

Abstract
Mitochondrial biogenesis protects metabolism from mitochondrial dysfunction produced by activation of innate immunity by lipopolysaccharide (LPS) or other bacterial products. Here we tested the hypothesis in mouse heart that activation of toll-like receptor-4 (TLR4), which induces early-phase genes that damage mitochondria, also activates mitochondrial biogenesis through induction of nitric oxide synthase (NOS2). We compared three strains of mice: wild type (Wt) C57BL/6J, TLR4−/−, and NOS2−/− for cardiac mitochondrial damage and mitochondrial biogenesis by real-time RT-PCR, Western analysis, immunochemistry, and isoform analysis of myosin heavy chain (MHC) after sub-lethal heat-killed E. coli (HkEC). After HkEC, Wt mice displayed significant myocardial mtDNA depletion along with enhanced TLR4 and NOS2 gene and protein expression that normalized in 72h. HkEC generated less cytokine stress in TLR4−/− and NOS2−/− than Wt mice, and NOS2−/− mice had mtDNA damage comparable to Wt, both knockout strains failed to restore mtDNA copy number because of mitochondrial transcriptosome dysfunction. Wt mice also showed the largest β-MHC isoform switch, but MHC recovery lagged in the NOS2−/− and TLR4−/− strains. The NOS2−/− mice also unexpectedly revealed the co-dependency of TLR4 expression on NOS2. These findings demonstrate the decisive participation of NOS2 induction by TLR4 in optimization of mitochondrial biogenesis and MHC expression after gram negative challenge.
Keywords: cardiomyocytes, lipopolysaccharide, mitochondrial biogenesis, nitric oxide, NOS II, TLR4, sepsis
Introduction
Cardiovascular collapse and microvascular deregulation are two major consequences of severe sepsis that lead to shock and poor tissue perfusion. Even after resuscitation, nearly half of patients with severe sepsis die from multiple organ dysfunction syndrome (MODS), the most common cause of death in the critically ill [1]. Earlier studies have implicated mitochondrial dysfunction in the pathogenesis of organ damage and failure in sepsis by mechanisms that are not yet well understood [2].
The innate immune response provides the initial defense against sepsis; for instance, gram-negative microbes activate Toll-like receptor-4 (TLR4) by recognition of lipopolysaccharide (LPS). LPS binds TLR4 in conjunction with LPS-binding protein and CD14 to activate nuclear factor-κB (NF-κB) and major pro-inflammatory pathways [3–6]. The early phase, mediated by Toll-IR-1R (TIR) and the MyD88 adaptor protein, promotes gene expression for inflammatory cytokines such as TNF-α and IL-1β, as well as the inducible nitric oxide synthase (NOS2) [6, 7]. These events generate reactive oxygen and nitrogen species (ROS, RNS) which inhibit respiration, damage mitochondrial DNA (mtDNA), and decrease mtDNA copy number [8–10]. The early phase of sepsis is also associated with cardiac depression, thus implicating TLR4 in the cardiovascular dysfunction [11, 12]. Affirmatively, TLR4-deficient mice are protected from mitochondrial dysfunction and cardiomyocyte impairment [13] and LPS-induced shock [14].
To counteract microbial challenges, multiple host genes are induced that are needed to maintain oxidative phosphorylation, mitochondrial DNA transcription and replication, and mitochondrial biogenesis. Under these conditions, compensatory mitochondrial biogenesis is driven in part by TLR4-dependent ROS production, consistent with the antipodal effects of oxidative stress on mitochondria [15]. Thus, mitochondrial ROS production is not only a cause of oxidative damage, but a signal to activate mitochondrial biogenesis. The molecular program is regulated at the transcriptional level by the nuclear respiratory factors (NRF-1 and NRF-2), the PGC-1 family of co-activators, and the mitochondrial transcription factor, Tfam [16, 17]. Its failure is presumed to lead to cell dysfunction and delayed organ recovery due to mtDNA damage and disturbances in oxidative phosphorylation [18, 19].
The main source of NO in sepsis is NOS2, and NO overproduction is known to depress mitochondrial and cardiac function, may impair cardiomyocyte survival, and has been linked to heart failure and poor clinical outcomes [20–22]. However, NOS2 also confers protective anti-apoptotic effects [23]; for instance, NOS2 induction by TLR4 is required for functional rescue and survival of cardiomyocytes after LPS [24]. In other settings, NOS3 and NOS1 protect the cell by initiating mitochondrial biogenesis by activation of guanylate cyclase and cGMP-dependent PGC-1α expression [25–29]. However, no comparable role for NOS2 in cardiac protection has been identified.
Because LPS stimulates NO production via TLR4 activation, we tested the hypothesis that TLR4 and NOS2 interact to provide cardiac mitochondrial protection in mice challenged with E. coli. We expected that TLR4-dependent NOS2 up regulation would improve the expression of critical proteins required for transcriptional regulation of mitochondrial biogenesis, including NRF-1, PGC-1α, and Tfam. Furthermore, we hypothesized that the mitochondrial response to TLR4 and NOS2 stimulation would be integrated functionally with isoform switching in the myosin heavy chain (MHC). To investigate these ideas, we evaluated cardiac mitochondrial damage and quantified mRNA and proteins involved in regulating mitochondrial biogenesis in wild type and TLR4- and NOS2-deficient mice before and after acute systemic administration of heat-killed E. coli (HkEC).
Materials and Methods
Animal studies
TLR4−/− mice were generously provided by Dr. S. Akira of Osaka University [30, 31] then speed congenically backcrossed onto a C57BL/6J background. NOS2−/− and Wt C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and the NOS2 mice bred at our institution. For immunofluorescence studies, we used transgenic reporter mice that express green fluorescent protein (GFP) exclusively in mitochondria (mtGFP-tg), a gift from Hiroshi Shitara and Hiromichi Yonekawa of Tokyo Metropolitan Institute of Medical Science [32]. Mice were kept in pathogen-free housing on α-dry bedding and studies were conducted with 20–25 g males on approved protocols that conform to the NIH Guide for the Care and Use of Laboratory Animals.
Live bacteria were prepared from lyophilized E. coli (serotype 086a:K61, American Type Tissue Culture Collection, Rockville, MD) as described [33], carefully heat-inactivated to avoid disruption, and stored at −80°C until use. These inactivated bacteria (HkEC) were thawed once and diluted with sterile 0.9% NaCl to a concentration of 1 × 107/ml and 0.5 ml administered by single intraperitoneal injection to groups of 3 to 6 mice per time point. Preliminary studies showed that this sub-lethal dose of bacteria produces minimal depression of peak respiration in isolated cardiac mitochondria and is below the threshold for cardiomyocyte necrosis in these mouse strains.
Cardiac harvest
Mice injected with HkEC were killed under anesthesia at 0, 6, 24, 48, and 72 hours and the hearts collected immediately. The hearts were flash frozen in liquid nitrogen for all mRNA and protein analysis and stored at −80°C until processed. Hearts for immunofluorescence studies were fresh-fixed in 10% formalin for 24 hours and then stored in 70% ethanol and phosphate buffered saline until processed as described below.
Real-time PCR and gene expression
Total cardiac RNA was isolated from tissue using Trizol Reagent (Invitrogen, Carlsbad, CA) and cDNA synthesized using the SUPERSCRIPT Choice System Kit (Invitrogen). Mouse specific primers and probes [Table 1] were designed using Primer Express (Applied Biosystems, Branchburg, NJ). Reactions to quantify per sample levels of TNF-α, IL-6, IL-1β, ICAM-1, NOS2, ND1, and COX1 were carried out on a 7700 Sequence Detector System (Applied Biosystems) as described [10]. β-MHC and TLR-4 mRNA expression was determined as described [34] using gene-specific primer pairs [Table 1]. Each sample was assayed in triplicate and mean values reported. The RNA data were normalized to 18S rRNA expression levels.
Table 1.
Gene Primers and Probe Sequences
| TNF-α | FW: ACGGCATGGATCTCAAAGAC RV: GTGGGTGAGGAGCACGTAGT Probe: FAM-5′-CTCTTCAAGGGACAAGGCTG-3′-MGB |
| IL-6 | FW: CCGGAGAGGAGACTTCACAG RV: TCCACGATTTCCCAGAGAAC Probe: FAM-5′-ACCACTTCACAAGTCGGAGG-3′-MGB |
| Cyt b | Cyt b-s: ACCCTAGTCGAATGAAT Cyt b-as: TCTGAGTTTAATCCTGT |
| WT mtDNA | Mt1F: AGGGATCCCACTGCACATAG Mt2R: CTCCTCATGCCCCTATGAAA Probe: FAM-5-TTTAATTCAAATTTACCCGCTACTCAACTCTACTATCATTTTAA-3′-MGB |
| Deleted mtDNA | Dl1F: TGGCCTACACCCAGAAGATT Dl2R: TGTTTTCTTAGGGCTTTGAAGG Probe: VIC-5′-TCATGACCAATGAACACTCTGACCCAACTAATTAC-3′-TAMRA |
| TLR-4 | FW: ACCTGGCTGGTTTACACGTC RV: CTGCCAGAGACATTGCAGAA |
| NOS2 | FW: CACCTTGGAGTTCACCCAGT RV: ACCACTCGTACTTGGGATGC |
| ND1 | FW: CACCCCCTTATCAACCTCAA RV: ATTTGTTTCTGCGAGGGTTG |
| COX I | FW: GGAGCAGTATTCGCCATCAT RV: GAGCACTTCTCGTTTTGATGC |
| 18S rRNA | FW: TCAATCTCGGGTGGCTGAACG RV: GGACCAGAGCGAAAGCATTTG |
Mitochondrial isolation, respiration, and DNA copy number
Intact mitochondria were isolated from fresh hearts using sucrose density centrifugation [16]. Respiration was measured at 35°C in water-jacketed cuvettes with calibrated polarographic oxygen electrodes (Diamond General, Ann Arbor, MI) using 0.5 mM ADP and either succinate (5mM) or malate + glutamate (2.5 mM each) as substrates. MtDNA was isolated from intact mitochondria [15, 34] and the copy number quantified using real-time PCR for cytochrome b (cyt b) as described [10]. Samples were analyzed in triplicate and the mtDNA copy number per nanogram total cellular DNA was reported by logarithmic expression relative to known DNA standards.
Western blot analysis
To prepare cardiac tissue for Western blotting, hearts were divided longitudinally so that atrial and ventricular tissues were represented in each sample. Proteins were separated by SDS-PAGE and transferred to PVDF membranes for Western analysis [16, 34]. Membranes were incubated with validated polyclonal or monoclonal antibodies against PGC-1α, NRF1, Tfam, Pol-γ, CD68, Akt, pAkt, AMPK, and p-AMPK (Santa Cruz Biotechnology, Santa Cruz, CA). Anti-tubulin or β-actin (1:1000; Sigma) was used as a loading control. After three washes in TBST, membranes were incubated in HRP-conjugated goat anti-rabbit or anti-mouse IgG (Amersham) at 1:2000 or 1:5000 dilutions, respectively. Blots were developed with ECL and proteins quantified by densitometry of digitized images from the mid-dynamic range and expressed relative to β-actin or tubulin.
Immunofluorescence microscopy
Four-micron, formalin-fixed, paraffin-embedded cardiac sections were processed for immunostaining as described [35]. Anti-TLR4 (StressGen) and anti-NOS2 (Upstate) primary antisera were used at a working dilution of 1:100. Anti-CD68 antibody (Santa Cruz) was used at 1:50 dilution. Conventional fluorescence images were obtained using a Nikon Microphot-FXA fluorescence microscope. MtGFP-tg was visualized in green fluorescence and all other proteins in red fluorescence.
MHC isoform analyses
Samples of the heart were prepared as described by Suliman et al [35]. MHC isoforms were separated via SDS-PAGE using 4% acrylamide-bis stacking gels and 8% separating gels. Gels were scanned using QuantityOne Software (Bio-Rad).
Statistics
Group values are expressed as means ± SD. The n values refer to independent samples. Data analyses were performed using commercial software and Student’s unpaired t-test or ANOVA followed by Fisher’s post hoc exact test. Regression analyses were performed using Statview (SAS, Version 5.0.1). A P < 0.05 was considered significant.
Results
TLR4 and NOS2-dependent cardiac cytokine responses to HkEC
The early induction of IL-1β, IL-6, ICAM-1 and TNF-α after HKEC in Wt mice was robust, but detectable changes in cytokine and chemokine expression had dissipated by 72 h. These effects were abrogated in TLR-4 −/−mice, and in the iNOS−/− mice, the induction of IL-1β, IL-6, ICAM-1 and TNF-α was delayed and dampened compared with Wt mice but statistically significant (Fig. 1). In TLR4−/− mice, the early (6 h) TNF-α response was blocked, but a minor late TNF-α response was seen that was not present in Wt (Fig. 1A). This suggested that HkEC activated more than one cardiac cytokine pathway, of which only the earliest is TLR4–dependent.
Figure 1. Cytokine expression in Wt, TLR4−/− and NOS2−/− mice after HkEC.

Hearts of Wt, TLR4−/−, and NOS2−/− mice were collected at 0, 6, 24, and 72 hours after HkEC. Real-time RT-PCR was performed to determine mRNA levels of A: IL-1β, B: IL-6, C: ICAM-1, and D: TNF-α in all three strains. †Value statistically different from Wt at 0h. *Value is statistically different from Wt 0h and other strains at same time (n = 3–5 mice per point).
Mitochondrial damage and recovery after HkEC challenge
Because contractile cardiac myocytes require ATP continuously, even moderate mitochondrial damage adversely affects cardiac performance. Real-time PCR confirmed that E. coli challenge induces mtDNA damage and decreases mtDNA copy number in the murine heart [16]. In Wt mice, mtDNA copy number fell significantly 24h after HkEC exposure but recovered fully by 72h (Fig. 2A). TLR4−/− mice, consistent with their limited cytokine response, demonstrated no significant change in mtDNA copy number until 72h, suggesting the late mtDNA damage is TLR4-independent. Conversely, NOS2−/− mice showed a fall in mtDNA copy number comparable to Wt but failed to recover at 72 h. This implies that NOS2 induction, although not crucial to mtDNA damage, is critical to restore mtDNA copy number in sepsis.
Figure 2. Analysis of mtDNA and mitochondrial gene expression after HkEC.

The recovery of mtDNA damage is impaired in TLR4- and NOS2-deficient strains. Real-time RT-PCR was performed in Wt, TLR4−/−, and NOS2−/− mice 0, 6, 24, and 72 hours after HkEC exposure. A: MtDNA copy number was determined by amplifying cytochrome b and the results were plotted logarithmically. Mitochondrial mRNA of B: COX1 and C: ND1 were quantified and plotted relative to 18S control. †Value is statistically different from 0h Wt. *Value is statistically different from 0h Wt and the other strains at same times (n = 3–5 mice per point). D: State 3 respiration in isolated cardiac mitochondria after HkEC challenge in Wt and NOS2-deficient mice. Peak respiration is depressed in both strains by 24h and recovers by 72h. The Western blot indicates the amount of outer membrane protein, porin, in the mitochondrial samples. †Value is statistically different from 0h Wt or NOS2−/− respectively.
Changes in mtDNA function were evaluated by real-time RT-PCR of mRNA expression for two mtDNA-encoded components of the electron transport chain, COX1 (Complex IV) and ND1 (Complex I). In Wt mice, exposure to HkEC up regulated COX1 and ND1 gene expression in both the mtDNA damage and the recovery phases (Fig. 2B, C). COX1 and ND1 mRNA expression continued to rise in Wt mice for 72h but did not increase significantly in TLR4−/− or NOS2−/− mice. This reflected the patterns of mtDNA damage, as mtDNA transcription is necessary to compensate for loss of mtDNA-encoded proteins. These data imply that cardiac mtDNA transcription is similarly impaired in TLR4-and NOS-deficient mice. Because TLR4-dependent NOS2 expression is involved in up-regulation of mitochondrial transcriptosome proteins (see below), we measured State 3 respiration in isolated cardiac mitochondria from Wt and NOS2−/− mice and noted comparable functional loss and reversibility after E. coli challenge (Fig. 2D); however recovery of respiration in the NOS2−/− mice occurs at a lower mtDNA copy number, indicating the mice still had the reserve to recover from a single limited insult.
TLR4-dependent NOS2 up-regulation supports mitochondrial transcriptosome expression in the heart
To understand how TLR4-dependent NOS2 up-regulation activates cardiac mitochondrial biogenesis we measured two regulatory proteins of the mitochondrial transcriptosome in the hearts of TLR4−/− and NOS2−/− mice. Figure 3A indicates that HkEC induces the accumulation of mitochondrial Tfam and Pol-γ protein by 24h in Wt but not TLR4−/− or NOS2−/−mice. These responses preceded recovery of mtDNA copy number in Wt mice at 72h (Fig. 2A). Moreover, nuclear protein for the key regulators of mitochondrial biogenesis, NRF-1 and the PGC-1α co-activator also increased significantly by 24h in Wt but not TLR4−/− or NOS2−/− mice (Fig. 3B).
Figure 3. Western analysis of proteins of mitochondrial biogenesis.

A. Mitochondrial protein content in Wt, TLR4- and NOS2-deficient mice after HkEC administration. Western blots of cardiac mitochondrial protein were performed using antibodies to Tfam and DNA polymerase-γ compared with porin. B. Western blots of nuclear NRF-1 and PGC-1α protein were compared to tubulin control. Representative blots are shown and densitometry is provided from samples of 3–5 mice per time point for each strain (* indicates P<0.05 compared with time 0 and with the other two strains).
Among the upstream regulators of NRF-1 and PGC-1α are two pro-survival kinases, Akt and AMPK. Increased cardiac AMPK and Akt activity in Wt mice 6h after HkEC was demonstrated by increases in phospho- to total protein by Western blot (data not shown); however, both TLR4−/−and NOS2−/− mice failed to activate these kinases significantly after HkEC challenge. Thus, NOS2 is integral to activation of important downstream pro-survival kinases for mitochondrial biogenesis after TLR4 activation.
Cardiac TLR-4 protein expression after HkEC-induced mitochondrial damage
To localize TLR4 expression in cardiac tissue and evaluate its association with loss of mitochondrial mass after HkEC, we used COX8 mitochondrial-targeted GFP transgenic reporter mice (green fluorescence) to visualize cardiac mitochondria and then stained the sections with anti-TLR4 polyclonal antibody (red fluorescence) as shown in Figure 4A. These reporter mice show a loss of cardiomyocyte mitochondrial mass at 6 and 24h after HkEC challenge which recovers by 72h (Fig. 4A, a–e). The GFP reporter is constitutively imported into mitochondria on a leader sequence for COX8. Because the mitochondria are green at the time of damage, and mitochondrial turnover is normally slow, the loss of GFP signal in 6 to 24 h reflects the damage and its clearance (mitophagy). And provided the myocyte nucleus is transcriptionally-competent and the mRNA translated, GFP signal recovery is a measure of effective mitochondrial biogenesis, including protein importation.
Figure 4. Immunofluorescence for COX8, TLR4, and NOS2 in mitochondrial reporter mice following HkEC exposure.
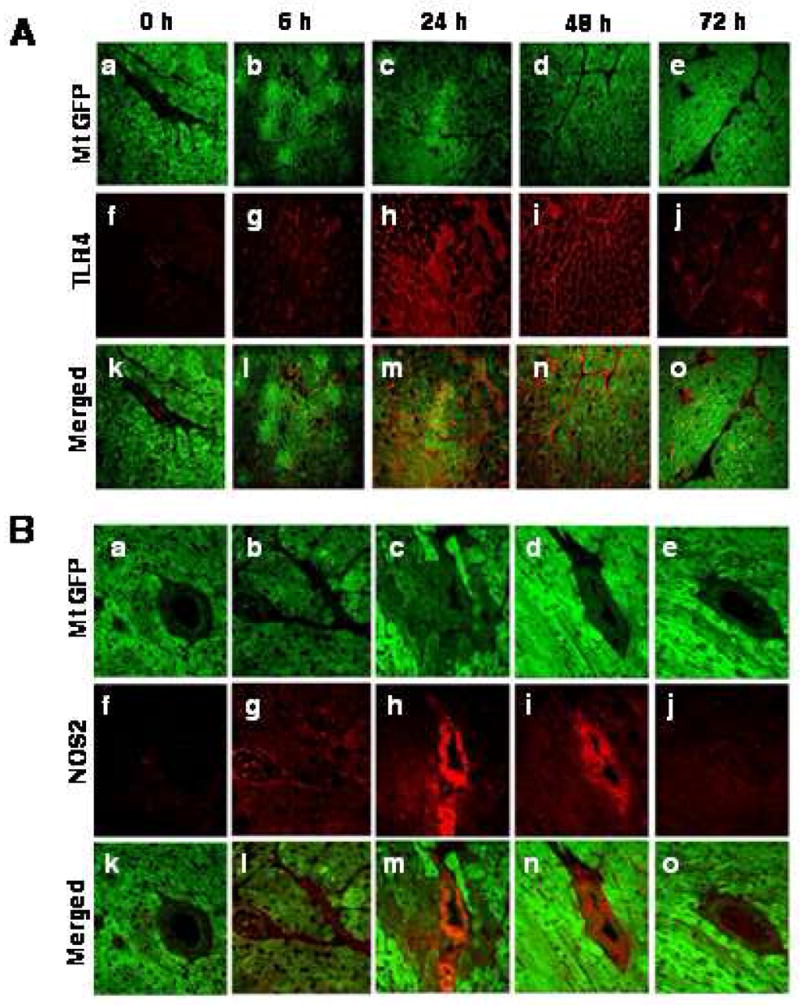
A: Cardiac tissue was harvested, fixed, sliced, and visualized for a–e: COX8 (green fluorescence), stained for f–j: TLR4 (red fluorescence), and superimposed in panels k–o. B: Tissue from transgenic mice was also prepared and stained for a–e: COX8 and f–j: NOS2, which are superimposed in panels k–o.
In the control hearts, TLR4 staining is seen at low intensity in vascular endothelium but not in deep myocardium (Fig. 4A, f). Six hours after HkEC administration, TLR4 localizes to the plasma membranes of mononuclear cells and the endothelial cells lining intramyocardial vessels (Fig. 4A, g). By 24 hours, TLR4 expression is extensive and observed primarily at the sarcolemma (Fig. 4A, h). Furthermore, scattered foci of intense TLR4 staining are noted in contiguous cardiomyocytes, which is absent in sections from control hearts. Not shown is that this focal staining is inhibited by pre-incubation with the hTLR4-peptide and not observed with control antiserum. In the reporter mice, the decrease in mitochondrial fluorescence at 6h (Fig. 4A, b) is accompanied by enhanced TLR4 fluorescence (Fig. 4A, h), which associates mitochondrial damage with TLR4 expression. Subsequently however, TLR4 is further up regulated as mitochondrial mass is restored (Fig. 4A, d–e), connecting TLR4 expression temporally with mitochondrial biogenesis after gram-negative challenge.
The NOS2 expression pattern in the mitochondrial reporter mice was also checked, and NOS2 was absent or minimally detectable in the vascular walls of controls (Fig. 4B, f). After HkEC, NOS2 expression substantially increased primarily in the vascular wall—endothelial and smooth muscle cells, peaking at 24h and reverting to baseline by 72h (Fig. 4B, f–j). Less prominent but clearly present NOS2 was found in the sarcoplasm at 6, 24 and 48 h. These increases in NOS2 corresponded temporally to the changes in TLR4 expression. No appreciable influx of activated macrophages was found to explain the NOS2 signal in Wt hearts by Western blotting for CD68 (data not shown).
NOS2 promotes cardiac TLR4 expression after HkEC-treatment
To determine if TLR4 up regulation requires NOS2, we measured TLR4 gene expression by semi-quantitative RT-PCR in Wt and NOS2−/− mice. In Wt mice, TLR4 mRNA increased within 6h of HkEC, peaked by 24h, and had returned toward baseline by 72h (Fig. 5A). However, NOS2−/− mice exhibited delayed TLR4 expression, suggesting that NO produced by vascular NOS2 induction is involved in the rapid and robust up regulation of TLR4 by HkEC challenge.
Figure 5. Transcriptional co-dependence of TLR4 and NOS2 mRNA after gram-negative challenge.

A: Conventional RT-PCR was performed for TLR4 and 18S in wild type and NOS2−/−mice. B: NOS2 gene expression relative to 18S was quantified by Real-time RT-PCR in Wt, TLR4−/−, and NOS2−/− mice after HkEC injection. †Value is statistically different from 0h Wt. *Value is statistically different from 0h Wt and other strains at same time.
To correlate NOS2 gene expression with mitochondrial biogenesis, we compared NOS2 mRNA levels in the mice by real-time RT-PCR. In Wt mice, cardiac NOS2 mRNA increased almost three-fold by 6h and seven-fold by 24h after HkEC (Fig. 5B). Conversely, NOS2 mRNA level remained at basal levels in the TLR4−/− mice and was expectedly undetectable in the NOS2−/− strain. A lack of NOS2 induction in TLR4−/− mice confirms regulation by TLR4, ostensibly via MyD88 and NF-κB.
HkEC increases β-MHC mRNA in the heart
The fetal-neonatal genes encoding β-MHC are pathological markers for functional myocardial damage [36]. To establish a MHC isoform switch, adult mice from the three strains were treated with HkEC and tissues processed for semi-quantitative RT-PCR or silver staining. In Wt mice, β-MHC mRNA content increased significantly 24h after HkEC administration (P<0.02, Fig. 6A). By comparison, the β-MHC response in NOS2−/− and TLR4−/−mice was diminished relative to Wt, but both knockout strains maintained a significant isoform switch (P<0.05), even 24h after HkEC (Fig. 6A). The changes in mRNA levels reflected MHC protein expression, as shown in Fig. 6B. In Wt hearts, β-MHC protein was strongly up regulated 24h after HkEC and returned to normal by 72h. In contrast, NOS2−/− and TLR4−/− mice showed less prominent increases in β-MHC at 24h that persisted after 72h. However, minimal variations in α-MHC expression were found in these mice after they had received HkEC (Fig. 6B). This implies that although TLR4-dependent NOS2 expression contributes to cardiac dysfunction in sepsis, NOS2 is essential to avoid compromising the energy balance.
Figure 6. β-MHC isoform expression in cardiac tissue of Wt, TLR4−/− and NOS2−/− mice after HkEC exposure.

Hearts from Wt, TLR4−/−, and NOS2−/− mice were obtained following HkEC exposure. A: Samples processed by conventional RT-PCR to amplify β-MHC mRNA relative to 18S. B: Protein expression by silver staining for α-MHC and β-MHC relative to tubulin.
Discussion
The pathogenesis of cardiac damage by activation of the TLR4-dependent host inflammatory response involves NF-κB-dependent release of early-phase cardio-depressive cytokines and excessive production of cellular ROS and RNS [11]. NOS2 is rapidly up-regulated, yet there is limited evidence that it protects the heart; instead nitrosative damage is thought to impair oxidative phosphorylation and reduce contractile function [11, 37]. Although NO is known to help regulate energy capacity through mitochondrial biogenesis, which is vital to avoid apoptosis and necrosis, the impact of inflammatory NOS2 induction has been unknown. Previously, it was known only that loss of TLR4 activation after LPS-mediated inflammation in vivo leads to impaired hepatic mitochondrial biogenesis [10], but no role for NOS2 was detailed.
Using combined molecular and histological approaches in another organ, the heart, this study confirms that TLR4 activation by LPS-bearing organisms contributes both to mitochondrial damage and biogenesis, and extends two important aspects of our understanding of molecular pathogenesis in the host. First, TLR4-dependent NOS2 expression participates in the activation of the nuclear transcription factors required for mitochondrial biogenesis. Second, NOS2 promotes TLR4 expression, and the absence of NOS2 delays the recovery of cardiac mitochondrial mass after mitochondrial damage and prolongs the persistence of the myocyte fetal β form of MHC relative to the wild type.
It is well known that LPS binding to TLR4 activates NF-κB, signaling, which regulates multiple downstream pro-inflammatory pathways, but not that several intersect with mitochondrial biogenesis. The normal heart responds to LPS by increasing pro-survival kinase activity and mRNA and protein expression for nuclear transcription factors (NRF-1, NRF-2), the PGC-1α co-activator, and mitochondrial transcriptosome proteins (Tfam, Pol-γ) [16]. These molecular events help restore mtDNA copy number and mitochondrial gene transcription in a TLR4-dependent manner [10]. Here in normal mice we confirm that gram negative challenge depletes cardiac mtDNA, in part through NOS2 up-regulation, and report that the loss of mtDNA copy number and fall in transcription are quickly reversed by the expression of genes involved in mitochondrial biogenesis, reflected by the timing of restoration of cardiac mitochondrial mass in reporter mice. Since this transient injury model produces little or no cardiac cell death, our interpretation of the reversible fall in the reporter signal is parsimonious. Moreover, the NOS2−/− mouse data provide the first indication that after activation of innate immunity the transcriptional program for mitochondrial biogenesis is optimized by NOS2. Although TLR4 does regulate NOS2 expression via MyD88-dependent NF-κB signaling, we also found that TLR4 is not fully expressed in NOS2-deficient mice after HkEC for reasons not elucidated in this study.
NOS2 over-expression protects serum-deprived cardiomyocytes against apoptosis [24], and protects against cardiac ischemia-reperfusion [38], and the mechanisms appear to involve NOS2 induction by NF-κB [5, 39], as well as TNF-α and certain interleukins [40]. Because LPS cytokine production lags in NOS2-deficient mice, direct inflammatory damage is limited, yet as reported by Zhu et al., TLR4-dependent stress survival in cardiomyocytes does require NOS2 [24]. Moreover, in this study, NOS2-deficient mice challenged with HkEC fail to up-regulate the transcriptional circuitry for mitochondrial biogenesis and mtDNA fails to recover within 72 hours. These observations thus extend and provide an explanation for the idea that NOS2 is essential for survival and functional rescue of cardiac cells from LPS [24, 41], and because TLR4-dependent NOS2 expression requires NF-κB, NOS2 is revealed as a component of the mitochondrial defense against gram-negative microbes in agreement with the participation of NO in mitochondrial biogenesis [28, 29].
The impairment of cardiac contractility by LPS, e.g. as demonstrated by Baumgarten et al., is abrogated by inhibition of TLR4 or NOS2 [42]. Accordingly, our new data link together the detrimental and positive aspects of TLR4-dependent NOS2 expression in vivo because their activities in tandem lead to both mitochondrial dysfunction and mitochondrial biogenesis. Also, the clear and specific shift in MHC, an important marker of heart muscle function directly related to changes in cardiac contractility [43], points logically to a connection between contractility and mitochondrial biogenesis. The fractional α-MHC decline of the failing heart [44], and the HkEC induced shift here to the β-MHC isoform imply deteriorating cardiac performance, which in the TLR4 and NOS2-deficient strains, is reversed only slowly. This result also offers the intriguing possibility that the innate immune response connects mitochondrial biogenesis to the essential expression of contractile proteins, such as titin, myogenin, and desmin, through a specific molecular program.
In summary, TLR4-dependent NOS2 expression makes an important contribution to the apparent heterogeneity of oxidative/nitrosative cardiac mitochondrial pathology after E. coli challenge in vivo in part through its previously unappreciated benefit on the accelerated restoration of lost cardiac mitochondrial mass. NOS2 up-regulation facilitates the expression of the major nuclear and mitochondrial transcription factors required for mtDNA replication. The expression of NOS2 to help reinstate mtDNA copy number and to support mitochondrial biogenesis would avert energy failure and impairment of cardiac contractility. The key implication is that strategies to preserve oxidative phosphorylation and maintain mitochondrial biogenesis could limit cardiac failure as well as help circumvent systemic organ dysfunction in severe sepsis.
Acknowledgments
The authors thank Craig Marshall, Susan Fields, Lynn Tatro, and Martha Salinas for excellent technical assistance.
Research Support: National Institutes of Health R01 AI064789 (CAP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garnacho-Montero J, Garcia-Garmendia JL, Barrero-Almodovar A, Jimenez-Jimenez FJ, Perez-Paredes C, Ortiz-Leyba C. Impact of adequate empirical antibiotic therapy on the outcome of patients admitted to the intensive care unit with sepsis. Critical Care Medicine. 2003;31:2742–2751. doi: 10.1097/01.CCM.0000098031.24329.10. [DOI] [PubMed] [Google Scholar]
- 2.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4:729–741. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Tobias PS, Soldau K, Gegner JA, Mintz D, Ulevitch RJ. Lipopolysaccharide binding protein-mediated complexation of lipopolysaccharide with soluble CD14. J Biol Chem. 1995;270:10482–10488. doi: 10.1074/jbc.270.18.10482. [DOI] [PubMed] [Google Scholar]
- 4.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science (New York, NY) 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 5.Yang H, Young DW, Gusovsky F, Chow JC. Cellular events mediated by lipopolysaccharide-stimulated toll-like receptor 4. MD-2 is required for activation of mitogen-activated protein kinases and Elk-1. J Biol Chem. 2000;275:20861–20866. doi: 10.1074/jbc.M002896200. [DOI] [PubMed] [Google Scholar]
- 6.Muller-Decker K, Manegold G, Butz H, Hinz DE, Huttner D, Richter KH, Tremmel M, Weissflog R, Marks F. Inhibition of cell proliferation by bacterial lipopolysaccharides in TLR4-positive epithelial cells: independence of nitric oxide and cytokine release. J Invest Dermatol. 2005;124:553–561. doi: 10.1111/j.0022-202X.2004.23598.x. [DOI] [PubMed] [Google Scholar]
- 7.Jerala R. Structural biology of the LPS recognition. Int J Med Microbiol. 2007;297:353–363. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Dhalla AK, Hill MF, Singal PK. Role of oxidative stress in transition of hypertrophy to heart failure. Journal of the American College of Cardiology. 1996;28:506–514. doi: 10.1016/0735-1097(96)00140-4. [DOI] [PubMed] [Google Scholar]
- 9.Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. doi: 10.1161/01.res.85.4.357. [DOI] [PubMed] [Google Scholar]
- 10.Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. Faseb J. 2005;19:1531–1533. doi: 10.1096/fj.04-3500fje. [DOI] [PubMed] [Google Scholar]
- 11.Stein B, Frank P, Schmitz W, Scholz H, Thoenes M. Endotoxin and cytokines induce direct cardiodepressive effects in mammalian cardiomyocytes via induction of nitric oxide synthase. J Molecular and Cellular Cardiology. 1996;28:1631–1639. doi: 10.1006/jmcc.1996.0153. [DOI] [PubMed] [Google Scholar]
- 12.Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92:2303–2312. doi: 10.1172/JCI116834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circulation Research. 2004;95:700–707. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 14.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) The Journal of Experimental Medicine. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suliman HB, Carraway MS, Piantadosi CA. Postlipopolysaccharide oxidative damage of mitochondrial DNA. American Journal of Respiratory and Critical Care Medicine. 2003;167:570–579. doi: 10.1164/rccm.200206-518OC. [DOI] [PubMed] [Google Scholar]
- 16.Suliman HB, Welty-Wolf KE, Carraway M, Tatro L, Piantadosi CA. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc Res. 2004;64:279–288. doi: 10.1016/j.cardiores.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Suliman HB, Carraway MS, Welty-Wolf KE, Whorton AR, Piantadosi CA. Lipopolysaccharide stimulates mitochondrial biogenesis via activation of nuclear respiratory factor-1. J Biol Chem. 2003;278:41510–41518. doi: 10.1074/jbc.M304719200. [DOI] [PubMed] [Google Scholar]
- 18.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nature Genetics. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 19.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nature Genetics. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 20.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–223. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 21.Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. doi: 10.1161/hc3201.092284. [DOI] [PubMed] [Google Scholar]
- 22.Ullrich R, Scherrer-Crosbie M, Bloch KD, Ichinose F, Nakajima H, Picard MH, Zapol WM, Quezado ZM. Congenital deficiency of nitric oxide synthase 2 protects against endotoxin-induced myocardial dysfunction in mice. Circulation. 2000;102:1440–1446. doi: 10.1161/01.cir.102.12.1440. [DOI] [PubMed] [Google Scholar]
- 23.Hatano E, Bennett BL, Manning AM, Qian T, Lemasters JJ, Brenner DA. NF-kappaB stimulates inducible nitric oxide synthase to protect mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis. Gastroenterology. 2001;120:1251–1262. doi: 10.1053/gast.2001.23239. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Zhao H, Graveline AR, Buys ES, Schmidt U, Bloch KD, Rosenzweig A, Chao W. MyD88 and NOS2 are essential for toll-like receptor 4-mediated survival effect in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;291:H1900–1909. doi: 10.1152/ajpheart.00112.2006. [DOI] [PubMed] [Google Scholar]
- 25.Clementi E, Nisoli E. Nitric oxide and mitochondrial biogenesis: a key to long-term regulation of cellular metabolism. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:102–110. doi: 10.1016/j.cbpb.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 26.Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015–2024. doi: 10.1523/JNEUROSCI.5654-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leary SC, Shoubridge EA. Mitochondrial biogenesis: which part of “NO” do we understand?[comment] Bioessays. 2003;25:538–541. doi: 10.1002/bies.10298. [DOI] [PubMed] [Google Scholar]
- 28.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide.[see comment] Science (New York, NY) 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 29.Nisoli E, Carruba MO. Nitric oxide and mitochondrial biogenesis. J Cell Sci. 2006;119:2855–2862. doi: 10.1242/jcs.03062. [DOI] [PubMed] [Google Scholar]
- 30.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 31.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 32.Shitara H, Kaneda H, Sato A, Iwasaki K, Hayashi J, Taya C, Yonekawa H. Non-invasive visualization of sperm mitochondria behavior in transgenic mice with introduced green fluorescent protein (GFP) FEBS letters. 2001;500:7–11. doi: 10.1016/s0014-5793(01)02574-1. [DOI] [PubMed] [Google Scholar]
- 33.Welty-Wolf KE, Carraway MS, Huang YC, Simonson SG, Kantrow SP, Piantadosi CA. Bacterial priming increases lung injury in gram-negative sepsis. American Journal of Respiratory and Critical Care Medicine. 1998;158:610–619. doi: 10.1164/ajrccm.158.2.9704064. [DOI] [PubMed] [Google Scholar]
- 34.Suliman HB, Carraway MS, Velsor LW, Day BJ, Ghio AJ, Piantadosi CA. Rapid mtDNA deletion by oxidants in rat liver mitochondria after hemin exposure. Free Radical Biology & Medicine. 2002;32:246–256. doi: 10.1016/s0891-5849(01)00797-3. [DOI] [PubMed] [Google Scholar]
- 35.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730–3741. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abraham WT, Gilbert EM, Lowes BD, Minobe WA, Larrabee P, Roden RL, Dutcher D, Sederberg J, Lindenfeld JA, Wolfel EE, Shakar SF, Ferguson D, Volkman K, Linseman JV, Quaife RA, Robertson AD, Bristow MR. Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Molecular Medicine (Cambridge, Mass. 2002;8:750–760. [PMC free article] [PubMed] [Google Scholar]
- 37.Nemoto S, Vallejo J, Knuefermann P, Misra A, Defreitas G, Carabello B, Mann D. Escherichia coli LPS-induced LV dysfunction: role of toll-like receptor-4 in the adult heart. Am J Physiol Heart Circ Physiol. 2002;282:H2316–H2323. doi: 10.1152/ajpheart.00763.2001. [DOI] [PubMed] [Google Scholar]
- 38.Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL, 2nd, Billiar TR. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase.[see comment] Circulation. 2000;101:2742–2748. doi: 10.1161/01.cir.101.23.2742. [DOI] [PubMed] [Google Scholar]
- 39.Taylor BS, Alarcon LH, Billiar TR. Inducible nitric oxide synthase in the liver: regulation and function. Biochemistry (Mosc) 1998;63:766–781. [PubMed] [Google Scholar]
- 40.Kurose I, Miura S, Higuchi H, Watanabe N, Kamegaya Y, Takaishi M, Tomita K, Fukumura D, Kato S, Ishiii H. Increased nitric oxide synthase activity as a cause of mitochondrial dysfunction in rat hepatocytes: roles for tumor necrosis factor alpha. Hepatology. 1996;24:1185–1192. doi: 10.1002/hep.510240534. [DOI] [PubMed] [Google Scholar]
- 41.Meng X, Ao L, Brown J, Meldrum D, Sheridan B, Cain B, Banerjee A, Harken A. LPS induces late cardiac functional protection against ischemia independent of cardiac and circulating TNF-alpha. Am J Physiol Heart Circ Physiol. 1997;273:H1894–H1902. doi: 10.1152/ajpheart.1997.273.4.H1894. [DOI] [PubMed] [Google Scholar]
- 42.Baumgarten G, Knuefermann P, Schuhmacher G, Vervölgyi V, von Rappard J, Dreiner U, Fink K, Djoufack C, Hoeft A, Grohé C, Knowlton A, Meyer R. Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock. 2006;25:43–49. doi: 10.1097/01.shk.0000196498.57306.a6. [DOI] [PubMed] [Google Scholar]
- 43.Herron T, McDonald K. Small amounts of alphamyosin heavy chain isoform expression significantly increase power output of rat cardiac myocyte fragments. Circ Res. 2002;90:1150–1152. doi: 10.1161/01.res.0000022879.57270.11. [DOI] [PubMed] [Google Scholar]
- 44.Miyata S, Minobe W, Bristow M, Leinwand L. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386–390. doi: 10.1161/01.res.86.4.386. [DOI] [PubMed] [Google Scholar]