

Abstract
Virtual screening was employed to identify new drug-like inhibitors of NAD synthetase (NADs) as antibacterial agents. Four databases of commercially available compounds were docked against three subsites of the NADs active site using FlexX in conjunction with CScore. Over 200 commercial compounds were purchased and evaluated in enzyme inhibition and antibacterial assays. 18 compounds inhibited NADs at or below 100 μM (7.6% hit rate), and two were selected for future SAR studies.
With the increasing threat of pathogens, such as Bacillus anthracis, being used as bioweapons,1 and the rise in the incidence of multi-drug resistant bacteria,2 the need for new antibiotics that act at novel targets has never been greater. Previous studies within this group3-5 have revealed that inhibition of one such target, the amidotransferase enzyme nicotinamide adenine dinucleotide (NAD) synthetase (NADs), could hinder both spore outgrowth and vegetative growth, which would provide antibacterial action at two different steps in the bacterial life cycle.6-10
The first class of NADs inhibitors designed by this group consisted of tethered dimers that contain two hydrophobic groups linked by a polymethylene tether, and a positively charged nitrogen on one end.3-5 These inhibitors were antibacterial, and there was a correlation between the potencies of enzyme inhibition and antibacterial effects. However, the permanent positive charge and detergent-like properties of this class of compounds were unattractive for further drug development.11,12 More drug-like lead inhibitors were, therefore, sought.
Virtual screening of compound databases using the detailed structure of the drug target can serve to greatly enhance success in the lead discovery process.13-17 Here we use the in silico screening program FlexX 1.20.1(BiosolveIT GmbH®) for the virtual screening of commercially available compounds within the catalytic site of NADs to identify new classes of lead inhibitors. In this study, four commercial compound databases were filtered according to Lipinski's rule of 5 using Tripos' program Unity: Maybridge (58,650 after filtering), ChemBridge (404,132), Tripos' LeadQuest (72,660), and ComGenex (82,737). Because these docking studies predate our solution of the crystal structure of B. anthracis NADs (PDB code 2PZB),18 the highest available resolution crystal structure of B. subtilis NADs,19 reported by our group, was utilized for docking (PDB code 1KQP19). The crystal structures of B. anthracis and B. subtilis NADs reveal that the binding sites are nearly identical, with all active site residues being conserved.18
NADs is a large homodimer of approximately 60 kDa that contains two identical binding sites. The crystal structure of the protein from B. subtilis reveals two identical long, linear binding sites containing the adenylated reaction intermediates lying partly within the dimer interface on the NaAD end, and in a buried cavity within one monomer on the ATP end. Due to the enormity of the NADs homodimer catalytic site, and considering our limited computational resources at that time, three smaller binding subsites were constructed to be used in the virtual screening study. To accomplish this, a sphere with radius 25 Å around one of the bound intermediates was extracted from the whole protein structure to produce a partial protein structure which consisted of the three shells of amino acid residues immediately surrounding the binding cavity and which fully contained one complete binding site. All crystallographic waters and metals were removed, hydrogens were added, and the protonation states of active site residues were adjusted to their dominant ionic forms assuming a local physiological pH. The “active site,” as needed for use by FlexX, was further defined by creating a smaller sphere of radius 17 Å which consisted of the first two shells of amino acids surrounding the bound substrate, resulting in a rather large active site: 31 Å in length, and a width ranging from 7 Å on the NaAD end to 16 Å on the ATP end.
As explained earlier, the complete catalytic site was then divided into three overlapping subsites: the NaAD binding subsite, the ATP subsite, and a center subsite which bridges the two end sites. The resulting NaAD binding subsite is the most confined and is approximately 16 Å long and 7 Å wide, appearing as a “canyon” near the homodimer interface; the center subsite is shaped like a tunnel, and is 14 Å long and 9 Å wide; the ATP subsite is buried within a single monomer and is the largest of the three at 21 Å long and 16 Å in width. The bound ligand was excluded from all docking runs.
Each of the four commercial databases was docked into each of the three subsites employing FlexX 1.20.1, which has been shown to be suitable for exploring many kinds of binding sites,14,20 and routinely produces hit rates comparable to other highly regarded programs.21-23 FlexX was accessed using the SYBYL 6.9 suite of programs (Tripos, Inc.®), and default parameters were used for each docking run. For our purposes, automatic base fragment selection was employed. Within each of the three subsites, the core subpocket was defined as all residues which interact directly with the bound substrate. Formal charges were assigned, and 5 poses for each ligand were saved. Docking began on a 64 bit dual processor SGI Octane computer running Unix, and was completed in parallel using a 64 bit PQS 4-processor Opteron Quantum Cube running Linux. After all databases were screened against all sites and ranked according to FlexX score, the best poses from each run were combined and re-ranked using a consensus scoring24 program, CScore.25 A total of 22,240 compounds were ranked with CScore, and all compounds with a CScore of 5 were reviewed according to several criteria: realistic orientation within the binding pocket, a predicted binding conformation that is energetically reasonable, structures that are chemically simple and can be easily modified synthetically, and compounds representative of chemically diverse structural classes that are considered medicinally interesting. Additionally, selected compounds with both a CScore of 4 and a good FlexX score were reviewed if they were structurally unique. Representatives from the most interesting structural classes were purchased and screened in our NADs enzyme inhibition and B. anthracis antibacterial assays.
The high-throughput assay utilized by us for previous synthetic NAD synthetase inhibitors11 monitored production of NAD via enzymatic conversion to NADH, and the latter was detected by both fluorescence and uv absorption. However, this assay was unsuitable for many commercial compounds because they interfered with the fluorescence and/or absorbance at the wavelengths observed. Further, some compounds gave false positives due to direct reaction with NADH. Therefore, an alternate HPLC assay was designed and is presented here for the first time.
In this new assay the reaction product NAD was directly monitored. Sample plates were prepared using a BioMek® FX liquid handling system and the reaction volume was 200 μL. The reaction mixture contained 60 mM HEPPS, pH 8.5, 0.5 mM NH4Cl, 20 mM KCl, 10 mM MgCl2, 0.1 mM NaAD, 0.2 mM ATP, 6 μg/ml purified B. anthracis NADS, 2.5% (v/v) DMSO, 0.3% BOG and inhibitors at various concentrations. Compounds were assayed beginning at 600 μM and at doubling dilutions down to 0.6 μM. The reaction was initiated by adding 0.2 mM ATP, and quenched after 10 minutes by adding 50 μL of 6 M guanidine-HCl. The plates were sealed by aluminum tape, and centrifuged at 2500 rpm for 10 minutes in order to pellet any precipitation that may have been caused by the inhibitors. Plates were stored at 4 °C prior to the HPLC analysis.
The HPLC procedure utilized a Gilson® 215 liquid handler, two Gilson® 306 pumps, and a Gilson® 170 diode array detector. A Phenomenex® Luna 5μm, C5, 100Å, 100 × 4.60 mm column was used for separations. The mobile phase was A: 20 mM NaH2PO4 pH 6.90 and B: acetonitrile. The gradient was 100% A from 0 – 3 minutes, to 5% A / 95% B from 3 – 4 minutes for each 20 μL injection. The flow rate was 1.0 mL/min and DAD detection was 190 – 400nm. Peak height estimation for NAD was based on baseline integration. The % inhibition at each inhibitor concentration was calculated by the difference in peak height of NAD compared to reactions without inhibitor. The IC50 was determined from the plot of NAD peak height vs. inhibitor concentration, and is defined as the concentration of inhibitor required to produce NAD peak height at 50% of the uninhibited reaction. Peak areas were used to calculate the IC50 for selected active compounds, and similar results were obtained. Each compound was tested in duplicate, and the IC50 is reported as the average IC50 obtained from duplicate runs. False positives due to promiscuous inhibition were excluded by including detergents in the inhibition assay.
All purchased commercial compounds were also screened against Bacillus anthracis Sterne in an antibacterial assay as previously reported8,11 with the following modifications. B. anthracis Sterne spores were subcultured from stock cultures into Luria-Bertani (LB) broth and incubated for 2-3 hours at 37 °C in ambient air until the OD600 measurement reached 0.5 to 0.6, when the bacteria were in mid-log phase. The cultures were diluted 1:1 into LB Broth with an absorbance at 600 nm measuring 0.25 to 0.3, then were added to plates containing 240 μM samples of the compounds to be tested. Compounds were tested at a final DMSO concentration of 1%. The plates were incubated at 37 °C, and absorbance at 600 nm was read at 0h and every hour for 5 hours. Any compounds which inhibited growth of the vegetative cell (as compared to the control containing only DMSO) were screened in a full MIC determination starting at 240 μM and creating doubling dilutions down to 1.88 μM in quadruplicate wells. A plot of cell density vs. time yields inhibition of growth results, and the MIC is defined as the lowest concentration of compound required to completely inhibit growth (100% inhibition). MIC100 is reported as the average of the four data points acquired for each compound. Controls for each assay measured sterility, B. anthracis Sterne viability, and included a commercial antibiotic positive control (ciprofloxacin hydrochloride from MP Biomedicals).
Among the NADs subsites, the best FlexX scores were obtained from docking in the larger ATP subsite, presumably due to the many residues capable of charge-charge interactions. A total of 211 commercial compounds were purchased based on the CScore rankings: 135 from the NaAD, 31 from the center and 45 from the ATP subsites; 43 (20%) of those compounds were found to have IC50's less than or equal to 300 μM against NADs (Table 1). It should be noted that ranking compounds solely by their FlexX scores produced fewer hits than when compounds were ranked using consensus scoring. At 100 μM or below, 16 compounds (7.6% hit rate) were active against NADs (a cutoff routinely used to define virtual screening hit rates)15,17,26, while 4 were active at or below 50 μM. The hit rate at 100 μM is similar to those obtained by other virtual screening studies against different enzymatic targets.17,26,27 Of these active compounds, 27 inhibitors resulted from their predicted binding in the NaAD subsite, while 9 and 7 were predicted to bind in the center and ATP sites, respectively. The hit rates (100 μM) based on the number of compounds purchased from the NaAD, center, and ATP subsites were 8.1%, 6.5%, and 8.9%, respectively. Only a few compounds scored well in more than one subsite, and none of those screened were enzyme inhibitors.
Table 1.
Commercial compounds identified by FlexX studies to be NAD synthetase inhibitors at or below 300 μM, the subsites in which they were predicted to bind, and their biological activities.
| ID | MW | NADs subsite | IC50 (μM) | MIC100 (μM) |
|---|---|---|---|---|
| 5379 | 278.27 | NaAD | 51 | 120 |
| 5588 | 466.84 | ATP | 78.5 | > 215 |
| 5589 | 378.34 | center | 136.6 | > 264 |
| 5591 | 364.32 | center | 160 | > 274 |
| 5597 | 446.48 | ATP | 86.1 | > 224 |
| 5599 | 356.40 | center | 168.1 | 3.75 |
| 5604 | 450.54 | ATP | 141 | > 222 |
| 5605 | 368.37 | ATP | 145.9 | > 259 |
| 5606 | 422.37 | center | 141.1 | > 237 |
| 5609 | 490.61 | ATP | 70 | > 204 |
| 5615 | 449.40 | ATP | 55.4 | > 223 |
| 5616 | 404.21 | center | 207.5 | > 247 |
| 5617 | 438.29 | center | 77.5 | 15 |
| 5660 | 258.23 | NaAD | 22.5 | > 387 |
| 5679 | 303.71 | NaAD | 262 | > 329 |
| 5684 | 440.26 | NaAD | 99.5 | > 227 |
| 5691 | 430.25 | NaAD | 106 | > 232 |
| 5707 | 424.43 | ATP | 253 | > 240 |
| 5710 | 327.39 | NaAD | 128.5 | > 240 |
| 5724 | 443.44 | NaAD | 290.6 | > 240 |
| 5731 | 506.92 | center | 270.7 | > 240 |
| 5737 | 354.39 | NaAD | 235.3 | > 240 |
| 5749 | 527.76 | NaAD | 219.8 | > 240 |
| 5763 | 472.89 | NaAD | 232.1 | > 240 |
| 5764 | 505.96 | NaAD | 97.2 | > 240 |
| 5768 | 455.50 | center | 170.5 | > 240 |
| 5775 | 432.33 | NaAD | 290 | > 240 |
| 5785 | 426.39 | center | 108.6 | > 240 |
| 5792 | 346.35 | NaAD | 76 | > 240 |
| 5793 | 465.52 | NaAD | 78.8 | > 240 |
| 5798 | 472.68 | NaAD | 61.8 | > 240 |
| 5799 | 479.45 | NaAD | 174.8 | > 240 |
| 5802 | 411.42 | NaAD | 225.2 | > 240 |
| 5806 | 413.44 | NaAD | 67.8 | > 240 |
| 5807 | 401.40 | NaAD | 123.9 | > 240 |
| 5815 | 404.47 | NaAD | 185.6 | > 240 |
| 5818 | 494.51 | NaAD | 65.7 | > 240 |
| 5821 | 411.80 | NaAD | 103.6 | > 240 |
| 5822 | 424.46 | NaAD | 107.1 | > 240 |
| 5824 | 481.32 | NaAD | 10 | 1.9 |
| 5830 | 441.49 | NaAD | 198.2 | > 240 |
| 5831 | 451.89 | NaAD | 243.3 | > 240 |
| 5833 | 483.51 | NaAD | 78.3 | 15 |
The most significant result of this study was the identification of drug-like compounds that have good activities against both NADs and B. anthracis: 5617, 5824, and 5833. However, unlike our earlier tethered dimer inhibitors, there is a poor correlation between enzyme inhibition and antibacterial effects. Several enzymatically inactive commercial compounds were found to behave as antibacterial agents, while only 4 compounds that inhibited NADs were also effective against the vegetative cell, with MIC's at or below 15 μM. As mentioned earlier, this is in contrast to our results for earlier libraries of tethered dimer NADs inhibitors, which exhibited a linear correlation between enzyme inhibition and antibacterial activity.10 Possible explanations for active enzyme inhibitors that do not show a good MIC include: (1) low permeability into the bacterial cell; (2) loss via efflux pumps;28 (3) metabolism by the bacterial cell into inactive forms. It can also be inferred that those compounds which confer antibacterial activity against the vegetative cell but do not inhibit NADs must be acting on a different target(s). Our preliminary studies support the identity of a second target that explains the antibacterials with no enzyme activity, and these results will be reported separately upon completion.
Among the enzyme inhibitors identified, several different structural classes have emerged (Table 2), and those that also inhibit bacterial growth are considered most interesting for further optimization. 5379 is an acrylonitrile – potentially a good Michael acceptor, and thus not an ideal drug candidate. Other structural classes that produced NADs inhibitors include sulfonamides (5599, 5617 and 5824), ureas (5609, 5617, and 5824), complex amides (5615, 5798, 5818 and 5833), and Schiff bases (5660). Except for 5833, all of the antibacterial inhibitors (5599, 5617 and 5824) contain a sulfonamide, a urea, or a combination of both. While all four of these antibacterial inhibitors meet the requirements for moderate molecular weight in a drug-like structure, with the possibility for further analog generation, we selected 5617 and 5824 as compounds that best meet these requirements. 5833 appears less suitable for facile synthetic modifications, and the o-nitronaphthylamine moiety of 5599 contains two lower ranking functionalities relative to drug potential (e.g., the nitro and naphthalene groups). Compounds 5617 and 5824 reveal some similarities; both contain three aryl rings linked by a urea and a sulfonamide, and both contain a 3,4-dichlorophenyl ring. This class of urea-sulfonamides was chosen for future SAR analysis via parallel library synthesis.
Table 2.
Examples of NADs inhibitors from the most common structural classes identified through in silico screening.
| Cmpd. ID | Structure | NADs IC50 (μM) | B.a. MIC (μM) |
|---|---|---|---|
| Cipro | 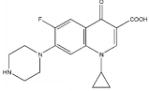 |
- | 0.5 |
| 5824 | 10 | 1.9 | |
| 5599 | 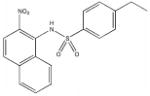 |
168.1 | 3.75 |
| 5617 | 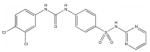 |
77.5 | 15 |
| 5833 | 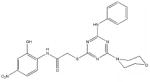 |
78.3 | 15 |
| 5660 | 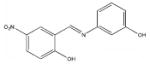 |
22.5 | > 387 |
| 5379 | 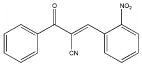 |
51 | 120 |
| 5615 |  |
55.4 | > 223 |
| 5798 | 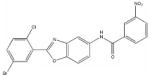 |
61.8 | > 240 |
| 5818 | 65.7 | > 240 | |
| 5609 | 70 | > 204 |
During submission of this report, a related online prepublication29 appeared describing modest inhibitors of NADs from mycobacteria – the only other reported inhibitors of NADs – although these compounds did not block mycobacterial growth.
In conclusion, virtual screening has provided the first reported drug-like small molecule inhibitors of NAD synthetase with antibacterial activity.
Supplementary Material
Acknowledgments
We thank Dr. Steve Harville for assistance with the HPLC assay, and Ms. Qingxian Zhou and Dr. Irina Protasevich for help with protein purification. Financial support was provided by the Department of Chemistry at UAB and by NIH (U01 AI056477 to WJB and U01 AI070386 to CGB).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data Supplementary data, including (a) structures of compounds from Table 1 not shown in Table 2, (b) graphical representations of the binding sites used and poses of selected docked ligands, and (c) a sample HPLC-chromatogram used in the enzyme assay, can be found in the online version.
References
- 1.a Smiley ST. Adv Exp Med Biol. 2007;603:376. doi: 10.1007/978-0-387-72124-8_35. [DOI] [PubMed] [Google Scholar]; b Santic M, Molmeret M, Klose KE, Abu Kwaik Y. Trends Microbiol. 2006;14:37. doi: 10.1016/j.tim.2005.11.008. [DOI] [PubMed] [Google Scholar]; c Knight J. Nature. 2001;414:837. doi: 10.1038/414837a. [DOI] [PubMed] [Google Scholar]
- 2.a Chopra I, Hodgson J, Metcalf B, Poste G. Antimicrob Agents Chemother. 1997;41:497. doi: 10.1128/aac.41.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Walsh CT. Nature. 2000;406:775. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]; c Pratt WB, Fekety R. The Antimicrobial Drugs. Chapter 7 New York: Oxford; 1986. [Google Scholar]
- 3.Velu SE, Cristofoli W, Garcia GJ, Brouillette CG, Pierson M, Luan CH, DeLucas LJ, Brouillette WJ. J Med Chem. 2003;46:3371. doi: 10.1021/jm030003x. [DOI] [PubMed] [Google Scholar]
- 4.a Brouillette WJ, Muccio D, Jedrzejas MJ, Brouillette CG, Devedjiev Y, Cristofoli W, DeLucas LJ, Garcia GJ, Schmitt L, Velu SE. U.S. Patent 6,500,852 B1. 2002; b Brouillette WJ, Brouillette CG, DeLucas LJ. U.S. Patent 6,673,827. 2004; c Brouillette WJ, Muccio D, Jedrzejas MJ, Brouillette CG, Devedjiev Y, Cristofoli W, DeLucas LJ, Garcia GJ, Schmitt L, Velu SE. U.S. Patent 6,727,237. 2004; d Brouillette WJ, DeLucas LJ, Brouillette CG, Velu SE, Kim YC, Mou L, Porter S. U.S. Patent 6,861,448. 2005
- 5.Moro WB. PhD Dissertation. University of Alabama; Birmingham: Nov, 2007. [Google Scholar]
- 6.Zalkin H. Adv Enzymol Relat Areas Mol Biol. 1993;66:203. doi: 10.1002/9780470123126.ch5. [DOI] [PubMed] [Google Scholar]
- 7.Jedrzejas MJ. Crit Rev Biochem Mol Biol. 2002;37:339. doi: 10.1080/10409230290771537. [DOI] [PubMed] [Google Scholar]
- 8.Tritz GJ. In: In Escherichia coli and Salmonella typhimurium Cellular and Molecular Biology. Neidhardt FC, Ingraham JL, Brooks Low K, Magasanik B, Schaechter M, Umbarger HE, editors. Vol. 1. Washington, D.C.: American Society for Microbiology; 1987. pp. 557–563. [Google Scholar]
- 9.Sutherland S. Drug Discovery Today. 2003;8:335. doi: 10.1016/s1359-6446(03)02665-5. [DOI] [PubMed] [Google Scholar]
- 10.Nessi C, Albertini AM, Speranza ML, Galizzi A. J Biol Chem. 1995;270:6181. doi: 10.1074/jbc.270.11.6181. [DOI] [PubMed] [Google Scholar]
- 11.Velu SE, Luan CH, DeLucas LJ, Brouillette CG, Brouillette WJ. J Comb Chem. 2005;7:898. doi: 10.1021/cc050063j. [DOI] [PubMed] [Google Scholar]
- 12.Velu SE, Mou L, Luan CH, DeLucas LJ, Brouillette CG, Brouillette WJ. J Med Chem. 2007;50:2612. doi: 10.1021/jm061349l. [DOI] [PubMed] [Google Scholar]
- 13.Rester U. Curr Opin Drug Discov Devel. 2008;11:559. [PubMed] [Google Scholar]
- 14.Lyne PD, Kenny PW, Cosgrove DA, Deng C, Zabludoff S, Wendoloski JJ, Ashwell S. J Med Chem. 2004;47:1962. doi: 10.1021/jm030504i. [DOI] [PubMed] [Google Scholar]
- 15.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. J Med Chem. 2002;45:2213. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 16.Guido RV, Oliva G, Andricopulo AD. Curr Med Chem. 2008;15:37. doi: 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- 17.Perola E, Xu K, Kollmeyer TM, Kaufmann SH, Prendergast FG, Pang YP. J Med Chem. 2000;43:401. doi: 10.1021/jm990408a. [DOI] [PubMed] [Google Scholar]
- 18.McDonald HM, Pruett PS, Deivanayagam C, Protasevich II, Carson WM, DeLucas LJ, Brouillette WJ, Brouillette CG. Acta Crystallogr Sect D. 2007;63:891. doi: 10.1107/S0907444907029769. [DOI] [PubMed] [Google Scholar]
- 19.Symersky J, Devedjiev Y, Moore K, Brouillette C, DeLucas L. Acta Crystallogr Sect D. 2002;58:1138. doi: 10.1107/s0907444902006698. [DOI] [PubMed] [Google Scholar]
- 20.a Stahl M, Rarey M. J Med Chem. 2001;44:1035. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]; b Luksch T, Chan NS, Brass S, Sotriffer CA, Klebe G, Diederich WE. Chem Med Chem. 2008;3:1323. doi: 10.1002/cmdc.200700270. [DOI] [PubMed] [Google Scholar]
- 21.Kontoyianni M, Sokol GS, McClellan LM. J Comput Chem. 2005;26:11. doi: 10.1002/jcc.20141. [DOI] [PubMed] [Google Scholar]
- 22.Bursulaya BD, Totrov M, Abagyan R, Brooks CL., III J Comput Aided Mol Des. 2003;17:755. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 23.Rarey M, Kramer B, Lengauer T. Bioinformatics. 1999;15:243. doi: 10.1093/bioinformatics/15.3.243. [DOI] [PubMed] [Google Scholar]
- 24.a Yang JM, Chen YF, Shen TW, Kristal BS, Hsu DF. J Chem Inf Model. 2005;45:1134. doi: 10.1021/ci050034w. [DOI] [PubMed] [Google Scholar]; c Wang R, Wang S. J Chem Inf Comput Sci. 2001;41:1422. doi: 10.1021/ci010025x. [DOI] [PubMed] [Google Scholar]
- 25.a Dessalew N, Bharatam PV. Biophys Chem. 2007;128:165. doi: 10.1016/j.bpc.2007.04.001. [DOI] [PubMed] [Google Scholar]; b Forino M, Jung D, Easton JB, Houghton PJ, Pellecchia M. J Med Chem. 2005;48:2278. doi: 10.1021/jm048962u. [DOI] [PubMed] [Google Scholar]
- 26.Shoichet BK, McGovern SL, Wei B, Irwin JJ. Curr Opin Chem Biol. 2002;6:439. doi: 10.1016/s1367-5931(02)00339-3. [DOI] [PubMed] [Google Scholar]
- 27.Bissantz C, Folkers G, Rognan D. J Med Chem. 2000;43:4759. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 28.Walsh CT, Wright GD. Chem Rev. 2005;105:391. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 29.Hegymegi-Barakonyi B, Székely R, Varga Z, Kiss R, Borbély G, Németh G, Bánhegyi P, Pató J, Greff Z, Horváth Z, Mészáros G, Marosfalvi J, Erös D, Szántai-Kis C, Breza N, Garavaglia S, Perozzi S, Rizzi M, Hafenbradl D, Ko M, Av-Gay Y, Klebl BM, Örfi L, Kéri G. Curr Med Chem. 2008;15:2760. doi: 10.2174/092986708786242886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.