAT-32 and NRP-E6 induced phospho-c-Jun accumulation only in differentiated T-cells. Analyses were done on gated CD8+ T-cells. Top panels show representative staining patterns. Bottom panels show averages ± s.e.m. of 3-4 experiments/peptide/time point. MFI values from TUM-challenged cells were subtracted. P values shown correspond to differences vs. TUM at 24 h (for naïve T-cells) or 5h (for differentiated T-cells) (Mann-Whitney U).
Weak agonists elicited Ca2+ responses of different magnitude in naïve vs. differentiated 8.3-CD8+ T-cells. Whereas naïve T-cells mobilized calcium in response to APCs pulsed with NRP-A7, significantly less with NRP-E6 and almost not at all with NAT-32, their differentiated counterparts mobilized calcium in response to all three peptides, albeit with different efficiency (NRP-A7 > NAT-32 or NRP-E6) (Fig. 2B). Similar results were obtained when the T-cells were challenged with soluble pMHC complexes (Fig. 2C).
These differences among pMHC agonists were also observed at the level of Erk1/2 phosphorylation. NRP-A7 was superior to NAT-32 and NRP-E6 at inducing phosphoErk1/2 in naïve T-cells (Fig. 2D), suggesting that weak agonists are less efficient than strong agonists at triggering Erk1/2 activation in naïve T-cells.
It has been reported that full T-cell activation requires intracellular accumulation of phospho c-Jun above a certain threshold (28). Whereas phospho c-Jun was already present in naïve 8.3-CD8+ T-cells within 1 hour of stimulation with NRP-A7, it remained low throughout the entire 24-hour stimulation period in naïve T-cells challenged with NAT-32 or NRP-E6, as compared to the negative control peptide TUM (Fig. 2E). In contrast, all three peptides fostered phospho c-Jun accumulation in pre-activated 8.3-CD8+ cells for at least 5 hours, although with different efficiency (NRP-A7 > NAT-32 > NRP-E6) (Fig 2E).
Taken together, these results (summarized in Table I) suggested that differential responsiveness of naïve vs. differentiated 8.3-CD8+ T-cells to weak agonists is regulated upstream of the Ca2+, Erk1/2 and c-Jun pathways, prompting us to examine proximal TCR signaling in detail.
Table I.
Summary of responses induced by primary vs. secondary agonists on naïve and differentiated 8.3-CD8+ T-cells
| ACTIVATION STATUS | RESPONSE | PRIMARY AGONISTS (NRP-A7) |
SECONDARY AGONISTS (NAT-32/NRP-E6) |
|---|---|---|---|
| Naive | IFNγ | Yes | No |
| Ca2+ | Yes | No or weak | |
| pErk1/2 | Yes | Yes | |
| p-c-Jun | Yes | No | |
| pCD3ζ | Yes | Yes | |
| pCD3ε | Yes | No | |
| pCD3ε-Nck | Yes | No | |
| Synapse | Yes | No | |
| Differentiated | IFNγ | Yes | Yes |
| Ca2+ | Yes | Yes | |
| pErk1/2 | Yes | Yes | |
| p-c-Jun | Yes | Yes | |
| pCD3ζ | Yes | Yes | |
| pCD3ε | Yes | Yes | |
| pCD3ε-Nck | Yes | Yes | |
| Synapse | Yes | Yes |
Lack of CD3ε but not CD3ζ phosphorylation in weak agonist-challenged naïve T-cells
Naïve and differentiated CD8+ T-cells expressed phosphorylated p23 CD3ζ upon stimulation with NRP-A7, NAT-32 and NRP-E6 (Fig 3A), suggesting that in weak agonist-stimulated naïve CD8+ T-cells, phosphorylation of CD3ζ does occur. In contrast, whereas weak agonists induced the phosphorylation of CD3ε in differentiated CD8+ T-cells, they were unable to do so in their naïve precursors (Fig 3B). We thus reasoned that differential T-cell responsiveness to strong vs. weak agonists might be regulated at the level of CD3ε activation.
Figure 3. Proximal TCR signaling events induced by strong and weak agonists.

CD3ζ (A) and CD3ε (B) phosphorylation in response to peptide-pulsed DCs. Cell lysates were immunoprecipitated with a hamster anti-mouse CD3ε mAb (clone 14C11). The Western blots were first probed with 4G10 (top panels in A and B), and then stripped and probed with anti-CD3ζ (bottom of A) or anti-CD3ε (bottom of B). Data are representative of 3 experiments. The densitometric values (normalized to total CD3ζ in A, or CD3ε in B) for NRP-A7, NAT-32 and NRP-E6 vs. TUM were 2.7, 3.4 and 2.2-fold (naïve T-cells) and 2.2, 1.7 and 1.9-fold (differentiated T-cells), respectively (A), and 2, 1, and 1-fold (naïve T-cells) and 4, 2 and 2.8-fold (differentiated T-cells), respectively (B). C, GST-Nck-SH3.1 beads were used to pull down (PD) ‘open’ CD3ε conformers. Proteins were resolved by SDS-PAGE and Western blot analysis using goat polyclonal anti-CD3ε Abs as probe. The samples in the far left lanes of each panel correspond to lysates pre-incubated with the CD3ε PRS-specific mAb APA1/1, as a specificity control. Data are representative of 2 experiments. The densitometric values corresponding to NRP-A7, NAT-32 and NRP-E6 vs. TUM were 2, 1 and 1-fold (naïve T-cells) and 2.1, 1.5 and 2-fold (differentiated T-cells), respectively. D, Recruitment of Nck to CD3ε in response to peptide stimulation. CD3ε immunoprecipitates were resolved by SDS-PAGE and subjected to Western blot analysis with anti-Nck- or anti-CD3ε Abs. Data are representative of 3 experiments. The densitometric values (normalized to total CD3ε) for NRP-A7 and NAT-32 vs. TUM were 2 and 1-fold (naïve T-cells) and 1.8 and 1.6-fold (differentiated T-cells), respectively. E, Presence of ‘open’ CD3ε conformers in T-cell–DC conjugates. Naïve or differentiated CD8+ cells were co-cultured with peptide-pulsed DCs, fixed, and stained with APA1/1 mAb and Alexa 488-conjugated anti-mouse Ig secondary Ab. Graphs show the % cells making contacts with DCs that are APA1/1+. Data correspond to average ± s.e.m. of at least 50 conjugates/experiment in 3 experiments. The photographs show representative images at 60x magnification. P values were calculated by Chi-Square analysis.
Weak agonists induce CD3ε's conformational change in differentiated but not naïve T-cells
TCR ligation induces a conformational change in CD3ε that exposes its Nck-binding PRS. Because this event precedes CD3ζ tyrosine phosphorylation and is independent of src kinase activation (7, 13), we reasoned that failure of weak agonists to trigger CD3ε's phosphorylation might be accompanied with a failure to elicit CD3ε's conformational change. To investigate this, we challenged naïve and differentiated 8.3-CD8+ T-cells with strong and weak agonists and analyzed cell lysates for presence of CD3ε molecules capable of binding a GST fusion protein containing the SH3.1 domain of Nck, which binds to the CD3ε PRS. Weak agonists exposed the CD3ε PRS in differentiated T-cells, but could not do so in their naïve precursors (Fig. 3C). As expected, incubation of lysates from NRP-A7-stimulated T-cells with the APA1/1 mAb, which specifically recognizes the ‘open’ conformation of CD3ε, abrogated the GST-Nck-SH3.1-induced precipitation of CD3ε (Fig 3C, right). These observations were confirmed for NRPA7 and NAT-32 by examining CD3ε immunoprecipitates from peptide-challenged T-cells for presence of Nck. Unlike NRP-A7, NAT-32 could only foster recruitment of Nck to CD3ε in differentiated 8.3-CD8+ T-cells (Fig. 3D). Furthermore, whereas the APA1/1 mAb only stained naïve T-cells conjugated with strong, but not weak agonist-pulsed DCs, it stained differentiated T-cells conjugated with DCs pulsed with both types of agonists (Fig. 3E). These data established a correlation between agonistic activity of pMHC ligands on naïve CD8+ T-cells and their ability to elicit CD3ε's conformational change.
Impaired recruitment of PKCθ by weak agonists in naïve T-cells
It has been shown that recruitment of Nck to the TCR contributes to synapse formation (7, 13, 29). We thus reasoned that impaired recruitment of Nck to CD3ε might impair the formation of mature synapses between naïve T-cells and weak agonist-pulsed DCs. We first investigated whether weak agonists could enhance the formation of stable conjugates between naïve T-cells and DCs, by flow cytometry. NAT-32 and NRP-E6 were both capable of enhancing the formation of conjugates between naïve 8.3-CD8+ T-cells and DCs within 30 minutes of co-culture, as compared to the negative control peptide TUM (Fig. 4A). To ascertain whether this was accompanied with impaired synapse formation, we examined PKCθ's redistribution in T-cell–DC conjugates (to the T-cell:DC contact site) within 30 minutes of co-culture. As shown in Figs. 4B and 4C, weak agonists were only able to induce redistribution of PKCθ in differentiated 8.3-CD8+ T-cells. Thus, in addition to failing to induce CD3ε's conformational change and phosphorylation, weak agonists fail to recruit PKCθ to the immune synapse in naïve T-cells.
Figure 4. Recruitment of PKCθ to the synapse by strong and weak agonists.
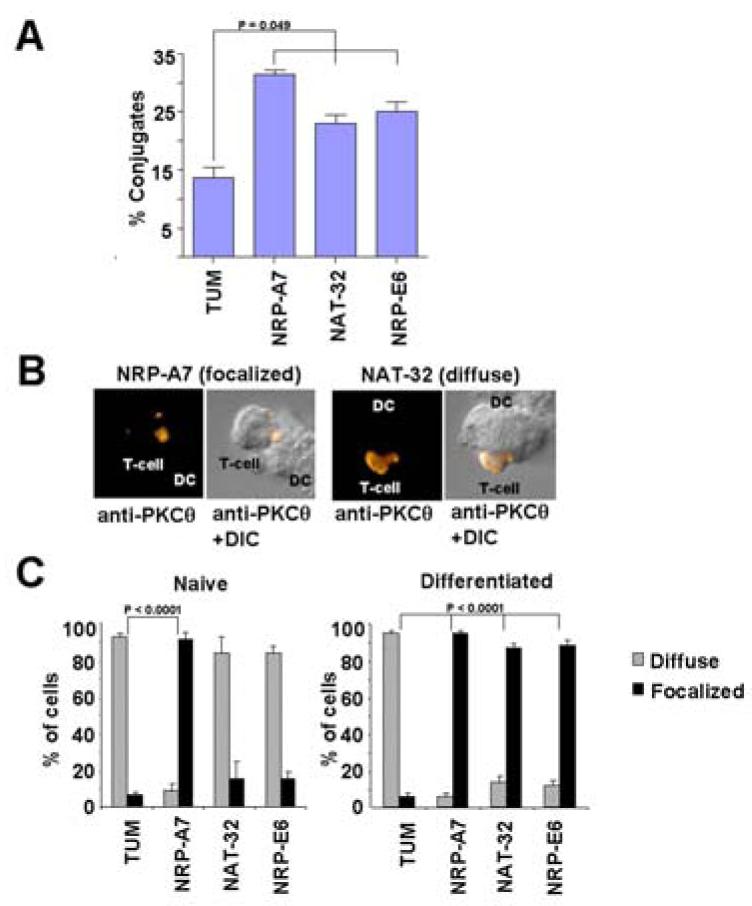
A, % stable conjugates formed between naïve 8.3-CD8+ cells and peptide-pulsed DCs. Results are average ± s.e.m. of 3 experiments. P values were calculated by Mann-Whitney U. B and C, Recruitment of PKCθ to the contact zone in T-cell–DC conjugates formed within 30 min. B shows representative images of T-cell–DC conjugates displaying focalized (left, with NRP-A7) or diffuse (right, with NAT-32) PKCθ staining. C shows the efficiency with which strong and weak agonists induce PKCθ recruitment in naïve and differentiated T-cells. T-cell–DC conjugates were scored for presence of PKCθ in the T cell–DC contact site (focalized staining) or throughout the cytosol (diffuse staining). Results are average ± s.e.m. of ∼100 conjugates/experiment in 3 experiments. P values were calculated by Chi-Square analysis.
PRS-dependent phosphorylation of CD3ε
Notwithstanding the fact that the CD3ε PRS and ITAM motifs are dispensable for T-cell development and function (20, 30, 31), our observation that responsiveness to weak pMHC agonists correlates with ligand-induced exposure of the CD3ε PRS suggested that this motif serves to amplify weak (low affinity/avidity) TCR signals. We reasoned that if this was true, the CD3ε PRS should control certain early TCR signaling events that enable responsiveness of differentiated T-cells to weak pMHC agonists, but are dispensable for responsiveness to strong ones.
To test this hypothesis, and to investigate the precise sequence of early TCR signaling events resulting from strong and weak pMHC agonist-induced TCR ligation, we expressed the 8.3-TCR-transgenes in CD3ε-deficient mice (24); CD3εΔP/ΔP, which also display significantly reduced expression of CD3γ and CD3δ. We then transduced hematopoietic stem cells from these mice with retroviruses encoding wild-type, PRS-mutant or ITAM-mutant CD3ε chains linked to wild-type CD3γ and CD3δ chains by ‘self-cleaving’ 2A peptides, which give rise to near absolute cleavage and expression of each CD3 chain with proper stoichiometry, and IRES-eGFP (20) (Fig. 5A), and used these stem cells to generate bone marrow-chimeras in irradiated wild-type hosts.
Figure 5. Role of the CD3ε PRS and ITAM in 8.3-CD8+ T-cell development.
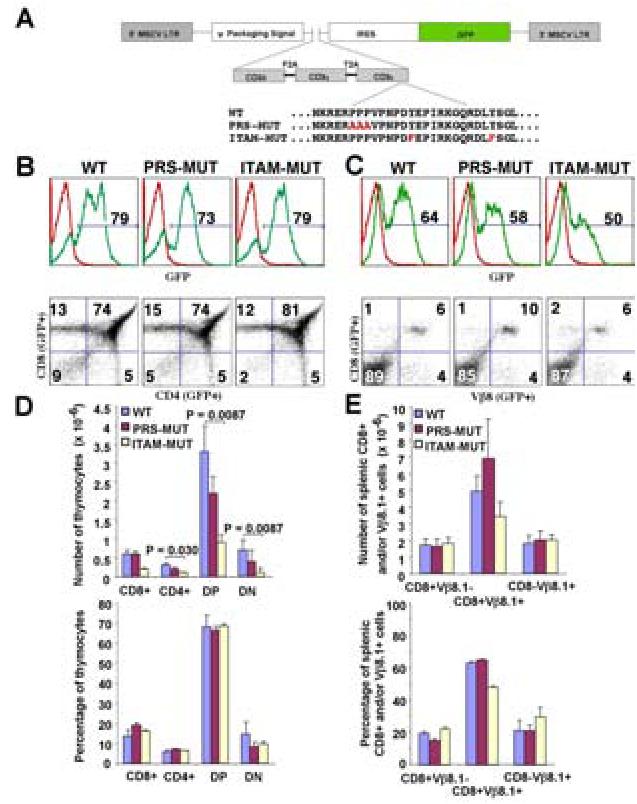
A. Schematic diagram showing the CD3ε wild-type and mutant retroviral constructs used in this study. B and C, eGFP profiles of thymocytes (B) and splenocytes (C) in mice expressing wild-type CD3ε or its PRS or ITAM-deficient counterparts. Bottom panels in B and C show the CD4/CD8 and CD8/Vβ8.1 profiles of gated eGFP+ cells. Profiles are representative of 4-7 mice/chimera type. D and E, Absolute (top) and relative numbers (bottom) of the different thymocyte (D) and splenocyte (E) eGFP+ lymphocyte subsets. Data correspond to averages ± s.e.m. of 5-7 mice/chimera type. P values were calculated with Mann-Whitney U.
More than 70% of thymocytes in all three types of chimeras were GFP+, and >70% of the chimeras' peripheral GFP+ T-cells expressed the transgenic Vb8.1+ TCR (Figs. 5B-5E). The peripheral GFP+CD8+ T-cells of all three types of chimeras displayed a naïve phenotype (CD62Lhigh, CD44int, CD69−, CD25−) (data not shown). The wild-type and PRS-deficient forms of CD3ε supported the development of 8.3-CD8+ T-cells with similar efficiency: the absolute and relative numbers of the different thymic (Figs. 5B and 5D) and splenic T-cell subsets (Figs. 5C and 5E) were statistically indistinguishable. Although mice expressing the ITAM-deficient form of CD3ε had significantly fewer thymocytes than mice expressing its wild-type counterpart (Fig. 5D), their spleens contained similar numbers of GFP+Vb8.1+CD8+ T-cells (Fig. 5D). These data indicated that the CD3ε PRS and ITAM motifs are dispensable for 8.3-CD8+ T-cell development (20).
CD3ε pull-down assays using GST-Nck SH3.1 beads confirmed that, unlike GFP+CD8+ T-cells expressing wild-type or ITAM-mutant CD3ε, differentiated GFP+CD8+ T-cells expressing PRS-mutant CD3ε could not recruit Nck to CD3ε upon stimulation with NRP-A7 (Fig. 6A). This not only indicated that the PRS mutation carried by the 8.3-CD8+ T-cells of these mice effectively precludes Nck recruitment to CD3ε, but also that the ITAM-mutation does not interfere with recruitment of Nck to the CD3ε PRS. Surprisingly, analysis of peptide-induced CD3ε phosphorylation revealed that, in the absence of the PRS motif, none of the peptides (NRP-A7, NAT-32 and NRP-E6) could induce phosphorylation of the CD3ε ITAM in differentiated 8.3-CD8+ T-cells (Fig. 6B, and data not shown).
Figure 6. Role of the CD3ε PRS and ITAM in responsiveness to strong and weak agonists.

A, GST-Nck-SH3.1 pull-down of ‘open’ CD3ε conformers from differentiated T-cells upon stimulation with peptide-pulsed DCs. Data shown correspond to two independent experiments (wild-type and PRS-mutant in one and ITAM-mutant in another). B, CD3ε phosphorylation in differentiated 8.3-CD8+ T-cells from chimeras in response to peptide-pulsed DCs. Cell lysates were immunoprecipitated with anti-CD3ε Abs. The blots were first probed with the 4G10 mAb (top) and then stripped and probed with anti-CD3ε (bottom; as loading control). Data are representative of 3 experiments. The densitometric values (normalized to total CD3ε) for NRP-A7 as compared to TUM for WT and PRS- or ITAM-mutant T-cells were 2, 1 and 1-fold, respectively. C, IFNγ production by naïve splenic 8.3-CD8+ T-cells from all three types of chimeras in response to peptide-pulsed DCs at four peptide concentrations. Graph shows IFNγ values normalized to the levels produced by wild-type T-cells at 1 μM of NRP-A7 (100% value). Data correspond to average ± s.e.m. of n=7, 7 and 5 mice per chimera type (wild-type, PRS-deficient and ITAM-deficient, respectively). P values were calculated via two-way ANOVA. D, Proliferative activity of naïve T-cells from the mice used in the experiments shown in C. E, IL-2 production by naive T-cells from the different chimeras in response to peptide-pulsed (1 μM) DCs. IFNγ values are normalized to those of wild-type CD8+ cells in response to NRP-A7 (100% value). Data correspond to average ± s.e.m. of 3 experiments. P values were calculated with Mann-Whitney U. F, IFNγ production by differentiated T-cells from the different chimeras in response to peptide-pulsed (1 μM) DCs. IFNγ values are normalized to those of wild-type CD8+ cells in response to NRP-A7 (100% value). Data correspond to average ± s.e.m. of 4 experiments. P values were calculated with Mann-Whitney U.
PRS-dependent TCR signal amplification via the CD3ε ITAM
To probe the functional implications of these biochemical observations we compared the responsiveness of FACS-sorted eGFP+CD8+ T-cells from the different chimeras to pMHC. Naive 8.3-CD8+ T-cells expressing the PRS- and, especially, the ITAM-mutant forms of CD3ε produced significantly lower levels of IFNγ in response to NRP-A7 than those expressing wild-type CD3ε, over a range of concentrations (Fig. 6C). Proliferation assays revealed a similar trend (Fig. 6D), although the observed differences only reached statistical significance for the ITAM-deficiency. PRS- and ITAM-mutant CD8+ T-cells also produced significantly less IL-2 than CD8+ T-cells expressing wild-type CD3ε (Fig. 6E). Importantly, the above differences in IFNg secretion where completely absent among differentiated 8.3-CD8+ T-cells from all three types of mice (Fig. 6F, left panel), presumably owing to the increased functional avidity of differentiated vs. naïve T-cells. In contrast, differentiated 8.3-CD8+ T-cells expressing the PRS- and ITAM-mutant forms of CD3ε responded significantly less to the weak agonists NAT-32 and NRP-E6 than those expressing wild-type CD3ε (Fig. 6F, middle and right panels), indicating that both motifs contribute to responsiveness to low-avidity pMHC ligands. When considered in the context of the biochemical data described above, these results suggest that the CD3ε PRS amplifies weak TCR signals, at least in part, by fostering the phosphorylation of the CD3ε ITAM. In naïve T-cells, however, signal amplification via the CD3ε ITAM would involve both PRS-dependent and PRS-independent events.
PRS- and ITAM-dependence of PKCθ recruitment
It has been shown that Nck links the TCR–CD3 complex with molecules necessary for the cytoskeletal rearrangements underlying synapse formation (13, 17-19). Accordingly, it has been postulated that recruitment of Nck to the CD3ε PRS contributes to the latter (13, 14). Although compelling, this hypothesis is at odds with our observation that recruitment of Nck to CD3ε is largely dispensable for responsiveness to high-avidity pMHC ligands, particularly if we consider the fact that Nck can be recruited to the TCR complex via SLP-76 or LAT, independently of CD3ε's conformational change (21).
To investigate whether CD3ε PRS-dependent phosphorylation of the CD3ε ITAM is necessary for mature synapse formation, we examined PKCθ's redistribution in differentiated 8.3-CD8+ T-cells (expressing wild-type or mutated CD3ε) engaging strong or weak agonist-pulsed DCs. As expected, 8.3-CD8+ T-cells expressing wild-type CD3ε redistributed PKCθ to the T-cell-DC synapse (Fig. 7A, left picture) in >70% of the conjugates in response to both strong and weak agonists (Fig. 7B). In contrast, PKCθ recruitment to the T-cell–DC synapse was significantly impaired in 8.3-CD8+ T-cells expressing the PRS- and ITAM-mutant forms of CD3ε, regardless of signal strength (i.e. TCR-binding avidity) (Fig. 7B), and despite the ability of ITAM-mutant CD3ε to recruit Nck (Fig. 6A). Additional analyses of T-cell–DC or T-cell–RMA-SKd conjugates with PKCθ- and talin-specific antibodies confirmed that whereas T-cells expressing wild-type CD3ε formed typical synapses, containing PKCθ and talin immunoreactivities in the center and periphery of the synapse, respectively, T-cells expressing mutant CD3ε chains did not (Figs. 7C-E). To ascertain if this was also the case in naïve T-cells, we compared PKCθ's redistribution in naïve 8.3-CD8+ T-cells expressing wild-type or PRS-mutant CD3ε in response to strong agonist-pulsed DCs. As expected, whereas naïve 8.3-CD8+ T-cells expressing wild-type CD3ε redistributed PKCθ to the T-cell-DC synapse in ∼80% of the conjugates, naïve 8.3-CD8+ T-cells expressing the PRS-mutant form of CD3ε only did so in ∼25% of the conjugates (Fig. 7F). Taken together, these data indicated that (i) CD3ε PRS and ITAM motifs play a major role in the formation of a mature synapse; (ii) responsiveness to strong agonists (unaffected by either mutation) is dissociated from PKCθ/talin recruitment to the synapse (impaired by both mutations); and (iii) responsiveness to weak agonists (impaired by both mutations) is associated with defective PKCθ/talin recruitment.
Figure 7. Recruitment of PKCθ and talin in T-cells expressing wild-type or mutant CD3ε upon stimulation with strong or weak agonists.

A, Representative anti-PKCθ Ab-stained images of conjugates between differentiated T-cells derived from the indicated chimera types and NRP-A7-pulsed DCs within 30 min. B, Efficiency of PKCθ re-distribution to the T-cell–DC contact zone. T-cell–DC conjugates were scored for the presence of PKCθ in the T cell–DC contact site (focalized staining) or throughout the cytosol (diffused staining). Results are average ± s.e.m. of 3 experiments scored by two different investigators (30-50 conjugates were examined per chimera type and experiment). P values were calculated by Chi-Square. C, Distribution of PKCθ and talin in differentiated T-cell–DC conjugates in response to NRP-A7. Note that the transgenic T-cells of the chimeras lose eGFP fluorescence upon antigen-induced differentiation such that all green fluorescence that is detectable in these experiments comes from the alexa-488-labeled talin/anti-talin complexes. D, Z axis image reconstruction of the T-cell–APC interface. Note the formation of disorganized synapses in conjugates of PRS-deficient T-cells and NRP-A7-pulsed RMA-SKd cells. E, Quantitative analyses of PKCθ/talin re-distribution in T-cell–DC conjugates. F, PKCθ re-distribution to the T-cell–DC contact zone in conjugates between NRP-A7-pulsed DCs and naïve 8.3-CD8+ T-cells from chimeras expressing wild-type or PRS-mutant CD3ε. Left panel: representative anti-PKCθ Ab-stained images of sorted eGFP+ T-cells. Right panel: average ± s.e.m. of conjugates displaying focalized or diffused PKCθ staning. P values were calculated by Chi-Square.
DISCUSSION
There is strong evidence suggesting that T-cell activation by pMHC requires the induction of conformational changes in the CD3 subunits of the TCR complex (32). However, the biochemical consequences and functional role of conformational changes in individual CD3 subunits such as CD3ε or CD3ζ remain unclear. Although there is evidence that pMHC-induced exposure of the CD3ε PRS correlates with the agonistic activity of pMHC ligands (33), whether recruitment of Nck to the PRS of CD3ε is necessary for, or even contributes to, T-cell activation is highly controversial (20, 21). Another issue that remains unresolved is whether the PRS contributes to CD3ε phosphorylation. pMHC-induced exposure of the CD3ε PRS is known to be independent of tyrosine kinase activity (13, 33) but direct evidence linking CD3ε phosphorylation to its conformational flip is lacking. Here, we provide significant observations that address some of these issues, reconcile previous observations that were seemingly at odds with one another, and uncover an unexpected role for PKCθ recruitment/synapse formation as a signal strength amplifier. We show that the downstream effects of TCR-CD3 conformational change either require or do not require the contribution of CD3ε PRS or ITAM motifs depending on the strength of the TCR-pMHC interaction. Our data support a model whereby the CD3ε PRS serves to amplify low-avidity TCR signals by promoting first ITAM phosphorylation and then PKCθ recruitment/synapse formation. The data further indicate that the latter event, while PRSand ITAM-dependent, is dispensable for responsiveness to high-avidity pMHC ligands. Taken together, these novel observations point to CD3ε as an amplifier of ‘weak’ TCR signals and provide a new mechanism for the increased ‘functional’ avidity of differentiated versus naïve T-cells. Fig. 8 shows a schematic view of these ideas.
Figure 8. A working model of PRS- and ITAM-dependent amplification of low-avidity TCR signals by CD3ε.
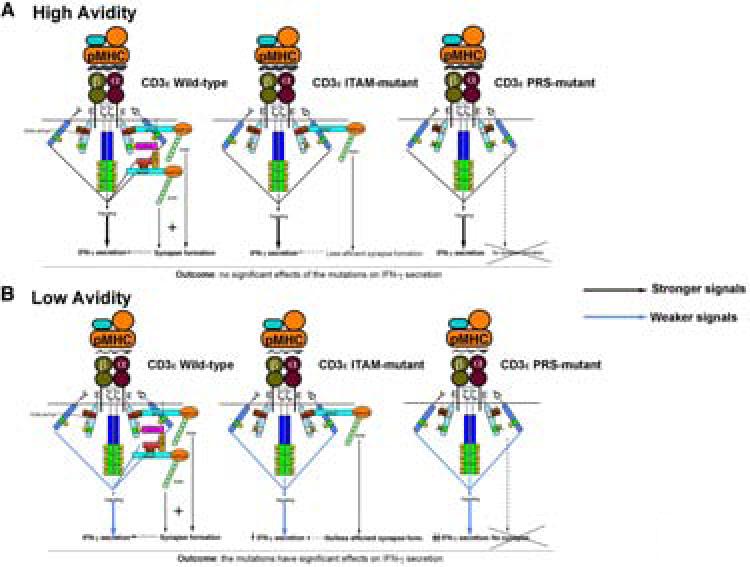
A, In differentiated cells expressing wild-type CD3ε, engagement of a high-avidity pMHC ligand induces (i) exposure of CD3ε's PRS and recruitment of Nck and other molecules involved in synapse formation; (ii) CD3ε's ITAM phosphorylation, leading to recruitment of additional Nck via phospho-ITAM-bound ZAP70-LAT-SLP76; (iii) efficient phosphorylation of CD3ζ and CD3γ which, in conjunction with CD3ε phosphorylation, contribute to downstream signaling promoting IFNγ secretion. The strength of signaling emanating from CD3ε and/or CD3ζ and CD3γ ITAMs in response to a high-avidity pMHC–TCR interaction would be such that synapse formation regulated by recruitment of Nck would not increase cytokine production any further. This would explain why T-cells expressing ITAM- or PRS-mutant CD3ε chains produce ‘normal’ levels of IFNγ, despite being unable to form synapses. B, In differentiated cells expressing wild-type CD3ε, engagement of a low-avidity pMHC ligand induces: (i) exposure of CD3ε's PRS and recruitment of Nck as in (A); (ii) CD3ε's ITAM phosphorylation, leading to recruitment of additional Nck, also as in (A); (iii) adequate (but suboptimal) phosphorylation of CD3ζ and CD3γ which, in conjunction with CD3ε phosphorylation, contribute to downstream signaling promoting IFNγ secretion. The suboptimal strength of signaling emanating from CD3ζ, CD3γ and CD3ε ITAMs in response to a low-avidity pMHC–TCR interaction would be such that synapse formation regulated by recruitment of Nck through (i) and (ii) would be required for optimal cytokine production. As a result, T-cells expressing ITAM- or PRS-mutant CD3ε chains (unable to form optimal synapses) would produce ‘suboptimal’ levels of IFNγ in response to low-avidity pMHC ligands.
Biochemical studies showed that in naïve 8.3-CD8+ T-cells weak/secondary (low-avidity) pMHC agonists could not induce CD3ε's conformational change. Pre-activation of these naïve T-cells with strong/primary (high-avidity) agonists caused a decrease in the threshold of CD3ε's flip, allowing responsiveness to weak/secondary agonists. Functional assays with 8.3-CD8+ T-cells expressing wild-type and PRS- or ITAM-mutant CD3ε later demonstrated that responsiveness of differentiated T-cells to weak agonists was, in fact, a function of both the CD3ε PRS and ITAM. Since phosphorylation of the CD3ε ITAM in these T-cells was PRS-dependent, we conclude that weak agonists can only trigger cytokine secretion from differentiated T-cells if they manage to expose the CD3ε PRS and to phosphorylate the CD3ε ITAM. Furthermore, since weak agonists induced CD3ζ phosphorylation in naïve T-cells, we can suggest that CD3ε phosphorylation occurs at a higher threshold of signal strength than CD3ζ phosphorylation. Alternatively, strong and weak agonists induce CD3ζ phosphorylation via different mechanisms, particularly since CD3ζ, unlike CD3ε, can be phosphorylated in a Lck-independent manner (34).
A recent report has suggested that phosphorylation of the CD3ε Y166 inhibits binding of Nck's SH3 domains to the CD3ε PRS, and that binding of the SH2 domain of ZAP-70 to the PxxDY motif of CD3ε competes with NcK for binding to the neighbouring PRS (35). Notwithstanding the fact that these data were obtained using a 293 cell-based transfection system over-expressing tagged domains of specific signaling molecules in isolation from other TCR components, they are compatible with our observation that the PRS facilitates ITAM phosphorylation if we consider a hierarchical binding model between ZAP-70, Nck and CD3ε. In this hypothetical model, PRS-dependent phosphorylation of the CD3ε ITAM would facilitate the binding of ZAP-70 and the subsequent dissociation of Nck from the CD3ε PRS.
Another important observation is that whereas strong agonist-induced CD3ε ITAM phosphorylation and PKCθ recruitment in differentiated CD8+ T-cells were CD3ε's PRS-dependent, T-cell function induced by the strong agonist was dissociated from both CD3ε motifs. This implies that, compared to low-avidity pMHC–TCR interactions, high-avidity interactions induce qualitatively and/or quantitatively superior downstream signaling events that obviate the requirement for CD3ε phosphorylation and synapse formation. The nature of these events is unknown but one possibility is that high-avidity ligands induce signal-transducing conformational changes in CD3ζ (36). The mechanisms by which the CD3ε PRS fosters ITAM phosphorylation are also uncertain, but it is tempting to speculate that recruitment of Nck to the CD3ε PRS via its SH3.1 domain allows the recruitment of a relevant, PRS-containing or phosphorylated tyrosine kinase via Nck's SH3.2-3 or SH2 domains, respectively.
In naïve 8.3-CD8+ T-cells, strong agonist-induced PKCθ recruitment also required CD3ε's PRS. In this case, however, the ITAM mutation had a more significant impact on T-cell responses than the PRS deficiency. Technical limitations associated with the relatively low yields of retrovirally-transduced (GFP+) 8.3-CD8+ T-cells from chimeric mice precluded us from formally establishing a link between CD3ε's PRS and phosphorylation of its ITAM, as was the case in differentiated CD8+ T-cells (requiring ∼90 donor mice and ∼30 chimeric mice for each chimera type). Nevertheless, the fact that the ITAM mutation has a more significant impact on naïve T-cell function than the PRS mutation suggests that, in naïve T-cells, signaling via CD3ε's ITAM in response to strong ligands would involve both PRS-dependent and -independent events. In differentiated T-cells, on the other hand, ligand discrimination of weak ligands and signal amplification via CD3ε's ITAM, would predominantly be PRS-dependent.
The finding that none of the pMHC agonists tested could effectively induce PKCθ recruitment in PRS- or ITAM-mutant differentiated T-cells was unexpected. Whether CD3ε's PRS and ITAM contribute to PKCθ recruitment/synapse formation by recruiting Nck or other SH3 domain-containing molecules remains to be determined. However, a role for Nck in this process is attractive, because it recruits molecules involved in TCR aggregation and cSMAC assembly (13, 17-19, 37-41). Since Nck can also be recruited to the TCR via ZAP70-LAT-SLP76 bound to phospho-CD3ε and CD3ζ (21, 42) it is reasonable to suspect that optimal synapse formation occurs only after a certain critical amount of Nck has been recruited (i.e. via both the CD3ε PRS and the CD3ε (and possibly CD3ζ's) ITAMs). This hypothesis is consistent with the observation that actin polymerization upon TCR ligation requires the phosphorylation of both LAT and SLP-76 (21, 43).
Another key implication of our work is that 8.3-CD8+ T-cells need not have to recruit PKCθ and form mature synapses with DCs to respond optimally to high-avidity pMHC ligands. This is not counterintuitive if we consider that T-cell activation is initiated and sustained in TCR/ZAP-70/SLP-76-containing microclusters generated at initial T-cell–APC contact sites in the periphery of the immune synapse (44); and that TCR signaling is terminated in the cSMAC (45). A corollary of this observation is that it may explain why T-cells can rapidly scan the surface of an APC and efficiently respond to high-avidity pMHC ligands without the need to form stable synapses (46, 47).
Lastly, our data imply that heightened susceptibility of differentiated 8.3-CD8+ T-cells to TCR triggering by low-avidity ligands can be explained, in part, by an activation-induced reduction in the threshold of CD3ε's flip. It is unclear why only antigen-activated CD8+ T-cells can generate ‘open’ CD3ε conformers in response to low-avidity agonists. This may be related to changes in the composition of the plasma membrane upon T-cell activation (48), or to established differences in the expression levels of TCRs, co-receptors and/or signaling molecules between naïve and differentiated T-cells.
In conclusion, we have shown that naïve 8.3-CD8+ T-cells use the ligand-induced conformational exposure of the CD3ε PRS as a sensor to discriminate between high- and low-avidity pMHC ligands, and have demonstrated that the CD3ε PRS is necessary for phosphorylation of the CD3ε ITAM and for formation of a mature immune synapse.
ACKNOWLEDGEMENTS
We thank S. Bou, M. DeCrom, M. Deuma, M. Foote, T. Irvine, S. Thiessen, H. Metselar, L. Kennedy, L. Robertson, Michael Schoel and P. Colarusso for technical assistance, and U. Walter, J. Yamanouchi, Y. Yang and Y. Shi for feedback.
Grant support: This work was supported by the Natural Sciences and Engineering Research Council of Canada and by a Core Facility supported by a Group Grant from the Canadian Institutes for Health Research. S.T., A.S. and P. Serra were supported by studentships from the Alberta Heritage Foundation for Medical Research (AHFMR). A.L.S and D.A.A.V. were supported by the NIH (AI52199), the St. Jude Cancer Center Support CORE grant (CA-21765) and the American Lebanese Syrian Associated Charities (ALSAC). P. Santamaria and S. R. are Scientists of the AHFMR. The JMDRC is supported by the Diabetes Association (Foothills).
Non-standard abbreviations
- DCs
dendritic cells
- LAT
linker for activation of T cells
- MFI
mean fluorescence intensity
- pMHC
peptide–MHC
- PLN
pancreatic lymph node
- PRS
proline-rich sequence
- PKC
protein kinase C
- SLP-76
SH2 domain-containing leukocyte protein
REFERENCES
- 1.Sloan-Lancaster J, Allen P. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Madrenas J, Schwartz R, Germain R. Interleukin 2 production, not the pattern of early T-cell antigen receptor-dependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc Natl Acad Sci USA. 1996;93:9736–9741. doi: 10.1073/pnas.93.18.9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monks C, Freibeg B, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular actvation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 4.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 5.Lanzavecchia A, Lezzi G, Viola A. From TCR engagement to T cell activation: a kinetic view of T cell behavior. Cell. 1999;96:1–4. doi: 10.1016/s0092-8674(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 6.Lanzavecchia A, Sallusto F. From synapses to immunological memory: the role of sustained T cell stimulation. Curr Opin Immunol. 2000;12:92–98. doi: 10.1016/s0952-7915(99)00056-4. [DOI] [PubMed] [Google Scholar]
- 7.Alarcon B, Gil D, Delgado P, Schamel W. Initiation of TCR signaling: regulation within CD3 dimers. Immunol Rev. 2003;191:38–46. doi: 10.1034/j.1600-065x.2003.00017.x. [DOI] [PubMed] [Google Scholar]
- 8.Levin S, Weiss A. Twisting tails exposed: the evidence for TCR conformational change. J Exp Med. 2005;201:489–492. doi: 10.1084/jem.20050179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee K, Holdorf A, Dustin M, Chan A, Allen P, Shaw A. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:39–42. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 10.Krogsgaard M, Huppa J, Purbhoo M, Davis M. Linking molecular and cellular events in T-cell activation and synapse formation. Semin Immunol. 2003;15:307–315. doi: 10.1016/j.smim.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, McCann F, Gordan J, Wu X, Raab M, Malik T, Davis D, Rudd C. ADAP-SLP-76 binding differentially regulates supramolecular activation cluster (SMAC) formation relative to T cell-APC conjugation. J. Exp. Med. 2004;200:1063–1074. doi: 10.1084/jem.20040780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chau L, Bluestone J, Madrenas J. Dissociation of intracellular signaling pathways in response to partial agonist ligands of the T cell receptor. J. Exp. Med. 1998;187:1699–1709. doi: 10.1084/jem.187.10.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gil D, Schamel W, Montoya M, Sanchez-Madrid F, Alarcon B. Recruitment of Nck by CD3 epsilon reveals a ligand-induced conformational change essential for T cell receptor signaling and synapse formation. Cell. 2002;109:901–912. doi: 10.1016/s0092-8674(02)00799-7. [DOI] [PubMed] [Google Scholar]
- 14.Gil D, Schrum A, Alarcon B, Palmer E. T cell receptor engagement by peptide-MHC ligands induces a conformational change in the CD3 complex of thymocytes. J Exp Med. 2005;201:517–522. doi: 10.1084/jem.20042036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lehmann J, Riethmuller G, Johnson J. Nck, a melanoma cDNA encoding a cytoplasmic protein consisting of the src homology unist SH2 and SH3. Nucl. Acids Res. 1990;18:1048–1056. doi: 10.1093/nar/18.4.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park D. Cloning, sequencing and overexpression of SH2/SH3 adaptor protein Nck from mouse thymus. Mol. Cell. 1997;7:231–239. [PubMed] [Google Scholar]
- 17.Gomez T, Hamann M, McCarney S, Savoy D, Lubking C, Heldebrant M, Labno C, McKean D, McNiven M, Burkhardt J, Billadeau DD. Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat. Immunol. 2005;6:261–270. doi: 10.1038/ni1168. [DOI] [PubMed] [Google Scholar]
- 18.Snapper S, Rosen F, Mizoguchi E, Cohen P, Khan W, Liu C, Hagemann T, Kwan S, Ferrini R, Davidson L, Bhan AK, Alt FW. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity. 1998;9:81–91. doi: 10.1016/s1074-7613(00)80590-7. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Shehabeldin A, Cruz L, Butler J, Somani A, McGavin M, Kozieradski I, Santos A, Nagy A, Grinstein S, Penninger JM, Siminovitch KA. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiscott-Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 1999;190:1329–1342. doi: 10.1084/jem.190.9.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szymczak A, Workman C, Gil D, Dilioglou S, Vignali K, Palmer E, Vignali D. The CD3e proline-rich sequence, and its interaction with Nck, is not required for T cell development and function. J Immunol. 2005;175:270–275. doi: 10.4049/jimmunol.175.1.270. [DOI] [PubMed] [Google Scholar]
- 21.Barda-Saad M, Braiman A, Titerence R, Bunnell S, Barr V, Samelson L. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat Immunol. 2005;6:80–89. doi: 10.1038/ni1143. [DOI] [PubMed] [Google Scholar]
- 22.Amrani A, Serra P, Yamanouchi J, Trudeau J, Tan R, Elliott J, Santamaria P. Expansion of the antigenic repertoire of a single T cell receptor upon T cell activation. J Immunol. 2001;167:655–666. doi: 10.4049/jimmunol.167.2.655. [DOI] [PubMed] [Google Scholar]
- 23.Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med. 1997;1186:11663–11676. doi: 10.1084/jem.186.10.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang B, Wang N, Whitehurst C, She J, Chen J, Terhorst C. T lymphocyte development in the absence of CD3e or CD3gqze. J. Immunol. 1999;162:88–97. [PubMed] [Google Scholar]
- 25.Han B, Serra P, Yamanouchi J, Amrani A, Elliott J, Dickie P, Dilorenzo T, Santamaria P. Developmental control of CD8 T cell-avidity maturation in autoimmune diabetes. J Clin Invest. 2005;115:1879–1887. doi: 10.1172/JCI24219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serra P, Amrani A, Yamanouchi J, Han B, Thiessen S, Utsugi T, Verdaguer J, Santamaria P. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity. 2003;19:877–889. doi: 10.1016/s1074-7613(03)00327-3. [DOI] [PubMed] [Google Scholar]
- 27.Anderson B, Park B, Verdaguer J, Amrani A, Santamaria P. Prevalent CD8+ T cell response against one peptide/MHC complex in autoimmune diabetes. Proc Natl Acad Sci USA. 1999;96:9311–9316. doi: 10.1073/pnas.96.16.9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosette C, Werlen G, Daniels M, Holman P, Alam S, Travers P, Gascoigne N, Palmer E, Jameson S. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15:59–70. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 29.Fuller C, Braciale V, Samelson L. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol Rev. 2003;191:220–236. doi: 10.1034/j.1600-065x.2003.00004.x. [DOI] [PubMed] [Google Scholar]
- 30.Sommers CL, Dejarnette JB, Huang K, Lee J, El-Khoury D, Shores EW, Love PE. Function of CD3 epsilon-mediated signals in T cell development. J Exp Med. 2000;192:913–919. doi: 10.1084/jem.192.6.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitcher L, van Oers N. T-cell receptor signal transmission: who gives an ITAM? Trends Immunol. 2003;24:554–560. doi: 10.1016/j.it.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Minget S, Swamy M, Alarcon B, Luescher I, Schamel W. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity. 2007;26:1–12. doi: 10.1016/j.immuni.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 33.Risueno R, Gil D, Fernandez E, Sanchez-Madrid F, Alarcon B. Ligand-induced conformational change in the T-cell receptor associated with productive immune synapses. Blood. 2005;106:601–608. doi: 10.1182/blood-2004-12-4763. [DOI] [PubMed] [Google Scholar]
- 34.Lysechko TL, Ostergaard HL. Differential Src Family Kinase Activity Requirements for CD3 Phosphorylation/ZAP70 Recruitment and CD3 Phosphorylation. J. Immunol. 2005;174:7807–7814. doi: 10.4049/jimmunol.174.12.7807. [DOI] [PubMed] [Google Scholar]
- 35.Kesti T, Ruppelt A, Wang J-H, Liss M, Wagner R, Tasken K, Saksela K. Reciprocal regulation of SH3 and SH2 domain binding via tyrosine phosphorylation of a common site in CD3e. J. Immunol. 2007;179:878–885. doi: 10.4049/jimmunol.179.2.878. [DOI] [PubMed] [Google Scholar]
- 36.Aivazian D, Stern L. Phosphorylation of T cell receptor ς is regulated by a lipid dependent folding transition. Nat. Struct. Biol. 2000;7:1023–1026. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 37.Zeng R, Cannon JL, Abraham RT, Way M, Billadeau DD, BudeckWardenberg J, Burkhardt JK. SLP-76 coordinates Nck-dependent Wiskott-Aldrich syndrome protein recruitment with vav-1/Cdc42-dependent Wiskott-Aldrich syndrome protein activation at the T cell-APC contact site. J. Immunol. 2003;171:1360–1368. doi: 10.4049/jimmunol.171.3.1360. [DOI] [PubMed] [Google Scholar]
- 38.Griffiths E, Penninger J. Communication between the TCR and integrins: role of the molecular adapter ADAP/Fyb/Slap. Curr. Opin. Immunol. 2002;14:317–322. doi: 10.1016/s0952-7915(02)00334-5. [DOI] [PubMed] [Google Scholar]
- 39.Krause M, Sechi AS, Konradt M, Monner D, Gertler FB, Wehland J. Fyn-binding protein (Fyb)/SLP-76-associated protein (SLAP), ena/vasodilator-stimulated phosphoprotein (VASP) proteins and the Arp2/3 complex link T cell receptor (TCR) signaling to the actin cytoskeleton. J. Cell. Biol. 2000;149:181–194. doi: 10.1083/jcb.149.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gelkop S, Isakov N. T cell stimulation stimulates the association of enzymatically-active tyrosine-phosphorylated ZAP-70 with the Crk adapter proteins. J. Biol. Chem. 1999;274:21519–21527. doi: 10.1074/jbc.274.31.21519. [DOI] [PubMed] [Google Scholar]
- 41.Sasahara Y, Rachid R, Byrne MJ, de la Fuente MA, Abraham RT, Ramesh N, Geha RS. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol. Cell. 2002;10:1269–1281. doi: 10.1016/s1097-2765(02)00728-1. [DOI] [PubMed] [Google Scholar]
- 42.Wunderlich L, Farago A, Downward J, Buday L. Association of Nck with tyrosine-phosphorylated SLP-76 in activated T lymphocytes. Eur. J. Immunol. 1999;29:1068–1077. doi: 10.1002/(SICI)1521-4141(199904)29:04<1068::AID-IMMU1068>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 43.Bezman N, Koretzky G. Compartmentalization of ITAM and integrin signaling by adapter molecules. Immunol. Rev. 2007;218:9–28. doi: 10.1111/j.1600-065X.2007.00541.x. [DOI] [PubMed] [Google Scholar]
- 44.Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin M, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of ZAP-70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- 45.Varma R, Campi G, Yokosuka T, Saito T, Dustin M. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gunzer M, Schafer A, Borgmann S, Grabbe S, Zanker K, Brocker E, Kampgen E, Friedl P. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived, and sequential. Immunity. 2000;13:323–332. doi: 10.1016/s1074-7613(00)00032-7. [DOI] [PubMed] [Google Scholar]
- 47.Sims T, Soos T, Xenias H, Dubin-Thaler B, Hofman J, Waite J, Cameron T, Thomas V, Varma R, WIggins C, Sheetz M, Littman D, Dustin M. Opposing effects of PKCθ and WASp on symmetry breaking and relocation of the immunological synapse. Cell. 2007;129:773–785. doi: 10.1016/j.cell.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 48.Kersh E, Kaech S, Onami T, Moran M, Wherry E, Miceli M, Ahmed R. TCR signal transduction in antigen-specific memory CD8 T cells. J Immunol. 2003;170:5455–5463. doi: 10.4049/jimmunol.170.11.5455. [DOI] [PubMed] [Google Scholar]