

Abstract
The LMO2 gene is activated by chromosomal translocations in human T cell acute leukemias, but in mouse embryogenesis, Lmo2 is essential for initiation of yolk sac and definitive hematopoiesis. The LMO2 protein comprises two LIM–zinc-finger-like protein interaction modules and functions by interaction with specific partners in DNA-binding transcription complexes. We have now investigated the role of Lmo2-associated transcription complexes in the formation of the vascular system by following the fate of Lmo2-null embryonic stem (ES) cells in mouse chimeras. Lmo2 is expressed in vascular endothelium, and Lmo2-null ES cells contributed to the capillary network normally until around embryonic day 9. However, after this time, marked disorganization of the vascular system was observed in those chimeric mice that have a high contribution of Lmo2-null ES cells. Moreover, Lmo2-null ES cells do not contribute to endothelial cells of large vessel walls of surviving chimeric mice after embryonic day 10. These results show that Lmo2 is not needed for de novo capillary formation from mesoderm but is necessary for angiogenic remodeling of the existing capillary network into mature vasculature. Thus, Lmo2-mediated transcription complexes not only regulate distinct phases of hematopoiesis but also angiogenesis, presumably by Lmo2 interacting with distinct partners in the different settings.
Keywords: hematopoiesis, leukemia, chromosomal translocation
Development of a vascular system is essential for embryos to grow after they reach a certain size to allow blood flow to the rapidly developing embryonic tissues. In embryogenesis, the vascular system is constructed by two distinct processes (1–3). The first is vasculogenesis, which forms a primary capillary network from hemangioblasts, which are putative precursors specified from mesoderm. The mature vascular structures are formed by a second process, called angiogenesis, which is a remodeling of the endothelial cells from the existing capillary network into mature blood vessels. There is a close relationship between endothelial cells and blood cell specification (4–8). Yolk sac hematopoiesis begins around embryonic day (E)7.5 in yolk sac blood islands, where centrally located cells give rise to blood cells and those on the periphery flatten and form vascular endothelial cells. On the other hand, definitive hematopoiesis initiates in the aorta-gonad-mesonephros region around E10.5 of mouse embryogenesis (9, 10). Moreover, in definitive hematopoiesis, blood cells are thought to be derived from endothelial cells of large arteries such as the dorsal aorta, umbilical artery, and vitelline artery in humans (11).
Transcription factors involved in these processes are important in control of both endothelial and hematopoietic cell fate. Few transcription factors that are involved in vasculogenesis or angiogenesis have been identified. Lmo2 was a candidate for such a role because of its role in hematopoiesis. LMO2 was first discovered by its homology with the T cell oncogene LMO1 (12), and it is also activated by chromosomal translocations in some T cell leukemias (12, 13). Normally, Lmo2 is an essential protein in both the primitive and definitive hematopoietic pathways (14, 15) and seems to mediate multimeric protein complexes through its LIM-domain zinc-finger-like structures (16, 17). The ability of the Lmo2 protein to interact with other proteins led to the identification of a multimeric protein complex in erythroid cells, comprising Tal1/Scl, E47, Ldb1, and GATA-1 in addition to Lmo2, which binds specifically to a bipartite DNA sequence (18). In view of the various roles for Lmo2 in hematopoiesis, it has been proposed that the Lmo2 acts as a bridging molecule that facilitates the formation of different multimeric complexes that regulate transcription of different genes at stages of hematopoiesis (15, 19, 20).
To assess a possible function of Lmo2 in transcriptional regulation of vascular development and consequent blood cell specification, the formation of the vascular system has been assessed in mouse chimeras generated with Lmo2-null embryonic stem (ES) cells. We have introduced the lacZ gene into the Lmo2 gene of ES cells by homologous recombination. The fate of ES-derived cells after injection into blastocysts was followed in embryonic vasculogenesis and angiogenesis via β-galactosidase expressed from the Lmo2 promoter. Comparing heterozygous and homozygous null Lmo2 ES cell fate, we observed that Lmo2 was expressed in endothelial cells during mouse embryogenesis and that vasculogenesis proceeded normally in the absence of Lmo2. However, the Lmo2 gene plays a critical role in the construction of the mature vascular network (angiogenesis), because this process did not occur in chimeras with a high contribution from Lmo2-null ES cells.
Materials and Methods
Homologous Recombination.
Construction of the Lmo2–lacZ targeting vector is described in Fig. 1. The linearized targeting vector DNA (25 μg) was used in the transfection to CCB ES cells by electroporation. Selection of resistant clones was done by cell growth in medium containing either 400 μg/ml G418 (GIBCO) or 300 μg/ml hygromycin B (Calbiochem). Targeted clones were found by sequentially hybridizing ES cell DNA [by standard filter hybridization (ref. 21) as described (ref. 22)] with flanking probes from both sides of the targeting region (ref. 14; see Fig. 1 legend) and by confirming the presence of a single insertion of the targeting fragment with an internal probe.
Figure 1.

Lmo2–lacZ fusion gene knock-in by homologous recombination. Two constructs were used to create the Lmo2-targeted ES cells used in this study. CCB ES cells were transfected with pKO5-lacZ-neo (A), and targeted events were detected by filter hybridization. Several targeted clones were initially analyzed, and one (designated KZ26) was chosen based on its ability to yield consistently high levels of chimerism in mice after injection into blastocysts. The KZ26 clone +/− was used for a second transfection with pKO5hygro(tk), and three clones (clones 1, 16, and 64) were studied in which the second allele of Lmo2 had been targeted to yield KZ26 −/− (Lmo2 −/−) ES cells. (A) Construction of the Lmo2–lacZ fusion gene targeting vector was done by cloning of 4.5-kilobase (kb) blunt-ended SfiI fragment of SfiI-lacZ-MC1neopA (38) into a blunt-ended BamHI site of gene targeting vector pKO5(tk) (14). In the resulting clone, KO5-lacZ-neo, the 24th codon of Lmo2 (exon 2) was linked to the 2nd codon of lacZ by a 12-bp linker sequence. The hygromycin-targeting vector pKO5hygro(tk) and the probes used to assess gene targeting, indicated on the map of pKO5-lacZ-neo, have been described (14). The targeting of the pKO5-lacZ-neo into Lmo2 yields a 6-kb SacI fragment with probe A (compared with a 9-kb germ-line band) and a 9.5-kb BamHI band with probe B (compared with a 12-kb germ-line band). Probe C is a neo probe used to verify a single insertion of the targeting vector. Targeting of pKO5hygro(tk) into Lmo2 yields a 10.8-kb SacI band with probe A and a 13.8-kb BamHI band with probe B. S, SacI; B, BamHI. (B) Detection of homologous recombination in ES clone DNA by Southern filter hybridization with probe A (3′ flanking), probe B (5′ flanking), and probe C (internal). Hybridization of representative +/+ (wild-type) and +/− (neo-targeted) clones is shown. Wt, wild-type hybridization band. (C) Identification of three independent double-targeted Lmo2 −/− clones (clones 1, 16, and 64) by filter hybridization with probe A. The integrity of these second targeted alleles was verified by using probe B and an internal hygromycin probe (data not shown).
Production of Chimeric Mice and Germ-Line Transmission.
ES cells were microinjected into C57/BL6 blastocysts and transferred to CBA/C57/BL6 recipients. For the embryonic studies, the day of injection was designated as E2.5. Germ-line transmission of the targeted allele was confirmed by Southern filter hybridization with tail DNA. The embryonic lethal phenotype of homozygous Lmo2 null −/− embryos was confirmed for the KZ26 germ-line transmitted allele by interbreeding heterozygous +/− carrier mice and examining the resulting embryos. This procedure showed that the KZ26 Lmo2–lacZ null mutation behaves the same as the previously described Lmo2 mutation (14).
Whole-Mount 5-Bromo-4-Chloro-3-Indolyl β-d-Galactoside (X-Gal) Staining.
Embryos at each stage were excised from the uterus, and any maternal decidual tissue was removed. Whole embryos were examined for X-Gal staining of β-galactosidase activity (resulting from Lmo2–lacZ gene expression) according to the procedure described in refs. 23 and 24. For histological studies, embryos were fixed in 10% (vol/vol) buffered formalin after X-Gal staining and embedded in paraffin. Sections were mounted on slides and counterstained with eosin.
Results
Construction of lmo2–lacZ Fusion Gene in ES Cells by Homologous Recombination.
Fig. 1A shows the structure of the Lmo2–lacZ fusion gene with lacZ incorporated into exon 2 of Lmo2. An ES clone was selected with the lacZ gene knock-in of one Lmo2 allele (Fig. 1A, KZ26 +/− ES cells), and this clone was retransfected with a Lmo2-hygromycin vector to create ES cells with a second targeted Lmo2 allele (KZ26 −/−; three independent clones were studied: clones 1, 16, and 64). Expression of β-galactosidase is controlled by the Lmo2 gene in these ES cells or their derivatives in vivo after injection of the ES cells into blastocysts and generation of embryos. In addition, the Lmo2 +/− ES cell (KZ26) was used to obtain germ-line transmission of the targeted allele to give KZ26 heterozygous mice.
Lmo2 Is Expressed in Endothelium.
We have used β-galactosidase to follow the fate of Lmo2-expressing ES cells in developing embryos. Initially, we used the KZ26 heterozygous mice to study the pattern of β-galactosidase expression from E8.5 to E14.5. During these embryonic stages, we found X-Gal staining chiefly in endothelial cells of the whole-body vascular system. From E11.5, X-Gal staining was also found just beneath the apical ectodermal ridge of limb buds, which is mesenchymal tissue called the progress zone. This tissue is avascular area and the field of sprouting angiogenesis. Additional sites were the tail bud and hippocampus. Fig. 2 A and B shows E10.5 and E12.5 Lmo2 +/− embryos, respectively. Expression of β-galactosidase in the limb buds can be seen in histological sections (Fig. 2D). The X-Gal staining of vessel endothelium is shown in a tissue section (Fig. 2C). These β-galactosidase expression data are compatible with Lmo2 expression patterns observed with RNA in situ hybridization (25–28), indicating that the expression of β-galactosidase in endothelial cells is also a true reflection of Lmo2 promoter activity in embryogenesis.
Figure 2.
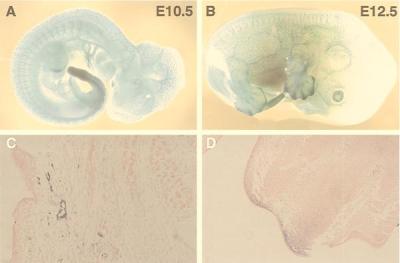
Whole-mount β-galactosidase staining of Lmo2 +/− heterozygous embryos. The targeted ES clone KZ26 (with one null Lmo2 allele) was injected into blastocysts; chimeric mice were obtained, and germ-line transmission of the Lmo2 null allele was obtained. Heterozygous KZ26 mice were crossed with C57/BL6 mice, and embryos were obtained at E10.5 and E12.5. These embryos were stained with X-Gal as a substrate for β-galactosidase activity. Blue staining denotes areas of β-galactosidase caused by Lmo2–lacZ gene expression. (A) E10.5 embryo. X-Gal staining was seen on major blood vessel walls and capillaries of whole body. (B) E12.5 embryo. In addition to β-galactosidase staining of vasculature, prominent staining was found in the limb buds and the tip of the tail. (C) Histological section of an E12.5 embryo stained for β-galactosidase and counterstained with eosin. Blood vessel endothelial cells can be observed in a background of eosin-stained tissue. (D) Histological section of the limb bud of an E12.5 embryo stained for β-galactosidase and counterstained with eosin. The region beneath the apical ectodermal ridge of a limb bud is positive for β-galactosidase.
Lmo2-Null ES Cells Cannot Contribute to Endothelial Cells of Large Vessels After E11.
As a method of following the fate of ES cells with the Lmo2–lacZ fusion gene and studying the consequence of the homozygous null mutation, we have compared the β-galactosidase staining patterns of chimeric embryos derived from injection of KZ26 +/− and −/− ES cells into blastocysts (Fig. 3). A number of findings emerged from this study. At E9.5, there was no obvious difference between +/− and −/− chimeric mice (data not shown). When E11.5 embryos of comparable size were stained with X-Gal from either KZ26 +/− or −/− injections (Fig. 3 A and B respectively), the β-galactosidase patterns were markedly different with respect of endothelial cell staining. KZ26 +/− E11.5 chimeric embryos have a staining pattern very like that of heterozygous embryos (Fig. 2). On the other hand, the E11.5 −/− chimeric embryos showed almost no contribution of ES-derived cells to the vascular system (Fig. 3B), although the ES cell contribution to the limb buds and hippocampus was retained. Peripheral scattered endothelial cells were observed by X-Gal staining, but there was no contribution of Lmo2 −/− ES cells to major vessel endothelial cells. The consistency of this observation was shown by examination of KZ26 +/− and −/− embryos at E12.5. Fig. 3C shows a KZ26 +/− chimeric embryo with an extensive staining pattern of endothelial cells and, in addition, limb bud and hippocampal staining. By contrast, E12.5 KZ26 −/− embryos from clone 1 (Fig. 3D), clone 16 (Fig. 3E), or clone 64 (Fig. 3F) did not have endothelial cell staining, but each had comparable limb bud, tail, and hippocampal staining. This lack of E12.5 endothelial cell staining parallels that of the E11.5 −/− chimeric embryos (Fig. 3B).
Figure 3.
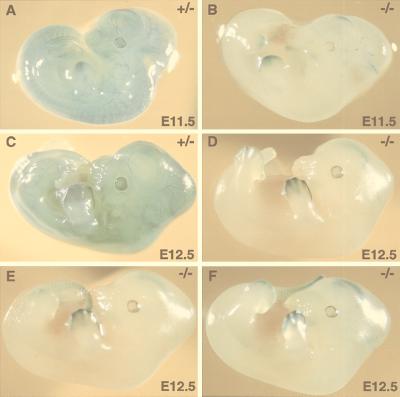
Comparison of whole-mount X-Gal staining of KZ26 +/− and KZ26 −/− chimeric embryos. ES cells with one (KZ26 +/−) or two (KZ26 −/−) Lmo2 null alleles were injected into C57/BL6 blastocysts and transferred to recipient females. At the indicated embryonic days, embryos were excised and whole-mount stained with X-Gal to test for β-galactosidase activity caused by Lmo2–lacZ gene expression. (A) E11.5 KZ26 +/− chimeric embryo with a staining pattern similar to that seen in heterozygous KZ26 mice. (B) E11.5 KZ26 −/− chimeric embryo derived from KZ26 −/− clone 1. In this embryo, ES cell contribution (blue) can be seen in the hippocampus and limb buds, and very few endothelial cells are stained blue (i.e., those of ES cell origin). (C) E12.5 KZ26 +/− chimeric embryo. Like the E11.5 KZ26 +/− embryo, the staining pattern was very similar to that seen in heterozygous KZ26 mice. (D–F) E12.5 KZ26 −/− chimeric embryos of three independent −/− clones. (D) An embryo derived from injection of KZ26 −/− clone 1. (E) An embryo from clone 16. (F) An embryo from clone 64. In KZ26 −/− chimeric embryos after E11.5, there was no contribution of −/− ES cells in endothelial cells of major vessels, whereas expression is maintained in the hippocampus, the limb bud, and tail. This selective loss of ES contribution in blood vessel endothelium suggests an essential role of Lmo2 protein in the maturation of vascular network (angiogenesis).
Vascular Disorganization and Growth Retardation in High-Level Lmo2-Null Chimeras.
Vascular endothelial remodeling begins at around E10.5 in mice. The effect of the null Lmo2 mutation was studied at this crucial stage. Embryos were obtained at E10.5 from a litter resulting from injection of KZ26 −/− clone 1 into blastocysts and were whole-mount stained with X-Gal. The staining patterns of seven embryos were compared (Fig. 4 B–H) with an E10.5 KZ26 +/− chimera (Fig. 4A). In the −/− chimeras, there was remarkable size variation, which was inversely proportional with the degree of ES cell contribution to endothelial cells (as judged by X-Gal staining). Mice with high X-Gal staining (i.e., high contribution of Lmo2 −/− ES-derived cells) were smaller than those with a low contribution in the same litters (Fig. 4). Normally sized Lmo2 −/− embryos had essentially no endothelial cells stained, although they retained limb bud and tail staining. In those small mice with high X-Gal staining, there was no well organized vascular system (Fig. 4 J and K), suggesting that when the chimerism of −/− ES cells was high, the endothelial remodeling fails because of a lack of Lmo2; these embryos are consequently destined to die (see below; Table 1). If there is relatively low chimerism in the embryos, they survive, presumably because blastocyst-derived cells can replace ES-derived ones in the remodeling process.
Figure 4.
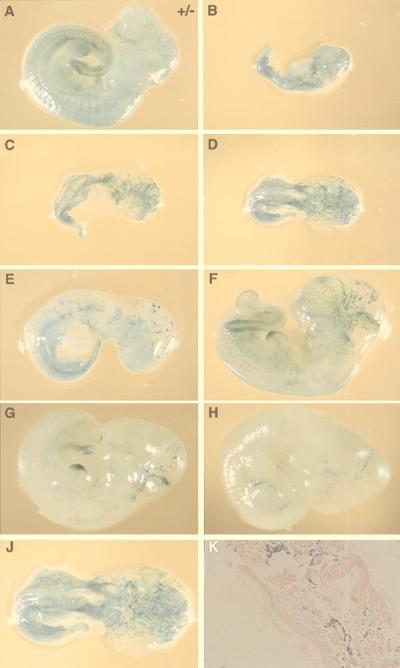
Growth retardation and disorganization of the vascular system in E10.5 Lmo2 −/− chimeric embryos. Chimeric embryos were generated by injection of KZ26 +/− ES cells (A) or KZ26 −/− ES cells (clone 1) (B–J) into blastocysts, obtaining embryos at E10.5. Embryos were subjected to whole-mount X-Gal staining. The embryos shown in B–J are litter mates. The difference in the size and X-Gal staining indicates an inverse relationship between size and level of ES cell contribution, as the higher chimeras (i.e., those with higher levels of β-galactosidase staining) are growth retarded. (A) E10.5 KZ26 +/− chimeric embryo. This photograph is of a representative embryo, and there was no marked size difference among the litter mates. (B–J) A series of whole-mount X-Gal staining of KZ26 −/− chimeric embryos. From A to H, the magnification rate is the same. (J) The enlarged figure (≈×1.7) of the embryo shown in D shows the major disorganization in vasculature of this high chimeric −/− embryo. No mature vessels were found, unlike the KZ26 +/− embryo (A). Histological section of the −/− chimeric embryo shown in D stained with X-Gal and counterstained with eosin.
Table 1.
Summary of chimeric mouse survival and LacZ expression
| Embryonic day | Uterine sacs (A) | Embryos (B) | B/A, % | LacZ positive (C) | C/B, % | No. of embryos per clone
|
||
|---|---|---|---|---|---|---|---|---|
| −/−Clone 1 | −/−Clone 16 | −/−Clone 64 | ||||||
| KZ26 (+/−) chimeric mice | ||||||||
| E9.5 | 30 | 30 | 100 | 15 | 50 | |||
| E10.5 | 18 | 18 | 100 | 13 | 72 | |||
| E11.5 | 15 | 15 | 100 | 11 | 73 | |||
| E12.5 | 15 | 15 | 100 | 10 | 67 | |||
| KZ26 (−/−) chimeric mice | ||||||||
| E9.5 | 12 | 12 | 100 | 10 | 83 | 10 | 2 | nd |
| E10.5 | 134 | 117 | 87 | 85 | 73 | 38 | 22 | 57 |
| E11.5 | 92 | 29 | 32 | 7 | 24 | 12 | 17 | nd |
| E12.5 | 71 | 22 | 31 | 5 | 23 | 16 | 0 | 6 |
| E14.5 | * | 20 | 8 | 40 | 13 | 7 | nd | |
Summary of chimeric mouse survival. Chimeric mice were produced from the injection into blastocysts of ES cells with a knock-in of lacZ into one allele of Lmo2 (KZ26 +/−) or a knock-in of lacZ into one allele of Lmo2 and a knock-out of the second allele by insertion of hygromycin (KZ26 −/−). These blastocysts were implanted into recipients. At the specified embryonic time points, uterine sacs were counted, and embryos were dissected and stained for β-galactosidase activity with X-Gal. The data are expressed as the percentage of live embryos per total uterine sacs (A/B) (those embryos with marked degeneration were not included) and the percentage of β-galactosidase-positive embryos (C/B). For the KZ26 −/− ES clones, the number of embryos examined for each clone is given (note that no clone 64 embryos were examined at E9.5, E11.5, or E14.5).
*Uterine sacs uncountable because of degeneration.
These data indicate a selective inability of Lmo2-null ES cells to contribute to endothelial cells after about E10.5–E11.5. The consistency of these observations has been confirmed by analysis of a large number of embryos from litters made by injecting the KZ26 −/− clones into blastocysts. These data are summarized in Table 1. Although the chimeric embryos made with the heterozygous ES cells KZ26 uniformly survived at all stages from E9.5 to E12.5 (Table 1), survival of the KZ26 −/− chimeras was 100% at E9.5 only. Thereafter, viability progressively decreased, concomitant with lower proportions of lacZ-positive chimeras. All together, 188 −/− chimeric embryos were analyzed, and a large gap in the survival ratio and X-Gal-positive ratio occurs between E10.5 and E11.5. There was no such relationship in +/− chimeric mice. Endothelial remodeling and survival do not occur if embryos have high proportions of Lmo2 −/− ES-cell derivatives, because they fail to form a mature vascular system. It is possible that Lmo2 −/− endothelial cells become selectively apoptotic after E10.5.
Discussion
The Lmo2 LIM-Only Protein Is Specifically Needed for Angiogenesis.
Vasculogenesis and angiogenesis are separate processes requiring separate signals and presumably separate transcriptional activity. Several tyrosine kinase-type cell surface receptors have been shown to be specifically expressed on endothelial cells, and they have important roles in either vasculogenesis or angiogenesis (3, 29). These signaling pathways are presumably responsible for activating transcription factor expression in the distinct vascular formation pathways. The requirement for Lmo2 in development of definitive hematopoiesis (15) suggested that this factor might be involved in endothelial cell differentiation. The data presented here confirm this possibility by showing a selective inability of Lmo2-null ES cells to contribute to vascular endothelial cells after about E10.5. The initial process of vasculogenesis, however, does not seem to require Lmo2, and thus Lmo2 is only necessary in vessel formation in angiogenesis. These findings suggest that at least two different functions of Lmo2 must now be considered, i.e., in angiogenesis and in hematopoiesis. These putative roles of Lmo2 may be enacted before the specification of the hematopoietic stem cells or in proliferation and/or further differentiation of hematopoietic stem cells. If the origin of hematopoietic stem cells occurs in the aorta-gonad-mesonephros region in the endothelium of large arteries, the role of Lmo2 in angiogenesis (before the specification of hematopoietic stem cells) may also explain the role in definitive hematopoiesis (although it would not explain that of yolk sac hematopoiesis). The expression of Lmo2 in hematopoietic progenitor cells, however, also suggests that specific roles may be associated with different hematopoietic lineages.
Lmo2-Containing Complexes in Vascular Formation.
The Lmo2 protein comprises two LIM domains that provide surfaces for protein interaction (30). Among proteins that can interact with Lmo2 is the basic helix–loop–helix T cell product oncogene Tal1/Scl (16, 17). This protein is expressed in endothelial cells (31) and has a role in hematopoiesis (32–35) akin to that observed for Lmo2 (14, 15). Thus, it is interesting that studies of null mutation of Tal1 gene showed that the gene has a role in yolk sac angiogenesis (36). Lmo2 binds to Tal1, in conjunction with E47, forming a DNA-binding element (18). A possible explanation for the Lmo2 function in specific embryological functions is that the Lmo2 molecule brings different sets of DNA-binding proteins together with the Tal1-E47 to create distinct bipartite DNA-binding complexes in different developmental cell types. This notion of pluralism for Lmo2 complexes (15, 18, 19) parallels the cocktail party model of hematopoiesis (37).
It is intriguing that Lmo2 is expressed in embryonic vasculogenesis but does not seem to have a role in differentiation of this mesoderm. It may be that Lmo2 can catalyze the formation of a multimeric complex, possibly with Tal1, E47, and Ldb1, at the vasculogenesis stage, but that this complex cannot function at that point in development, perhaps because of the lack of other interacting partners. As differentiation proceeds, new transcription factors may be expressed (perhaps activated by angiogenic growth factors) that could contribute to the Lmo2-associated complex and thereby create a DNA-binding complex that controls the target gene expression required for angiogenesis to proceed. The variation in composition of these putative complexes is an interesting area of investigation, as are the target genes regulated (positively or negatively) by the different Lmo2-containing complexes.
Finally, the role of Lmo2 in embryonic angiogenesis has implications in adult angiogenesis. There are a number of clinical conditions in which angiogenesis is important, such as tumor neovascularization, burn recovery, wound healing, and diabetic retinopathy. Lmo2 is expressed in the endothelial cells of adult mice (Y.Y. and T.H.R., unpublished observation) and humans (M. McCormack and T.H.R., unpublished observation), and TAL1 is also expressed in endothelial cells (31). The regulation of angiogenesis by these nuclear factors, therefore, has potential application to the treatment of angiogenesis-associated diseases in humans.
Acknowledgments
We would like to thank Gareth King, Theresa Langford, and Angela Middleton for expert help. Y.Y. was supported by an award from the National Foundation for Cancer Research.
Abbreviations
- En
embryonic day n
- ES
embryonic stem
- X-Gal
5-bromo-4-chloro-3-indolyl β-d-galactoside
- kb
kilobase
References
- 1.Hanahan D, Folkman J. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 2.Risau W. Nature (London) 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 3.Gale N W, Yancopoulos G D. Genes Dev. 1999;13:1055–1066. doi: 10.1101/gad.13.9.1055. [DOI] [PubMed] [Google Scholar]
- 4.Dieterlen-Lièvre F, Martin C. Dev Biol. 1981;88:180–191. doi: 10.1016/0012-1606(81)90228-1. [DOI] [PubMed] [Google Scholar]
- 5.Pardanoud L, Yassine F, Dieterlen-Lievre F. Development (Cambridge, UK) 1989;105:473–485. doi: 10.1242/dev.105.3.473. [DOI] [PubMed] [Google Scholar]
- 6.Jaffredo T, Gautier R, Eichmann A, Dieterlen-Lievre F. Development (Cambridge, UK) 1998;125:4575–4583. doi: 10.1242/dev.125.22.4575. [DOI] [PubMed] [Google Scholar]
- 7.Shalaby F, Ho J, Stanford W L, Fischer K-D, Schuh A C, Schwartz L, Bernstein A, Rossnat J. Cell. 1997;89:981–990. doi: 10.1016/s0092-8674(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 8.Choi K, Kennedy M, Kazarov A, Papadimitriou J C, Keller G. Development (Cambridge, UK) 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- 9.Müller A M, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Immunity. 1994;1:291–301. doi: 10.1016/1074-7613(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 10.Medvinsky A, Dzierzak E. Cell. 1996;86:897–906. doi: 10.1016/s0092-8674(00)80165-8. [DOI] [PubMed] [Google Scholar]
- 11.Tavian M, Hallais M-F, Peault B. Development (Cambridge, UK) 1999;126:793–803. doi: 10.1242/dev.126.4.793. [DOI] [PubMed] [Google Scholar]
- 12.Boehm T, Foroni L, Kaneko Y, Perutz M P, Rabbitts T H. Proc Natl Acad Sci USA. 1991;88:4367–4371. doi: 10.1073/pnas.88.10.4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Royer-Pokora B, Loos U, Ludwig W-D. Oncogene. 1991;6:1887–1893. [PubMed] [Google Scholar]
- 14.Warren A J, Colledge W H, Carlton M B L, Evans M J, Smith A J H, Rabbitts T H. Cell. 1994;78:45–58. doi: 10.1016/0092-8674(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 15.Yamada Y, Warren A W, Dobson C, Forster A, Pannell R, Rabbitts T H. Proc Natl Acad Sci USA. 1998;95:3890–3895. doi: 10.1073/pnas.95.7.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valge-Archer V E, Osada H, Warren A J, Forster A, Li J, Baer R, Rabbitts T H. Proc Natl Acad Sci USA. 1994;91:8617–8621. doi: 10.1073/pnas.91.18.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wadman I, Li J, Bash R O, Forster A, Osada H, Rabbitts T H, Baer R. EMBO J. 1994;13:4831–4839. doi: 10.1002/j.1460-2075.1994.tb06809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wadman I A, Osada H, Grutz G G, Agulnick A D, Westphal H, Forster A, Rabbitts T H. EMBO J. 1997;16:3145–3157. doi: 10.1093/emboj/16.11.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rabbitts T H. Genes Dev. 1998;12:2651–2657. doi: 10.1101/gad.12.17.2651. [DOI] [PubMed] [Google Scholar]
- 20.Rabbitts T H, Bucher K, Chung G, Grütz G G, Warren A J, Yamada Y. Cancer Res. 1999;59:1794S–1798S. [PubMed] [Google Scholar]
- 21.Southern E M. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 22.LeFranc M-P, Forster A, Baer R, Stinson M A, Rabbitts T H. Cell. 1986;45:237–246. doi: 10.1016/0092-8674(86)90388-0. [DOI] [PubMed] [Google Scholar]
- 23.Allen N D, Cran D G, Barton S C, Hettle S, Reik W, Surani M A. Nature (London) 1988;333:852–855. doi: 10.1038/333852a0. [DOI] [PubMed] [Google Scholar]
- 24.Shalaby F, Rossant J, Yamaguchi T P, Gertsenstein M, Wu X-F, Breitman M L, Schuh A C. Nature (London) 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 25.Foroni L, Boehm T, White L, Forster A, Sherrington P, Liao X B, Brannan C I, Jenkins N A, Copeland N G, Rabbitts T H. J Mol Biol. 1992;226:747–761. doi: 10.1016/0022-2836(92)90630-3. [DOI] [PubMed] [Google Scholar]
- 26.Hinks G L, Shah B, French S J, Campos L S, Staley K, Hughes J, Sofroniew M V. J Neurol. 1997;17:5549–5559. doi: 10.1523/JNEUROSCI.17-14-05549.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenny D A, Jurata L W, Saga Y, Gill G N. Proc Natl Acad Sci USA. 1998;95:11257–11262. doi: 10.1073/pnas.95.19.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng C, Justice N J, Abdelilah S, Chan Y-M, Jan L Y, Jan Y N. Proc Natl Acad Sci USA. 1998;95:10637–10642. doi: 10.1073/pnas.95.18.10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mustonen T, Alitalo K. J Cell Biol. 1995;129:895–898. doi: 10.1083/jcb.129.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Garcia I, Rabbitts T H. Trends Genet. 1994;10:315–320. doi: 10.1016/0168-9525(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 31.Hwang L-Y, Siegelman M, Davis L, Oppenheimer-Marks N, Baer R. Oncogene. 1993;8:3043–3046. [PubMed] [Google Scholar]
- 32.Shivdasani R A, Mayer E, Orkin S H. Nature (London) 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- 33.Robb L, Lyons I, Li R, Hartley L, Kontgen F, Harvey R P, Metcalf D, Begley C G. Proc Natl Acad Sci USA. 1995;92:7075–7079. doi: 10.1073/pnas.92.15.7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt F W, Orkin S H. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 35.Robb L, Elwood N J, Elefanty A G, Kontgen F, Li R, Barnet L D, Begley C G. EMBO J. 1996;15:4123–4129. [PMC free article] [PubMed] [Google Scholar]
- 36.Visvader J E, Fujiwara Y, Orkin S H. Genes Dev. 1998;12:473–479. doi: 10.1101/gad.12.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sieweke M H, Graf T. Curr Opin Genet Dev. 1998;8:545–551. doi: 10.1016/s0959-437x(98)80009-9. [DOI] [PubMed] [Google Scholar]
- 38.Dear T N, Colledge W H, Carlton M B L, Lavenir I, Larson T, Smith A J H, Warren A J, Evans M J, Sofroniew M V, Rabbitts T H. Development (Cambridge, UK) 1995;121:2909–2915. doi: 10.1242/dev.121.9.2909. [DOI] [PubMed] [Google Scholar]