

Abstract
Tie-1 is an endothelial specific receptor tyrosine kinase that is upregulated in diseases such as atherosclerosis and rheumatoid arthritis. We recently demonstrated that Tie-1 induced a proinflammatory response when overexpressed in endothelial cells. Here, we used a complementary approach and suppressed endogenous Tie-1 expression in endothelial cells to examine its function by microarray analysis. Tie-1 appeared to govern expression of many genes involved in inflammation. Expression knockdown of Tie-1 significantly reduced endothelial conditioned medium ability to stimulate MCP-1 production in U937 cells. Collectively, our results support the notion that Tie-1 has an inflammatory function in endothelial cells.
Keywords: Tie-1, endothelial inflammation, microarray, U937
1. Introduction
Tie-1 is a receptor tyrosine kinase specifically expressed in endothelial cells. Genetic deletion of Tie-1 induced embryonic lethality with extensive hemorrhage and edema. However, it is believed that Tie-1 is not required for vasculogenesis but essential in maintenance of microvasculature in murine embryonic development [1,2].
Tie-1 may play a role in inflammatory diseases. Endogenous Tie-1 mRNA is upregulated in atherosclerotic lesions from ApoE-null mice [3]. Immunostaining reveals that Tie-1 protein is expressed in endothelial cells in samples of rheumatoid arthritis [4,5]. Jin et al. showed that adenoviral overexpression of a soluble extracellular form of Tie-1 could reduce inflammation and destruction of joints in a collagen-induced arthritis model in mice [6], suggesting that Tie-1 may play a functional role in this disease.
Functional analysis of Tie-1 has been hampered by the fact that a high affinity binding, signaling ligand has not been conclusively identified. We recently showed that overexpressing mouse Tie-1 in human endothelial cells in vitro induced Tie-1 phosphorylation and an inflammatory response [7]. In this report, we used a complementary approach and examined the function of endogenous Tie-1 in endothelial cells by siRNA knockdown and microarray analysis. Our results indicate that suppression of endogenous Tie-1 expression in endothelial cells downregulates multiple proinflammatory genes, supporting the notion that Tie-1 activity induces inflammation in endothelial cells.
2. Materials and methods
2.1. Cell culture
HUVECs and U937 cells were maintained as described [8].
2.2. siRNA knockdown
Three siRNA duplexes against human Tie-1 were purchased from Invitrogen: #1—GGCCUCAGAACUGGAGUUCAACUUA; #2—GGGCCUGUAAGAACCGAGGUUACUU; #3—GCAGCCUCGAAACUGUGACGAUGAA. A non-targeting control stealth siRNA (cat. # 12935-300) was also purchased (Invitrogen). HUVECs (300,000 cells/2ml) were seeded on Matrigel coated (1:500 dilution) 6xwell plates in EBM-2 supplemented with additional VEGF (10 ng/ml, R and D) and EGF (50 ng/ml, Invitrogen). Next day, cells were transfected with 70 nM siRNA in the presence of 0.09% (v/v) Silentfect (Bio-Rad). After overnight incubation, cells were grown in fresh medium for 24 hours and processed for total RNAs using RNeasy kits (Qiagen). Western blots of Tie-1 were performed as described [7].
2.3. Microarray
HUVECs were transfected with either a control siRNA or human-Tie-1 specific siRNA#1 in triplicate. RNA samples were submitted to Cogenic (Morrisville, NC), a service contract company, for microarray analysis. Briefly, total RNA (500ng) was converted into labeled cRNA with nucleotides coupled to fluorescent dye Cy3 using the Low RNA Input Linear Amplification Kit (Agilent Technologies, Palo Alto, CA) following the manufacturer’s protocol. Quality of the samples was verified by using an Agilent Bioanalyzer. Cy3-labeled cRNA (1.65 μg) from each sample was hybridized to an Agilent Human microarray in 4×44K format (whole genome). The hybridized arrays were then washed and scanned, and data were extracted from the scanned images using Feature Extraction version 9.5 (Agilent Technologies). Data were combined into two groups: Tie-1-siRNA-treated samples and control-siRNA-treated samples. A Ratio Experiment Definition (ED) was built using these two groups in Rosetta Resolver. In this ED, the control group was considered the denominator. Using this ratio ED, genes were considered differentially expressed if the log (Ratio) p-value was <0.001 and the absolute fold change was >2.5 fold. This list of genes was adjusted for multiple testing hypothesis using the one-way ANOVA analysis and the Benjamin-Hochberg False Discovery Rate calculation (p-value < 0.01).
2.4. Real-time PCR
Real-time (Taq-man) PCR protocols and primer sequences for VCAM-1, ICAM-1, and E-selectin were reported previously [7]. The following real time PCR probes were purchased from Applied Biosystems:
Tie-1 5′-CTCATGTGGGCGCGGCGGTGGACCT -3′;
CXCL5 5′-AGAAAATTTTGGACGGTGGAAACAA -3′;
Complement 3 5′-GGACAAGAAAGGGATCTGTGTGGCA -3′;
DARC 5′-TCTGGACCTGCTGCTGAACCTGGCA -3′;
C15orf48 5′-GGAAAACCGATGTGATCCTTGATCG -3′;
TNFRSF9 5′-GTCGACCCTGGACAAACTGTTCTTT -3′;
TLR2 5′-GGAGTTCTCCCAGTGTTTGGTGTTG -3′;
GM-CSF 5′-GAAATGTTTGACCTCCAGGAGCCGA -3′;
IL-1 β 5′-ACAGCTGGAGAGTGTAGATCCCAAA -3′;
Tie-2 5′-AGCTTCTATCGGACTCCCTCCTCCA -3′;
eNOS 5′-GGAGAATGGAGAGAGCTTTGCAGCT -3′;
TGF-β 5′-GACATCAACGGGTTCACTACCGGCC -3′.
The primer for a gene of interest was multiplexed with a GAPDH primer for normalization.
2.5. U937 stimulation
U937 (200,000 cells/0.5ml) in 0.5%FBS RPMI (ATCC) was seeded in a 24xwell plate and serum starved overnight. Next morning, cells were stimulated with HUVEC conditioned medium (300μl used per well) for four hours prior to harvesting RNAs.
3. Results and Discussion
3.1. Identification of genes regulated by Tie-1 expression in HUVECs using siRNA and microarray analysis
Previously, we demonstrated that adenoviral-induced overexpression of Tie-1 in human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells induced significant upregulation of ICAM-1, VCAM-1, and E-selectin in vitro [7]. Therefore, Tie-1 may be proinflammatory in endothelial cells. However, due to high efficiency of transgene expression mediated by adenoviral transduction, the consequent Tie-1 expression level was extremely high in the system and may not accurately reflect in vivo expression level. In this study, we sought to address this issue by determining the function of endogenous Tie-1 using siRNA and microarray analysis. Confluent HUVECs were transfected with either a control siRNA duplex or a human-Tie-1-specific siRNA (#1). Treatment with Tie-1 siRNA effective suppressed Tie-1 protein levels (Fig. 1). Total RNAs were collected 48 hours post transfection and submitted for microarray analysis. Genes were considered to be Tie-1 targets if the average absolute fold change was equal to or larger than 2.5. As expected, Tie-1 expression was suppressed significantly (>7 fold) by the siRNA treatment (Table 1). In addition, we identified 56 genes that were downregulated and 34 genes that were upregulated upon Tie-1 knockdown.
Fig. 1.
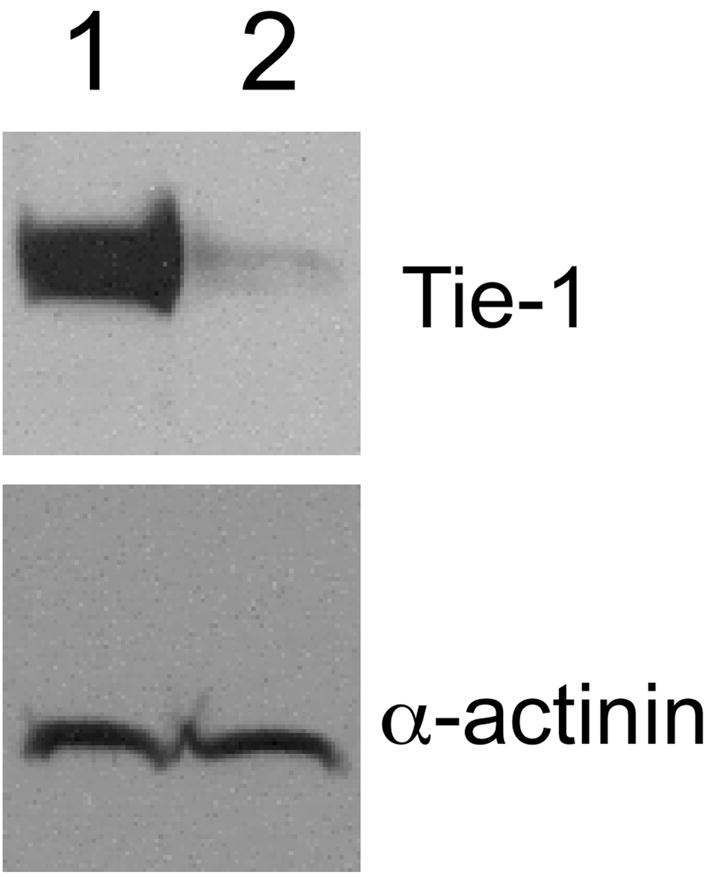
Western blots showing effective knockdown of Tie-1 protein by siRNA. (1) Control siRNA; (2) Tie-1 siRNA#1.
Table 1.
Genes differentially regulated upon Tie-1 knockdown. Bold: endogenous Tie-1. (−) and (+) indicate down- and up-regulation, respectively.
| Gene Name | Gene Symbol | Accesion Number | Fold change |
|---|---|---|---|
| Complement component 3 | C3 | NM_000064 | −7.5 |
| Tyrosine kinase with immunoglobulin-like and EGF- like domains | TIE1 | NM_005424 | −7.2 |
| Chromosome 15 open reading frame 48 | C15or48 | NM_032413 | −5.6 |
| Duffy blood group, chemokine receptor | DARC | NM_002036 | −5.5 |
| Toll-like receptor 2 | TLR2 | NM_003264 | −4.7 |
| Colony stimulating factor 3 (granulocyte) | CSF3 | NM_000759 | −4.7 |
| Tumor necrosis factor receptor superfamily, member 9 | TNFRSF9 | NM_001561 | −4.6 |
| Chemokine (C-C motif) ligand 23 | CCL23 | NM_005064 | −4.6 |
| Tumor necrosis factor, alpha-induced protein 6 | TNFAIP6 | NM_007115 | −4.5 |
| Zinc finger protein 143 | ZNF143 | U09850 | −4.4 |
| C-type lectin domain family 4, member E | CLEC4E | NM_014358 | −4.0 |
| Colony stimulating factor 2 (granulocyte-macrophage) | CSF2 | NM_000758 | −4.0 |
| Ubiquitin D | UBD | NM_006398 | −3.9 |
| Chemokine (C-X-C motif) ligand 5 | CXCL5 | NM_002994 | −3.9 |
| Complement component 1, s subcomponent | C1S | NM_001734 | −3.7 |
| P70872 (P70872) FliY protein (Fragment), partial (5%) | THC2440027 | XM_373971 | −3.7 |
| Epstein-Barr virus induced gene 3 | EBI3 | NM_005755 | −3.6 |
| Lipocalin 2 | LCN2 | NM_005564 | −3.4 |
| Arachidonate 5-lipoxygenase | ALOX5 | NM_000698 | −3.4 |
| Oxytocin receptor | OXTR | NM_000916 | −3.4 |
| Serpin peptidase inhibitor, clade A member 3 | SERPINA3 | NM_001085 | −3.4 |
| Complement component 1, r subcomponent | C1R | NM_001733 | −3.3 |
| Papilin, proteoglycan-like sulfated glycoprotein | PAPLN | NM_173462 | −3.3 |
| Kynureninase (L-kynurenine hydrolase) | KYNU | NM_003937 | −3.3 |
| Hypothetical gene supported by BC008048 | LOC440934 | CR593560 | −3.2 |
| Chemokine (C-C motif) ligand 3-like 3 | CCL3L3 | NM_001001437 | −3.2 |
| Phospholipase A2, group IVC (cytosolic, calcium-independent) | PLA2G4C | NM_003706 | −3.2 |
| High density lipoprotein-binding protein | LOC338328 | NM_178172 | −3.2 |
| TNFAIP3 interacting protein 3 | TNIP3 | NM_024873 | −3.2 |
| Selectin E (endothelial adhesion molecule 1) | SELE | NM_000450 | −3.1 |
| Interleukin 1, beta | IL1B | NM_000576 | −3.1 |
| Unknown | THC2280799 | -- | −3.1 |
| MHC class I mRNA fragment 3.8-1 (3.8-1) on chromosome 6. | 3.8-1 | NR_002812 | −3.1 |
| Family with sequence similarity 135, member A | FAM135A | NM_020819 | −3.1 |
| Likely ortholog of mouse lung-inducible Neutralized-related C3HC4 RING domain protein | LINCR | BC012317 | −3.0 |
| Guanine nucleotide binding protein (G protein) | GNG2 | NM_053064 | −3.0 |
| Superoxide dismutase 2, mitochondrial (SOD2), nuclear gene encoding mitochondrial protein | SOD2 | NM_000636 | −3.0 |
| RCSD domain containing 1 | RCSD1 | NM_052862 | −3.0 |
| Solute carrier family 26 (sulfate transporter), member 2 | SLC26A2 | NM_000112 | −2.9 |
| Complement factor B | CFB | NM_001710 | −2.9 |
| Unknown | A_24_P767901 | -- | −2.9 |
| Sex comb on midleg-like 1 (Drosophila) | SCML1 | NM_006746 | −2.9 |
| Similar to RIKEN cDNA 8030451K01 | RP11-50D16.3 | AL536879 | −2.9 |
| PDZK1 interacting protein 1 | PDZK1IP1 | NM_005764 | −2.9 |
| Chemokine (C-X-C motif) ligand 6 | CXCL6 | NM_002993 | −2.8 |
| Chemokine (C-C motif) ligand 8 | CCL8 | NM_005623 | −2.8 |
| full-length cDNA clone CS0DL003YC11 of B cells (Ramos cell line) Cot 25-normalized of Homo sapiens | ENST00000361204 | CR614040 | −2.8 |
| Similar to RIKEN cDNA 8030451K01 | RP11-50D16.3 | NM_001012754 | −2.7 |
| Phosphatidic acid phosphatase type 2B | PPAP2B | NM_003713 | −2.7 |
| UBX domain containing 2 | UBXD2 | NM_014607 | −2.7 |
| Chemokine (C-X-C motif) ligand 2 | CXCL2 | NM_002089 | −2.7 |
| Interleukin 7 receptor | IL7R | NM_002185 | −2.7 |
| Cathepsin S | CTSS | NM_004079 | −2.6 |
| Phospholipase A1 member A | PLA1A | NM_015900 | −2.6 |
| Chemokine (C-X-C motif) ligand 3 | CXCL3 | NM_002090 | −2.6 |
| Superoxide dismutase 2, mitochondrial | SOD2 | BC016934 | −2.6 |
| Hypothetical LOC646999 | LOC646999 | AK096857 | −2.6 |
| Carnitine O-octanoyltransferase | CROT | NM_021151 | 2.5 |
| Keratin 7 | KRT7 | NM_005556 | 2.7 |
| v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), transcript variant 1 | KIT | NM_000222 | 2.7 |
| Secretory protein LOC348174 | LOC348174 | AK096051 | 2.7 |
| Stanniocalcin 1 | STC1 | NM_003155 | 2.7 |
| Mesoderm specific transcript homolog | MEST | NM_002402 | 2.7 |
| Collagen, type XXI, alpha | COL21A1 | NM_030820 | 2.8 |
| Homo sapiens H19, imprinted maternally expressed untranslated mRNA (H19) on chromosome 11 | H19 | NR_002196 | 2.8 |
| chromosome 18 open reading frame 20 | C18orf20 | NM_152728 | 2.9 |
| Hypothetical LOC147710 | LOC147710 | XM_097278 | 2.9 |
| Cytokine-like 1 | CYTL1 | NM_018659 | 2.9 |
| Leucine rich repeat containing 17 | LRRC17 | NM_005824 | 2.9 |
| Mal, T-cell differentiation protein 2 | MAL2 | NM_052886 | 2.9 |
| Adenomatosis polyposis coli down-regulated 1 | APCDD1 | NM_153000 | 2.9 |
| Corticotropin releasing hormone binding protein | CRHBP | NM_001882 | 3.0 |
| G0/G1switch 2 | G0S2 | NM_015714 | 3.1 |
| Inhibin, beta B | INHBB | NM_002193 | 3.1 |
| Brain-derived neurotrophic factor (BDNF), transcript variant 1 | BDNF | NM_170735 | 3.2 |
| Unknown | THC2279735 | -- | 3.4 |
| KIAA1377 | KIAA1377 | NM_020802 | 3.4 |
| Unknown | A_24_P489649 | XM_067448 | 3.7 |
| Adenomatosis polyposis coli down-regulated 1-like | APCDD1L | NM_153360 | 3.7 |
| Hyaluronan and proteoglycan link protein 1 | HAPLN1 | NM_001884 | 3.8 |
| chromosome 1 open reading frame 187 | C1orf187 | AK075558 | 3.9 |
| Angiopoietin-like 5 | ANGPTL5 | NM_178127 | 4.1 |
| if20d02.x1 Melton Normalized Human Islet 4 N4-HIS 1 Homo sapiens cDNA clone IMAGE:5677082 3′ | BM129308 | BM129308 | 4.2 |
| Periostin, osteoblast specific factor | POSTN | NM_006475 | 4.4 |
| Nance-Horan syndrome (congenital cataracts and dental anomalies) | NHS | NM_198270 | 4.5 |
| Intelectin 1 (galactofuranose binding) | ITLN1 | NM_017625 | 4.6 |
| Prion protein 2 (dublet) | PRND | NM_012409 | 4.7 |
| Cytochrome P450, family 1, subfamily B | CYP1B1 | NM_000104 | 4.7 |
| Collagen, type III, alpha 1 (Ehlers-Danlos syndrome type IV, autosomal dominant) | COL3A1 | NM_000090 | 4.8 |
| Homo sapiens cDNA FLJ30847 fis, clone FEBRA2002792. [AK055409] | ENST00000381158 | AK055409 | 4.9 |
| Transmembrane protein 46 | TMEM46 | NM_001007538 | 5.2 |
Next, we performed real-time PCR analysis to validate some of the results from the microarray experiment. To demonstrate specificity of siRNA knockdown, we tested two additional non-overlapping Tie-1 siRNA duplexes (#2 and #3). All three siRNAs efficiently suppressed Tie-1 expression at the protein and RNA levels (Fig. 2 and data not shown). Real time PCRs showed that VCAM-1, ICAM-1, and E-selectin were significantly upregulated when Tie-1 was overexpressed, although only E-selectin was identified as a Tie-1 target gene in our microarray. We reasoned that the average downregulation of VCAM-1 and ICAM-1 resulted from Tie-1 knockdown might not have been large enough and thus filtered out in the array analysis. These results, together with the Tie-1 overexpression results we reported previously, strongly suggest that Tie-1 modulates VCAM-1, ICAM-1, and E-selectin expression in endothelial cells.
Fig. 2.
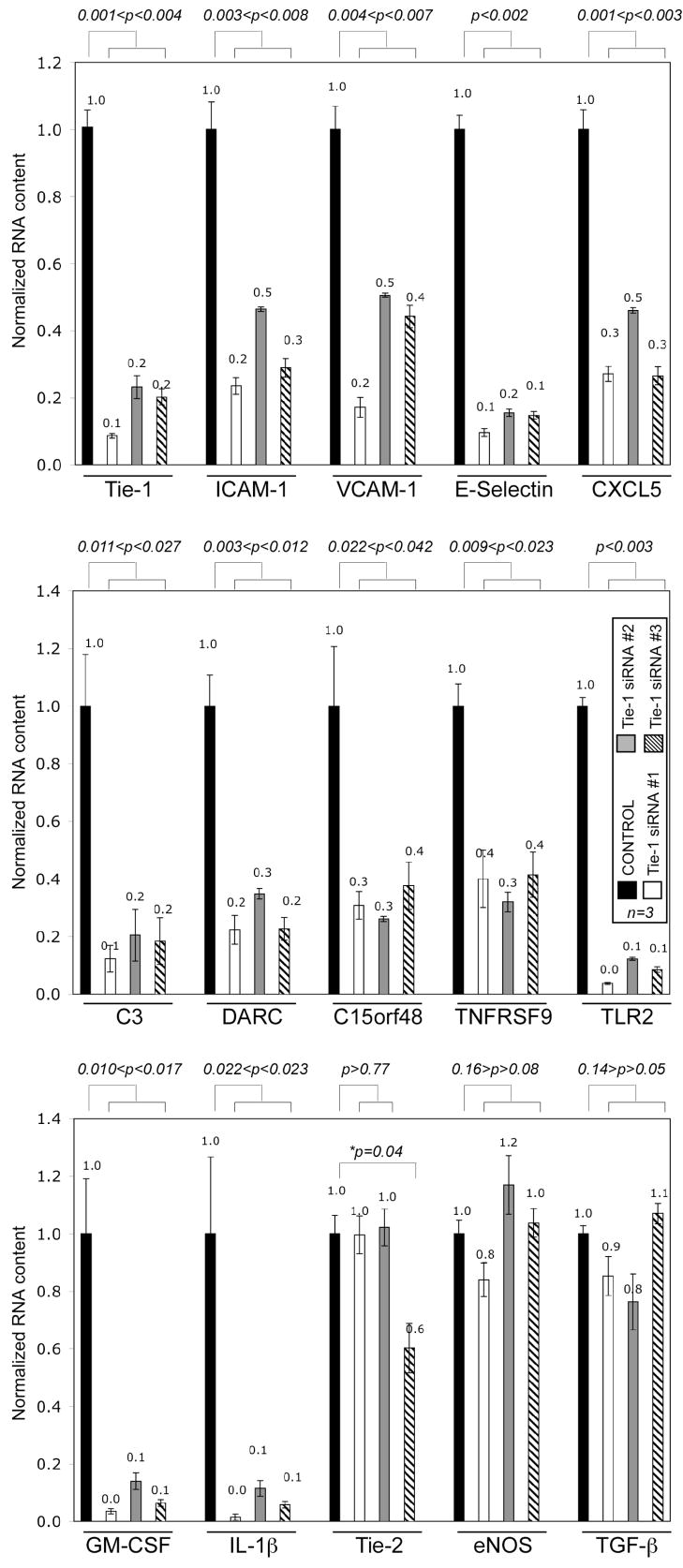
Real-time PCR validation of genes regulated by Tie-1 expression. Gene expression, normalized to GAPDH level, was determined by real-time PCRs and presented as mean±SD. Expression level of the gene of interest when HUVECs were treated with the control siRNA was arbitrarily set to one and used for normalization. *Tie-1 siRNA#3 appeared to partially knock down Tie-2 expression (see text for explanations). RNAs from three siRNA transfections were tested (n=3). P values were determined by t-tests.
Eight additional genes from Table 1 were chosen to be validated next (Fig. 2). They were complement component 3 (C3), Duffy blood group chemokine receptor (DARC), chromosome 15 open reading frame 48 (c15orf48), tumor necrosis factor receptor superfamily member 9 (TNFRSF9), toll-like receptor 2 (TLR2), granulocyte-macrophage colony stimulating factor (GM-CSF), interleukin-1β (IL-1β), and chemokine CXCL5. All these genes were concomitantly downregulated as Tie-1 was knocked down by all three siRNAs tested. As controls, we showed that expression of Tie-2, eNOS, and TGFβ were not significantly suppressed by the three siRNAs used, with one exception. Tie-1 siRNA #3, but not #1 or #2, appeared to reduce Tie-2 expression by approximately 40%. This knockdown might be due to the fact that the region of Tie-1 targeted by siRNA#3 had a high sequence homology to Tie-2. It was possible that Tie-1 siRNA#3 could partly anneal to Tie-2 mRNA, resulting in reduced Tie-2 expression.
3.2. Suppression of Tie-1 expression reduced endothelial cells ability to stimulate monocytes
Next, we examined whether expression of Tie-1 would affect the ability of endothelial cells to stimulate monocytes. HUVEC conditioned medium at basal level stimulated expression of cytokine MCP-1 in U937 cells, a monocytic cell line, in a time dependent manner (not shown). Four hours of stimulation with basal HUVEC conditional medium resulted in a modest but statistically significant upregulation of MCP-1 in U937 (Fig. 3). This stimulatory effect was completely abrogated when conditioned medium of Tie-1-siRNA#1-treated HUVECs was used, indicating that Tie-1 was critical for this inflammatory property. Treatment of HUVECs with Tie-1 siRNA #2 or #3 also significantly reduced the endothelial conditioned medium ability to stimulate MCP-1 production in U937. In contrast, endothelial conditioned medium was unable to stimulate IL-1β synthesis in U937, regardless of whether Tie-1 had been knockdown by siRNAs. We have not identified the stimulant responsible for this phenotype. A neutralizing anti-GM-CSF antibody inhibited MCP-1 production in U937 stimulated with HUVEC conditioned medium only by 25% (data not shown). Probably multiple agents were responsible for the stimulation, since many proinflammatory cytokines were present in HUVEC conditioned medium. Nonetheless, results of this loss-of-function study collectively support the notion that Tie-1 is proinflammatory.
Fig. 3.

Tie-1 in HUVECs was essential in stimulation of MCP-1 expression in U937 cells. Conditioned medium of HUVECs treated with control or Tie-1 siRNA was used to stimulate U937 cells for four hours. Expression level in unstimulated cells was arbitrarily set to one and used for normalization. MCP-1 (A) or IL-1β (B) expression in U937 was determined by real-time PCRs. Conditioned media from three siRNA transfections were tested (n=3). P values were determined by t-tests.
3.3. Microarray profiling reveals relevance of Tie-1 in inflammatory diseases
To gain insights into the role of Tie-1 may play in human diseases, we queried the gene profile depicted in Table 1 using the Ingenuity Pathway Analysis program. Of the 91 input genes (from Table 1), 70 were eligible for functional analysis and 59 were identified to have relevance to known diseases. These 59 genes were further divided according to specific function annotations and are shown in Table 2. The top ten scoring functions suggest that Tie-1 may play a role in autoimmune diseases and inflammatory disorders, consistent with our hypothesis that Tie-1 is proinflammatory in endothelial cells. We are particularly interested in atherosclerosis and rheumatoid arthritis, because Tie-1 is upregulated in these diseases [3–5].
Table 2.
Disease relevance of genes regulated by Tie-1 expression in HUVECs. Genes identified in Table 1 were analyzed by the Ingenuity Pathway Analysis Program using the Ingenuity Knowledge Base as the reference set. Of the 91 input genes (including Tie-1), 70 were eligible for functional analysis, and 59 were identified to have functions/diseases relevance. The top 10 functions are presented here. Detailed explanation of the method used to calculate p-values can be obtained from Ingenuity’s Website (https://analysis.ingenuity.com/pa/info/help/help.htm).
| Function Annotation | P-value | Molecules | # Molecules (of 59) |
|---|---|---|---|
| Connective tissue disorder | 6.80E-14 | ALOX5, BDNF, C1R, CCL8, CCL23, CFB, CLEC4E, COL3A1, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, RCSD1, SLC26A2, SOD2, TLR2, TNFAIP6, TNFRSF9 | 24 |
| Rheumatic disease | 5.45E-13 | ALOX5, BDNF, C1R, CCL8, CCL23, CFB, CLEC4E, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, RCSD1 SOD2, TLR2, TNFAIP6, TNFRSF9 | 22 |
| Arthritis | 7.19E-13 | ALOX5, BDNF, C1R, CCL8, CCL23, CFB, CLEC4E, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, RCSD1, SOD2, TLR2, TNFAIP6 | 21 |
| Autoimmune disease | 3.16E-12 | ALOX5, C1R, C1S, CCL8, CCL23, CFB, CLEC4E, CSF2, CSF3, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, KIT, RCSD1, TLR2, TNFAIP6, TNFRSF9 | 21 |
| Rheumatoid arthritis | 7.35E-12 | ALOX5, C1R, CCL8, CCL23, CFB, CLEC4E, CSF2, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, RCSD1, TLR2, TNFAIP6 | 17 |
| Skeletal and muscular disorder | 1.31E-11 | ALOX5, BDNF, C1R, CCL8, CCL23, CFB, CLEC4E, COL3A1, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, RCSD1, SLC26A2, SOD2, TLR2, TNFAIP6, TNFRSF9 | 24 |
| Immunological disorder | 6.58E-11 | ALOX5, BDNF, C1R, C1S, CCL8, CCL23, CFB, CLEC4E, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, G0S2, IL1B, IL7R, KIT, RCSD1, TLR2, TNFAIP6, TNFRSF9 | 23 |
| Inflammatory disorder | 1.78E-10 | ALOX5, BDNF, C1R, CCL8, CCL23, CFB, CLEC4E, CSF2, CSF3, CTSS, CXCL2, CXCL3, CXCL5, CXCL6, EBI3, G0S2, IL1B, IL7R, KIT, POSTN, RCSD1, SELE, SOD2, TLR2, TNFAIP6, TNFRSF9 | 26 |
| Inflammatory response | 2.44E-10 | ALOX5, CCL8, CCL23, CCL3L3, CSF2, CSF3, CXCL2, CXCL3, CXCL5, CXCL6, DARC, IL1B, LCN2, SELE, SERPINA3, TLR2, TNFAIP6 | 17 |
| Cell movement of phagocytes | 1.40E-09 | CCL8, CCL23, CFB, CSF2, CSF3, CTSS, CXCL3, CXCL5, CXCL6, IL1B, SELE, SERPINA3, TLR2 | 13 |
Atherosclerosis is a chronic inflammatory disease [9,10]. Interestingly, Tie-1 is specifically expressed in atherosclerosis-prone sites in the vasculature [3]. Since Tie-1 is proinflammatory in endothelial cells in vitro, we postulate that Tie-1 may play a role in atherogenesis by promoting endothelial inflammation. The gene profiling reported in the current study provides some supporting evidence for this hypothesis. In additional to the adhesion molecules, expressions of many genes important in atherosclerosis development were also altered when Tie-1 was knocked down (Table 1). For example, IL-1β is a well-known inflammation inducer and plays an instrumental role in the development of atherosclerosis [11–14]. Genetic knockout of TLR2 has been shown to be atheroprotective in several mouse models [15–17]. Interestingly, TLR2 is expressed in endothelial cells at lesion-prone sites [17]. Other genes that have also been implicated in atherosclerosis and were identified as Tie-1 targets in this study include TNFRSF9 (CD137), GM-CSF (CSF2), and G-CSF (CSF3). TNFRSF9 was detected at the vessel walls of sites of inflammation in vivo in mice [18] and was upregulated in atherosclerotic arteries in patient samples [19]. Furthermore, injection of TNFRSF9 agonistic antibody in ApoE null mice enhanced lesion formation [19]. Administration of exogenous GM-CSF or C-CSF also significantly upregulated atherosclerosis development in ApoE null mice maintained in high fat diet [20]. Therefore, endogenous Tie-1 in endothelial cells appears to regulate multiple genes that are critical in atherosclerosis development.
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease that primarily affects the joints. It is characterized by hypertrophy of intima of the synovium, leukocyte infiltration into the synovial tissue, and intensive angiogenesis, resulting in the formation of synovial pannus and ultimately cartilage destruction. As in atherosclerosis, inflammation of endothelial cells is believed to be crucial in the development of the disease [21].
Tie-1 is known to be upregulated in the endothelial cells in lesions of rheumatoid arthritis [4,5]. A recently identified alternatively spliced soluble Tie-1 ectodomain variant was shown to reduce clinical signs of arthritis in a collagen-induced arthritis mouse model [6]. However, the exact mechanism underlying this inhibitory effect was not addressed. Perhaps the soluble Tie-1 prevented endogenous Tie-1 activation and blocked endothelial inflammation by binding to a Tie-1 ligand yet to be identified. If our in vitro results could be extended to in vivo systems, suppressing Tie-1 activity would lead to downregulation of IL-1β VCAM-1, ICAM-1, E-selectin, CXCL5, and DARC. IL-1β is central in RA development [22]. The adhesion molecules are known to play a crucial role in mediating leukocyte infiltration [23], whereas CXCL5 and DARC are upregulated in the endothelial cells in RA lesions [24,25]. Therefore, blockade of Tie-1 function may reduce RA by suppression of the production of these proteins and factors influential in the development of the disease.
In conclusion, results presented in this report support the hypothesis that Tie-1 induces inflammation in endothelial cells in vitro. Experiments using transgenic mice with endogenous Tie-1 expression level altered will be needed to investigate the in vivo functions of Tie-1 in different disease models. Such experiments are currently underway.
Acknowledgments
This work was supported in part by a NIDDK Mentored Research Scientist Development Award (1K01DK077727) and seed funds from Beth Israel Deaconess Medical Center.
Abbreviations
- HUVEC
human umbilical vein endothelial cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sato TN, et al. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–4. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 2.Puri MC, et al. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. Embo J. 1995;14:5884–91. doi: 10.1002/j.1460-2075.1995.tb00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porat RM, et al. Specific induction of tie1 promoter by disturbed flow in atherosclerosis-prone vascular niches and flow-obstructing pathologies. Circ Res. 2004;94:394–401. doi: 10.1161/01.RES.0000111803.92923.D6. [DOI] [PubMed] [Google Scholar]
- 4.Shahrara S, et al. Differential expression of the angiogenic Tie receptor family in arthritic and normal synovial tissue. Arthritis Res. 2002;4:201–8. doi: 10.1186/ar407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchida T, et al. Immunohistochemical localisation of protein tyrosine kinase receptors Tie-1 and Tie-2 in synovial tissue of rheumatoid arthritis: correlation with angiogenesis and synovial proliferation. Ann Rheum Dis. 2000;59:607–14. doi: 10.1136/ard.59.8.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin P, et al. Novel splice variants derived from the receptor tyrosine kinase superfamily are potential therapeutics for rheumatoid arthritis. Arthritis Res Ther. 2008;10:R73. doi: 10.1186/ar2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan B, et al. Receptor tyrosine kinase Tie-1 overexpression in endothelial cells upregulates adhesion molecules. Biochem Biophys Res Commun. 2008;371:475–9. doi: 10.1016/j.bbrc.2008.04.091. [DOI] [PubMed] [Google Scholar]
- 8.Chan B, Sukhatme VP. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb Res. 2008 doi: 10.1016/j.thromres.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138:S419–20. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- 10.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 11.Dinarello CA. Interleukin-1. Cytokine Growth Factor Rev. 1997;8:253–65. doi: 10.1016/s1359-6101(97)00023-3. [DOI] [PubMed] [Google Scholar]
- 12.von der Thusen JH, et al. Interleukins in atherosclerosis: molecular pathways and therapeutic potential. Pharmacol Rev. 2003;55:133–66. doi: 10.1124/pr.55.1.5. [DOI] [PubMed] [Google Scholar]
- 13.Kirii H, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3. [DOI] [PubMed] [Google Scholar]
- 14.Elhage R, et al. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–4. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, et al. Toll-like receptor 2 plays a critical role in the progression of atherosclerosis that is independent of dietary lipids. Atherosclerosis. 2008;196:146–54. doi: 10.1016/j.atherosclerosis.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madan M, Amar S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: proteomic findings. PLoS ONE. 2008;3:e3204. doi: 10.1371/journal.pone.0003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mullick AE, et al. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. 2008;205:373–83. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drenkard D, et al. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. Faseb J. 2007;21:456–63. doi: 10.1096/fj.05-4739com. [DOI] [PubMed] [Google Scholar]
- 19.Olofsson PS, et al. CD137 is expressed in human atherosclerosis and promotes development of plaque inflammation in hypercholesterolemic mice. Circulation. 2008;117:1292–301. doi: 10.1161/CIRCULATIONAHA.107.699173. [DOI] [PubMed] [Google Scholar]
- 20.Haghighat A, et al. Granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor exacerbate atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2007;115:2049–54. doi: 10.1161/CIRCULATIONAHA.106.665570. [DOI] [PubMed] [Google Scholar]
- 21.Middleton J, et al. Endothelial cell phenotypes in the rheumatoid synovium: activated, angiogenic, apoptotic and leaky. Arthritis Res Ther. 2004;6:60–72. doi: 10.1186/ar1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology (Oxford) 2003;42(Suppl 2):ii3–10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- 23.Szekanecz Z, Koch AE. Cell-cell interactions in synovitis. Endothelial cells and immune cell migration. Arthritis Res. 2000;2:368–73. doi: 10.1186/ar114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch AE, et al. Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J Clin Invest. 1994;93:921–8. doi: 10.1172/JCI117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patterson AM, et al. Expression of the duffy antigen/receptor for chemokines (DARC) by the inflamed synovial endothelium. J Pathol. 2002;197:108–16. doi: 10.1002/path.1100. [DOI] [PubMed] [Google Scholar]