

Abstract
Objective
Neonatal seizures occur frequently, are often refractory to anticonvulsants, and are associated with considerable morbidity and mortality. Genetic and electrophysiological evidence indicates that KCNQ voltage-gated potassium channels are critical regulators of neonatal brain excitability. This study tests the hypothesis that selective openers of KCNQ channels may be effective for treatment of neonatal seizures.
Methods
We induced seizures in postnatal day 10 rats with either kainic acid or flurothyl. We measured seizure activity using quantified behavioral rating and electrocorticography. We compared the efficacy of flupirtine, a selective KCNQ channel opener, with phenobarbital and diazepam, two drugs in current use for neonatal seizures.
Results
Unlike phenobarbital or diazepam, flupirtine prevented animals from developing status epilepticus (SE) when administered prior to kainate. In the flurothyl model, phenobarbital and diazepam increased latency to seizure onset, but flupirtine completely prevented seizures throughout the experiment. Flupirtine was also effective in arresting electrographic and behavioral seizures when administered after animals had developed continuous kainate-induced SE. Flupirtine caused dose-related sedation and suppressed EEG activity, but did not result in respiratory suppression or result in any mortality.
Interpretation
Flupirtine appears more effective than either of two commonly used anti-epileptic drugs, phenobarbital and diazepam, in preventing and suppressing seizures in both the kainic acid and flurothyl models of symptomatic neonatal seizures. KCNQ channel openers merit further study as potential treatments for seizures in infants and children.
Epileptic seizures occur commonly in the first days after birth.1 Such neonatal seizures are usually symptomatic, arising as a result of developmental abnormalities, in utero injuries, perinatal hypoxia-ischemia, infection, and other causes. Patients experiencing neonatal seizures are at substantial risk of mortality and long-term morbidity, including static encephalopathy, cerebral palsy, and chronic epilepsy.2, 3 The first-line drugs given to neonates, phenobarbital and phenytoin, are effective in less than 50% of cases4. Moreover, phenobarbital, benzodiazepines and phenytoin have been shown to cause widespread neuronal apoptosis when given to young rodents, raising concerns about administration of these drugs in human infants.5-7 The need for new treatments for neonatal seizures that are both safer and more effective has been widely acknowledged.1, 7, 8
Genetic, physiological, and pharmacological evidence calls attention to KCNQ potassium channels (also termed Kv7 and M-channels) as potential molecular targets for treatment of neonatal seizures.9 Loss-of-function mutations in the KCNQ2 and KCNQ3 genes cause benign familial neonatal seizures (BFNS), an uncommon but highly penetrant, dominantly-inherited syndrome characterized by seizures that recur frequently over the first weeks of life.10-12 Experimental inhibition of KCNQ channels in rodents, by pharmacological and genetic methods, dramatically promotes neuronal and network hyperexcitability and seizures in neonatal animals and in slices from neonatal brain; these effects diminish or disappear progressively with maturation. KCNQ2 and KCNQ3 are highly expressed on both myelinated and unmyelinated axons, and are strictly colocalized with clusters of voltage-gated sodium channels in the proximal axon (where action potentials initiate) and at the nodes of Ranvier.14, 17-19 It has been proposed that this strategic localization at the exact sites where action potentials arise and are conducted allows KCNQ channels to exert strong control over neuronal firing.17, 18
Selective neuronal KCNQ channel openers have been identified among drugs already in human use, including diclofenac (a widely used anti-inflammatory agent), flupirtine (a non-opioid analgesic in approved use in Europe) and retigabine (which is currently in phase 3 clinical trials for adult partial epilepsy in the U.S. and Europe).20-22 Additional potent and selective KCNQ channel openers have recently been discovered through directed high-throughput screening and have begun to be subjected to preclinical testing for adult epilepsy and pain.23-25 KCNQ openers increase channel activity significantly above normal levels by speeding opening rates, slowing closing rates, and reducing the membrane depolarization required for activation. Flupirtine and retigabine are close structural analogues (Figure 1). Flupirtine and retigabine have been shown to exhibit neuroprotective activity in several experimental systems, including models of infantile neuronal ceroid lipofuscinosis.26-28 Remarkably, KCNQ channel opener compounds have not been evaluated as a potential treatments for neonatal seizures or status epilepticus (SE). The human KCNQ2 and KCNQ3 channel loss-of-function phenotype, BFNS, suggests the channels are present, active and important during the neonatal period. Thus, augmenting KCNQ channel activity is a rational strategy for safe suppression of hyperexcitability during the neonatal period.
Figure 1. Flupirtine and retigabine are close analogues.
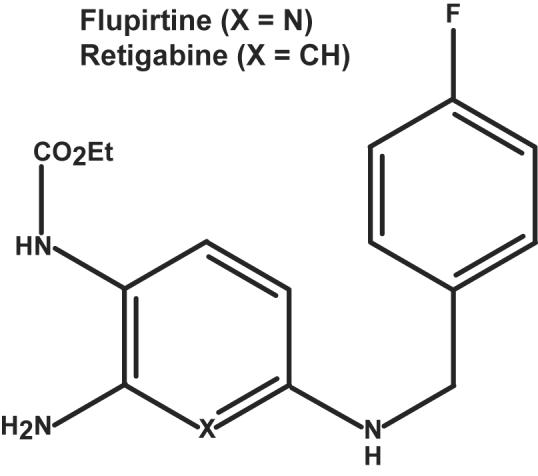
“X” indicates the position varied in the two compounds; substitutions are as indicated.
In early trials using adult rodent models and adult human patients, flupirtine showed evidence of anti-epileptic efficacy.29 Here, we have tested flupirtine's utility for treatment of neonatal seizures and SE. We have compared flupirtine to two drugs widely used for neonatal seizures, phenobarbital and diazepam. In two well-established models, using the glutamate receptor agonist kainic acid or the convulsant gas flurothyl, flupirtine appeared more effective than either standard agent in preventing the onset of seizures in rat pups. In the kainic acid model, which produces seizures that evolve into SE lasting hours, flupirtine was more effective than either standard agent in diminishing the severity of electrographic and behavioral seizures. Indeed, flupirtine was effective even when its administration was withheld until after kainate-induced seizure activity had progressed to continuous SE. These findings indicate that KCNQ channel opener therapy for neonatal seizures deserves further investigation.
Materials and Methods
Chemicals and drugs
Kainic acid (Sigma, St. Louis, MO) stocks dissolved in water were stored at -20° C; aliquots were diluted in normal saline solution immediately before use. Flupirtine maleate (Sigma) was freshly dissolved in 1:2 (vol/vol) mixture of DMSO and saline solution on the day of use. Solutions of diazepam (Hospira) and phenobarbital (Baxter) for injection were purchased from the pharmacy of the Hospital of the University of Pennsylvania, and where needed, serially diluted in either DMSO/saline 3:7 (vol/vol, diazepam) or saline (phenobarbital). All drugs were administered by IP injection. All experiments were performed in parallel on littermates randomly assigned to receive vehicle or one or more therapeutic drugs, in equal volumes.
Status epilepticus induction
The Institutional Animal Care and Use Committees of the University of Pennsylvania and Children's Hospital of Philadelphia approved all procedures used. Experimental design emphasized use of a minimal number of animals, and minimization of potential discomfort. Animals were warmed under an incandescent lamp during all experimental procedures to maintain body temperature. Pregnant Sprague-Dawley rats were obtained (Charles River laboratory, Kingston, PA) and allowed to give birth. Postnatal day 10 (P10) pups of both sexes were removed from home cages, weighed, placed in an observation cage, given intraperitoneal (IP) injections of kainic acid, and visually monitored for seizure activity by an observer blinded to treatment group. In preliminary experiments, 2 mg/kg IP kainic acid consistently induced seizures that progressed to status epilepticus (SE) in > 90% of the rat pups within 40-50 minutes with < 10% mortality.
Seizure induction using flurothyl
Flurothyl-induced seizure latencies were measured using procedures previously described.30, 31 P10 rat pups were placed in an air-tight cylindrical plastic chamber (radius, 7.5 cm; height, 18 cm) within a fume hood. Using a syringe pump (Harvard Apparatus, Holliston, MA), 50 μl/min of the volatile liquid 2,2,2-trifluoroethyl ether (flurothyl; Sigma) was introduced into the chamber. Latency to development of forelimb and hindlimb tonic extension was measured by an observer blinded to treatment group.
Electrocorticography
Brain electrical activity was monitored using bilateral screw electrodes placed over the motor cortices.32 P10 rats were anaesthetized with continuous inhalation using an isoflurane dispenser (System Specialties Inc.). A screw electrode behind the lambda served as ground and reference. To reduce noise contamination, EEG signals were directed to miniature preamplifiers glued with dental adhesive to the skull, powered through the EEG cable. Pups were allowed to recover from surgery and anesthesia for one hour. EEG was recorded using a custom-designed 16-channel EEG machine (ALA Scientific Instruments, Westbury, NY) and house-written software generated using MatLab (MathWorks, Natick, MA). Baseline EEG was recorded for approximately 30 minutes before kainic acid injection. The onset of stage 5 behavioral seizures was well correlated with EEG showing continuous seizure activity (see Results). Anti-epileptic drug or vehicle-only control solutions were injected 15 minutes after the onset of stage 5 seizures, determined by both behavioral and electrographic criteria (see below). The EEG was recorded for a minimum of 2 hours after the drug or vehicle treatment, except in uncommon cases where the animal died during SE.
Power and power spectrum analysis
EEG data were digitized at 250 Hz. Power calculation was performed and graphically displayed using MatLab software. For analysis of total power, the EEG was filtered using 5 Hz high-pass and 60 Hz low-pass filters to remove ambient noise, binned in 1 minute intervals, and power within that minute was determined. For spectral analysis, 1 minute periods of EEG data were subjected to a fast Fourier transform algorithm.
Statistical analysis
Fisher's exact test and Kaplan-Meier and Maltel-Cox logrank analysis were performed using MedCalc (v. 9.5.2.0, www.medcalc.be).
In order to compare the ability of test drugs to suppress EEG power, the average power within 2 minute time windows (±standard errors) was first computed for each treatment condition using Matlab. Testing for significance of differences in EEG power between groups was performed using a custom-designed Multicomparison for Correlated Repeated Measures test (Javier Echauz, Atlanta, GA), described further in supplementary material available online (see http://www.mrw.interscience.wiley.com/suppmat/0364-5134/suppmat/) . This test accommodates appropriately for multiple simultaneous comparisons and inclusion of partially correlated data acquired during time series using a permutation test33. Although this test results in higher estimated p-values than classical analysis of variance (ANOVA), it is more appropriate for analysis of our data set because, unlike ANOVA, it is not dependent on assumptions of normality and independence.
Results
Flupirtine, but not phenobarbital or diazepam, prevents progression to continuous status epilepticus after kainic acid
To model symptomatic human neonatal seizures, we induced seizures in rats at postnatal age 10 days (P10), an age established by numerous previous studies as comparable to the human neonatal period.34 Kainic acid injection caused behavioral seizures that progressively led to status epilepticus (SE). A behavioral rating scale, modified from previous work35, was used to quantify responses to kainic acid. At stage 1 onset, pups ceased normal exploratory behavior and remained immobile; at stage 2, frequent, sustained hindlimb scratching began; at stage 3, episodes of tonic extension of one side resulted in loss of balance; at stage 4, animals completely lost balance resulting in falling onto their backs, but with recovery; at stage 5, continuous clonic seizures involving all limbs and persistent loss of righting were present. As illustrated (Figure 2), animals passed predictably through this series of steps, but the rate at which animals progressed showed considerable variability, especially at later stages (i.e., stage 5 latency was 39.5 min +/- 12 min, s.d.).
Figure 2. Seizure manifestations develop predictably in P10 rats after kainic acid.

Animal behaviors were monitored after injection of 2 mg/kg kainic acid IP. The time from injection until first onset of behavioral immobility (stage 1), episodes of rapid hind limb scratching (stage 2), unilateral falling due to loss of motor control and extensor posturing (stage 3), bilateral loss of motor and postural control with posturing (stage 4), and sustained, irreversible loss of motor control with tonic and tonic/clonic activity (stage 5) were noted. A. Representative time courses for 11 animals show that animals progressed similarly through seizure stages, but latencies differed. B. Summary of results for 49 animals. At each seizure stage, diamonds indicate mean latency, lighter and darker boxes indicate second and third quartiles, respectively, and bars indicate maximal and minimal latencies observed.
To identify dose-response relationships for seizure prevention by flupirtine, phenobarbital and diazepam, we used a pretreatment protocol. Animals received one of the three anti-epileptic drugs, at various doses. After 15 minutes, animals received 2 mg/kg kainic acid, and were observed to see whether the drugs delayed or prevented the development of seizure behaviors. In each experimental trial, littermate controls received vehicle injections, followed by kainic acid. All three drugs showed evidence of sedative effects during the 15 minute wash-in period, manifested as dose-related reduction in spontaneous and stimulation-induced motor activity and hypotonia. At the highest doses tested, all three drugs resulted in essentially complete immobility within ten minutes, i.e.,prior to kainate administration. Because of these dose-related sedative effects, we used kainate-induced progression to seizure stage 5, a robust behavioral endpoint, for comparison between drug treatment groups (Figure 3). As expected, all control rats (n = 12) given vehicle followed by kainic acid showed signs of seizures, and 91% progressed to stage 5 (SE, Figure 3A-C). Phenobarbital, at doses up to 100 mg/kg, was ineffective in preventing the development of SE (Figure 3B). The serum concentrations of phenobarbital, 30 min after IP administration of 25 and 50 mg/kg, were 20.6 +/- 2.8 (s.e.m., n=3) and 38.4 +/- 2.2 μg/ml (s.e.m, n = 4), respectively, indicating that the upper clinical therapeutic range was attained. Diazepam pretreatment at doses as high as 16 mg/kg was also ineffective at preventing progression of kainic acid induced seizures to SE (Figure 3C). Unlike the standard agents, flupirtine suppressed kainic acid induced seizures in P10 rats potently and dose-dependently. Indeed, none of the young rats that received 50 mg/kg flupirtine (n = 7) developed stage 5 seizures (Figure 3A) within the two hour observation period, and all flupirtine treated animals survived. In spite of the small group size of these dose-finding experiments, flupirtine was significantly better than vehicle (p = 0.004) or either of the two approved drugs (p = 0.003) in prevention of progression to stage 5 seizures (Fisher exact test).
Figure 3. Pretreatment with flupirtine, but not phenobarbital or diazepam, prevents the progression to stage 5 seizures after kainic acid.

Animals were injected either with vehicle or the indicated doses of flupirtine, phenobarbital, or diazepam. After 15 minutes, kainic acid was given (2 mg/kg IP), and animals were monitored for 120 min. for evidence of behavioral seizures. Flupirtine pretreatment resulted in dose-dependent suppression of behavioral seizures, but phenobarbital and diazepam did not. Numbers above each bar indicate number of animals showing stage 5 seizures (numerator), and number of animals tested at the indicated dose (denominator). Height of bar indicates percentage of animals in each treatment group exhibiting stage 5 seizures (continuous clonic activity of all 4 limbs with loss of righting). The suppression of progression to stage 5 seizures by all doses of flupirtine pooled was statistically superior to control (p=0.004) or either diazepam or phenobarbital (p =0.003). The highest flupirtine dose (50 mg/kg) group taken alone was also superior to control (p = 005) or to 50-100 mg/kg phenobarbital or 8-16 mg diazepam (p = 0.0005; Fisher's exact test).
Flupitine is more effective than phenobarbital or diazepam in preventing flurothyl-induced seizures
Inhalation of flurothyl vapors is potently convulsant, and has been used as a model of recurrent seizures in neonates and for quantification of seizure susceptibility changes resulting from status epilepticus and brain injury.30, 36, 37 To assess the efficacy of flupirtine in a second neonatal seizure model, we pretreated P10 rats with high doses of either flupirtine, phenobarbital, or diazepam, then monitored the time between first flurothyl exposure and development of tonic limb extension seizures (Figure 4). Control rats not given any antiepileptic drug developed tonic limb extension 2.22 ± 0.14 minutes (s.d., n = 7) after flurothyl exposure. Phenobarbital and diazepam delayed average latency to 5.67 ± 0.39 minutes (s.d., n = 4) and 7.16 ± 1.28 minutes (s.d., n = 4), respectively. Rats given flupirtine (n = 3) failed to exhibit tonic limb extension during exposure to flurothyl for 18 minutes, the entire planned duration of the experiment. The observed ranked differences in seizure-free survival were strongly significant (P < 0.0001, logrank test, Kaplan-Meier survival analysis).
Figure 4. Flupirtine prevents development of flurothyl-induced seizures more effectively than phenobarbital or diazepam.

Comparison of latency to first tonic limb extension seizure in control P10 rats (n = 7), and rats pretreated 15 minutes before flurothyl exposure with phenobarbital (50 mg/kg, n = 4), diazepam (8 mg/kg, n =4), or flupirtine (50 mg/kg, n =3). No flupirtine-treated animals developed seizures during 18 minutes of exposure to flurothyl. Phenobarbital and diazepam increased the mean latency, but did not fully prevent seizures.
Flupirtine reverses kainic acid induced status epilepticus rapidly and persistently
Although pretreatment with anti-epileptic drugs is routinely used in preclinical testing of experimental seizure therapies38, 39, clinical treatment of neonatal seizures is nearly always begun after seizures occur and are diagnosed by clinical criteria and/or EEG1. Arresting an established pattern of recurring seizures may require different mechanisms than preventing the initiation of such seizures. We therefore induced a hyperexcitable state with kainate, allowed seizures to worsen until stage 5 persisted for 15 minutes, and then administered experimental therapeutic agents. Kainic acid injection led to progressive appearance of ictal EEG changes, including predominant fast sharp rhythmic activity, which correlated with gradual worsening in seizure behavior (Figure 5A). Such electrographic and behavioral seizures persisted throughout the recording (approximately 2 hours) in the vehicle treated animals (Figure 5A). Flupirtine (50 mg/kg) abolished both seizure behavior and associated EEG changes (Figure 5B). These effects occurred rapidly--suppression of electrographic seizure activity by flupirtine began within the first minute after IP injection, was maximal by 5-8 minutes, and was sustained for the entire observation period. Flupirtine resulted in a suppressed background with isolated large amplitude sharp transients (or less frequently, brief runs of sharp transients (Figure 5B, insets d-e). Phenobarbital (50 mg/kg) or diazepam (8 mg/kg) also reduced EEG ictal activity and diminished behavioral seizures. Compared to flupirtine treated animals, the suppression of ictal activity by phenobarbital appeared slightly slower in onset. Furthermore, all phenobarbital treated animals manifested persistent though reduced clonic seizure behavior after treatment, and the EEG showed essentially continuous fast sharp rhythmic activity after treatment (Figure 5C). Inhibition of seizure activity by diazepam was rapid in onset, but behavioral seizures that correlated with electrographic activity returned prominently within about 30 minutes after treatment (Figure 4D).
Figure 5. When given after onset of kainic acid-induced SE, flupirtine stops electrographic seizure activity more effectively than phenobarbital or diazepam.

P10 rat pups were monitored continuously for seizure behaviors and by electrocorticography. In each case, kainic acid was given to elicit seizures. Once stage 5 seizures were observed behaviorally and continuous electrographic seizure activity was seen by EEG for 15 minutes, either vehicle (A), 50 mg/kg flupirtine (B), 50 mg/kg phenobarbital (C), or 8 mg/kg diazepam (D) was given. For each condition, the continuous EEG trace is shown. In A, the duration of behavioral seizure stages 1-5 are indicated above the EEG trace. Each continuous EEG trace is labeled indicating time of injection of kainic acid (KA) and therapeutic drugs (FLUP, flupirtine; PHB, phenobarbital; DIAZ, diazepam). Below each continuous EEG trace, 2 sec intervals of EEG are shown at higher time resolution (a, baseline; b, during stage 5 seizures before therapeutic drug treatment; c, d, e, 15 min, 30 min and 60 min after therapeutic drug administration). Although all three drugs cause an initial reduction in electrographic seizure activity, the effects of flupirtine appeared more rapid in onset than phenobarbital and more complete and sustained than those of phenobarbital or diazepam.
To allow quantitative comparisons between the drug responses, we determined the electrical power in EEGs of control and therapeutic drug-treated animals. Representative individual EEG power time courses are shown in Figure 6. Kainic acid-induced behavioral seizures and ictal EEG changes were correlated with progressive increases in EEG power, initially greatest at lower frequencies but spreading steadily to higher frequencies (Figure 6A-D). Kainate-induced increases in EEG power peaked about 30 minutes after clinical and electrographic SE onset and, in control animals, remained high throughout the recording period (Figure 6A). Inspection of plots of EEG power of individual animals exposed to the therapeutic drugs revealed characteristic differences in the responses to the three agents (Figs. 6B-D). Flupirtine caused a rapid and sustained reduction of EEG power to below baseline levels (Figure 6B). Phenobarbital reduced total power to near baseline levels by 10-15 minutes after treatment, but spectral analysis revealed persistently elevated power in the 15-30 Hz range. Phenobarbital treated animals also showed recurrent episodes of more greatly increased power that correlated with the breakthrough seizure activity seen in the EEG and behaviorally (Figure 5C). Diazepam power analysis revealed both an initial favorable drug response and the partial reversal of this response by about 30 minutes after treatment (Figure 6D). Thus, EEG power plots captured in a quantified form many important features of the electrographic and behavioral responses we observed to the therapeutic drugs.
Figure 6. Kainate-induced increases in EEG power parallel clinical seizure activity, and are reversed by flupirtine, phenobarbital or diazepam.

A, Control, B, flupirtine, C, phenobarbital, D, diazepam. EEG data were analyzed for total power and power at various frequencies (see Materials and Methods). For each experimental condition, the upper panel shows the variation in total EEG power within one minute intervals before and after the administration of kainic acid and the indicated therapeutic drug (or vehicle). Time intervals containing EEG artifacts due to handling for injection are shown (red). The lower panels show power per minute within one Hz frequency bins, calculated by Fourier transformation of the EEG and displayed in color according to the scale bar (inset in A). For each experimental condition, kainic acid treatment results in progressive increase in total power; in spectral plots this is first detected at lower frequencies and later at higher frequencies. Note the rapid onset and sustained reduction in power after flupirtine administration. Phenobarbital shows a persistent band (light blue) of increased power at 15-30 Hz. Diazepam is effective at power suppression in the first 20 minutes of treatment, but power increases prominently at later time points.
Figure 7 shows average (+/- s.e.m.) EEG power in 2 minute time intervals for groups of control, flupirtine-treated, phenobarbital-treated, and diazepam-treated rats. The three drug-treated groups differed in several respects: onset of the effects of flupirtine was more rapid than that of phenobarbital, and the effect of diazepam was biphasic, with an initial strong reduction and subsequent increase in average power. We developed a rigorous statistical approach for analyzing whether apparent differences among the four averaged EEG power time courses were statistically significant (α = 0.05, see Methods and supplementary material available online at http://www.mrw.interscience.wiley.com/suppmat/0364-5134/suppmat/). We performed a global analysis (in which the entire time courses were compared), and also assessed the significance of differences detected at individual 2 minute time points after therapeutic drug treatment (Figure S1). The global analysis showed that each of the three therapeutic drugs significantly reduced EEG power compared to controls (p < 0.025). Although average EEG power after flupirtine treatment was less than after phenobarbital in 29 of 30 time bins (mean EEG power after flupirtine/mean after phenobarbital = 0.57), these differences did not achieve statistical significance in the global analysis of the entire time course (p = 0.3318; see Discussion). However, EEG power after flupirtine was significantly lower than after phenobarbital during the first 4 minutes of treatment ((p < 0.05; Supplementary Figure S1), supporting the assertion that flupirtine has a faster therapeutic onset than phenobarbital. In the entire time course analysis, power after flupirtine treatment was significantly lower than after diazepam (Figure 6B; p = 0.0357) but phenobarbital and diazepam did not differ significantly (p = 0.3006). Analysis of individual time windows showed that EEG power after diazepam was significantly lower than control during the first 40 minutes after treatment, but not during the period 40-60 minutes after treatment (Figure S1). This quantitative analysis of our grouped results supports the conclusion that, unlike either diazepam or phenobarbital, suppression of kainate-induced seizures by flupirtine is both rapid in onset and sustained.
Figure 7. Averaged kainate-induced EEG power increases are reversed by flupirtine, phenobarbital, and diazepam.

Mean power time courses for control animals (n=5), and those treated with diazepam (n = 8), phenobarbital (n =11), or flupirtine (n =8) are shown (+/- S.E.M). Power was sampled either before kainate injection (B, baseline), after kainate (at 25, 50, and 75% of the time interval between injection and SE onset, and 5 minutes after SE onset), or at the indicated time intervals after injection of the treatment drug or vehicle (control). A. EEG power after kainate-induced SE is significantly reduced by 50 mg/ml phenobarbital (p = 0.0006) or 50 mg/ml flupirtine (p = 0.0001). Although 29 of 30 individual flupirtine mean measurements are lower than phenobarbital, result are not statistically different (p = 0.3318). B. EEG power after kainate-induced SE is reduced by 8 mg/ml diazepam (p = 0.0242), with a biphasic time course (identical control and flupirtine data to panel A). Diazepam appears effective transiently after injection, but flupirtine power is significantly lower (p = 0.0357 for entire time course).
Discussion
To assess the utility of flupirtine for neonatal seizures, we have used two animal models of induced seizures that have been extensively employed in previous neonatal seizures studies. Either flurothyl or kainic acid robustly induced seizures in rats at P10, an age thought comparable in developmental maturity to human neonates. When give prior to seizure induction by kainic acid, flupirtine was significantly more effective than either diazepam or phenobarbital in preventing progression to status epilepticus (Figure 3). Flupirtine was also significantly more effective than either approved drug in preventing seizure induction by flurothyl (Figure 4). Even when administerered after kainic acid seizures that were allowed to progress to a state of continuous status epilepticus, flupirtine was very effective, by behavioral measures, qualitative EEG, and several tests of EEG power (Figures 5-7). In this post-treatment paradigm, suppression of total EEG power by flupirtine and high dose phenobarbital (50 mg/kg, yielding a serum level of approximately 40 μg/ml) did not differ significantly, but the effect of phenobarbital treatment was significantly delayed in onset and was associated with persistent continuous high frequency EEG activity and brief behavioral seizures that were not seen after flupirtine.
The current study confirms results of an earlier study by Dzhala et al. that showed that phenobarbital pretreatment was relatively ineffective for prevention of kainic acid induced seizures in neonatal rodents,38 and extends the findings to a second agent, diazepam. Dzhala used analysis of EEG power to compare the ability of phenobarbital and the transporter blocking agent, bumetamide, to suppress kainate induced seizures in neonatal rats. Although our approach was similar in many respects, there are some salient differences. In the current study, phenobarbital was used at a higher dose (50 mg/kg versus 25 mg/kg), and trial drugs were given after seizures had progressed to SE (rather than prior to kainate injection). We used a test of significance that imposed several statistical power-weakening corrections (see Methods), where the previous study used a less stringent, ANOVA-based approach. Indeed, pair-wise comparison of flupirtine and phenobarbital treatment using ANOVA and data from Figure 7 yields strong numerical support for superior power reduction by flupirtine (p = 0.0033). Because of these methodological differences, and the limitations of power measurement as a marker of more important clinical efficacy endpoints, drawing conclusions about the relative merits of bumetamide and flupirtine must await additional studies. At present, it is clear that flupirtine is strongly anticonvulsant in P10 rats.
Although our kainate induction protocol was designed to limit mortality to ~10%, it is noteworthy that 8 of 55 control animals (14.5%), 11 of 35 diazepam-treated animals (31.4%), 2 of 48 phenobarbital treated animals (4.2%), and 0 of 56 flupirtine-treated animals (0%) died within the experimental observation period. Factors underlying these observed mortality rate differences may include, for example, respiratory and hemodynamic effects we have not yet characterized. These may be clinically significant and deserve further study.
Flupirtine has not been approved for use in the United States. However, retigabine is a close structural analogue (Figure 1) that is currently undergoing phase 3 clinical trial as adjunctive therapy for adult partial epilepsy, after a large stage 2 trial showed tolerability and dose-dependent suppression of seizure frequency.21 Our findings do not imply that KCNQ openers such as retigabine and flupirtine are more likely to be effective in neonates than in older patients. KCNQ channels continue to be expressed throughout childhood and adulthood 40-42. Although neonatal brain is unique in its high degree of seizure susceptibility after blockade of KCNQ channels, 13-16, 43 this finding does not imply progressive diminishment in the experimental or clinical utility of openers with age. Evidence has been presented that flupirtine and retigabine are capable of antagonizing glutamate induced toxicity and enhancing inhibitory synaptic currents,27, 44 in addition to their well-established direct effects as KCNQ openers. Additional studies are required to better specify the mechanisms relevant to the anti-epileptic effects of these drugs. As noted above, in vivo experiments have raised concerns about potential pro-apoptotic effects of anticonvulsants currently utilized in neonates.6-8 Although in vitro results suggesting anti-apoptotic and anti-oxidative effects of flupirtine and retigabine are encouraging26, 27, it will be important in future studies to directly examine the effects of flupirtine and other KCNQ openers on apoptosis and other aspects of developmental plasticity in the neonatal brain.
The current translational investigation was made possible by earlier genetic and basic research9. The benign familial neonatal seizures syndrome was the first idiopathic epilepsy for which genetic loci were identified.45-47 Cloning of KCNQ2 and KCNQ3, two potassium channel genes, at these loci has allowed these novel channels to be studied both using in vitro and in vivo methods.48 Such studies have shown that the channels mutated in BNFS are widely expressed in the central nervous system, are critical regulators of excitability not only in infancy but throughout life, and can be pharmacologically activated by many drugs.41, 49-51 It is somewhat paradoxical that KCNQ2 and KCNQ3 mutations are rare and typically cause a phenotype restricted to the first weeks of life, but KCNQ channels exhibit widespread, lifelong expression and pivotal importance for brain function. This offers an important general lesson for translational neuroscientists: clinical disease can offer clues allowing the discovery of essential brain proteins and pathways, without fully revealing the range of functional activity of these components. This is the case with BFNS and the underlying KCNQ channels.
Neuronal KCNQ channels remain quite poorly understood, and many questions regarding their subunit composition, biological roles, and pharmacology are unanswered. Continued development of KCNQ channel openers with distinctive subunit specificity and potency profiles will be critical for achieving better understanding of channel functions. Flupirtine is a low potency KCNQ channel opener22, and is representative of a class of drugs that exert their greatest effects on the KCNQ3 subunit.52 Currently disclosed KCNQ channel openers include compounds of greater overall potency (e.g., retigabine, various ethyl acrylamides, ICA-27243), and agents that differ from flupirtine in their kinetic mechanisms and selectivity among the neuronal KCNQ subunits (e.g., zinc pyrithione).50 Further in vitro and in vivo testing of novel openers will help clarify differences in biological functions between the KCNQ subunits. In turn, this may allow drug action upon these channels to be rationally tailored to maximize therapeutic benefits while reducing unwanted side effects, for various indications in pediatric and adult patients.
Our studies join recent efforts to translate understanding of the distinctive cellular and molecular features of developing brain into mechanistically novel neonatal seizure therapies. Thus, developmental differences in GABA(A) receptor subunit expression and the transmembrane Cl- gradient have been found to make synaptic inhibition relatively less effective in the immature compared to the adult brain.38, 53 As noted above, the use of bumetamide, which promotes a more negative Cl- equilibrium potential and thereby enhances the inhibitory effects of GABAergic neurotransmission, has been investigated to circumvent this.38, 54 In contrast to the functional diminishment of GABAergic inhibition at birth, some excitatory glutamate receptors are overexpressed during early development, making blockade of these receptors another rational treatment approach. The AMPA receptor antagonist NBQX and kainate receptor antagonist topiramate have shown efficacy in the treatment of experimental neonatal seizures1, 36, 39, and do not cause enhanced apoptosis.55 Our results further validate the notion that neonatal seizure therapy may be improved through targeting molecules and mechanisms confirmed by laboratory or genetic studies to be of particular importance during early brain development.
Supplementary Material
Acknowledgements
We thank Dr. Javier Echauz for expert statistical consultation, and Brian Litt for helpful discussions of EEG power analysis. This research was supported by NINDS R21 NS55765 and the Miles Family Fund.
References
- 1.Silverstein FS, Jensen FE. Neonatal seizures. Ann Neurol. 2007;62:112–120. doi: 10.1002/ana.21167. [DOI] [PubMed] [Google Scholar]
- 2.Pisani F, Cerminara C, Fusco C, Sisti L. Neonatal status epilepticus vs recurrent neonatal seizures: clinical findings and outcome. Neurology. 2007;69:2177–2185. doi: 10.1212/01.wnl.0000295674.34193.9e. [DOI] [PubMed] [Google Scholar]
- 3.Ronen GM, Buckley D, Penney S, Streiner DL. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816–1822. doi: 10.1212/01.wnl.0000279335.85797.2c. [DOI] [PubMed] [Google Scholar]
- 4.Painter MJ, Scher MS, Stein AD, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med. 1999;341:485–489. doi: 10.1056/NEJM199908123410704. [DOI] [PubMed] [Google Scholar]
- 5.Olney JW, Young C, Wozniak DF, et al. Do pediatric drugs cause developing neurons to commit suicide? Trends Pharmacol Sci. 2004;25:135–139. doi: 10.1016/j.tips.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Bittigau P, Sifringer M, Genz K, et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci U S A. 2002;99:15089–15094. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sankar R, Painter MJ. Neonatal seizures: after all these years we still love what doesn't work. Neurology. 2005;64:776–777. doi: 10.1212/01.WNL.0000157320.78071.6D. [DOI] [PubMed] [Google Scholar]
- 8.Silverstein FS, Jensen FE, Inder T, et al. Improving the treatment of neonatal seizures: National Institute of Neurological Disorders and Stroke workshop report. J Pediatr. 2008;153:12–15. doi: 10.1016/j.jpeds.2008.01.041. [DOI] [PubMed] [Google Scholar]
- 9.Cooper EC, Jan LY. M-channels: Neurological diseases, neuromodulation, and drug development. Archives of Neurology. 2003;60:494–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- 10.Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- 11.Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–2737. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- 12.Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73:245–249. doi: 10.1016/j.eplepsyres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Okada M, Zhu G, Hirose S, et al. Age-dependent modulation of hippocampal excitability by KCNQ-channels. Epilepsy Res. 2003;53:81–94. doi: 10.1016/s0920-1211(02)00249-8. [DOI] [PubMed] [Google Scholar]
- 14.Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pena F, Alavez-Perez N. Epileptiform activity induced by pharmacologic reduction of M-current in the developing hippocampus in vitro. Epilepsia. 2006;47:47–54. doi: 10.1111/j.1528-1167.2006.00369.x. [DOI] [PubMed] [Google Scholar]
- 16.Peters HC, Hu H, Pongs O, et al. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- 17.Schwarz JR, Glassmeier G, Cooper EC, et al. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Z, Kao T, Horvath Z, et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasmussen HB, Frokjaer-Jensen C, Jensen CS, et al. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- 20.Peretz A, Degani N, Nachman R, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- 21.Porter RJ, Partiot A, Sachdeo R, et al. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology. 2007;68:1197–1204. doi: 10.1212/01.wnl.0000259034.45049.00. [DOI] [PubMed] [Google Scholar]
- 22.Martire M, Castaldo P, D'Amico M, et al. M channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wickenden AD, Krajewski JL, London B, et al. N-(6-chloro-pyridin-3-yl)-3,4-difluorobenzamide (ICA-27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol Pharmacol. 2008;73:977–986. doi: 10.1124/mol.107.043216. [DOI] [PubMed] [Google Scholar]
- 24.Wickenden AD, Roeloffs R, McNaughton-Smith G, Rigdon GC. KCNQ potassium channels: drug targets for the treatment of epilepsy and pain. Expert Opinion Therapeutic Patents. 2004;14:2–13. [Google Scholar]
- 25.Wua YJ, Dworetzky SI. Recent developments on KCNQ potassium channel openers. Curr Med Chem. 2005;12:453–460. doi: 10.2174/0929867053363045. [DOI] [PubMed] [Google Scholar]
- 26.Dhar S, Bitting RL, Rylova SN, et al. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann Neurol. 2002;51:448–466. doi: 10.1002/ana.10143. [DOI] [PubMed] [Google Scholar]
- 27.Seyfried J, Evert BO, Rundfeldt C, et al. Flupirtine and retigabine prevent L-glutamate toxicity in rat pheochromocytoma PC 12 cells. Eur J Pharmacol. 2000;400:155–166. doi: 10.1016/s0014-2999(00)00397-6. [DOI] [PubMed] [Google Scholar]
- 28.Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology. 2004;62:714–718. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- 29.Porter RJ, Gratz E, Narang PK, et al. Effect of Flupirtine on Uncontrolled Partial or Absence Seizures. Epilepsia. 1983;24:253–254. [Google Scholar]
- 30.Jensen FE, Holmes GL, Lombroso CT, et al. Age-dependent changes in long-term seizure susceptibility and behavior after hypoxia in rats. Epilepsia. 1992;33:971–980. doi: 10.1111/j.1528-1157.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann AF, Zhao Q, Holmes GL. Cognitive impairment following status epilepticus and recurrent seizures during early development: support for the “two-hit hypothesis”. Epilepsy Behav. 2004;5:873–877. doi: 10.1016/j.yebeh.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Raol YS, Budreck EC, Brooks-Kayal AR. Epilepsy after early-life seizures can be independent of hippocampal injury. Ann Neurol. 2003;53:503–511. doi: 10.1002/ana.10490. [DOI] [PubMed] [Google Scholar]
- 33.Good P. Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses. Springer; New York: 2000. [Google Scholar]
- 34.Jensen FE, Baram TZ. Developmental seizures induced by common early-life insults: short- and long-term effects on seizure susceptibility. Ment Retard Dev Disabil Res Rev. 2000;6:253–257. doi: 10.1002/1098-2779(2000)6:4<253::AID-MRDD4>3.0.CO;2-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunson KL, Schultz L, Baram TZ. The in vivo proconvulsant effects of corticotropin releasing hormone in the developing rat are independent of ionotropic glutamate receptor activation. Brain Res Dev Brain Res. 1998;111:119–128. doi: 10.1016/s0165-3806(98)00130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jensen FE, Blume H, Alvarado S, et al. NBQX blocks acute and late epileptogenic effects of perinatal hypoxia. Epilepsia. 1995;36:966–972. doi: 10.1111/j.1528-1157.1995.tb00954.x. [DOI] [PubMed] [Google Scholar]
- 37.McCabe BK, Silveira DC, Cilio MR, et al. Reduced neurogenesis after neonatal seizures. J Neurosci. 2001;21:2094–2103. doi: 10.1523/JNEUROSCI.21-06-02094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 39.Koh S, Jensen FE. Topiramate blocks perinatal hypoxia-induced seizures in rat pups. Ann Neurol. 2001;50:366–372. doi: 10.1002/ana.1122. [DOI] [PubMed] [Google Scholar]
- 40.Maljevic S, Kempfle J, Weber YG, et al. Developmental expression of KCNQ2 and KCNQ3 potassium channels at the axon initial segments of the mouse brain. Society for Neuroscience Annual Meeting; San Diego. 2007. [Google Scholar]
- 41.Cooper EC, Aldape KD, Abosch A, et al. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–4919. doi: 10.1073/pnas.090092797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geiger J, Weber YG, Landwehrmeyer B, et al. Immunohistochemical analysis of KCNQ3 potassium channels in mouse brain. Neurosci Lett. 2006;400:101–104. doi: 10.1016/j.neulet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 43.Qiu C, Johnson BN, Tallent MK. K+ M-current regulates the transition to seizures in immature and adult hippocampus. Epilepsia. 2007;48:2047–2058. doi: 10.1111/j.1528-1167.2007.01193.x. [DOI] [PubMed] [Google Scholar]
- 44.Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- 45.Zimprich F, Ronen GM, Stogmann W, et al. Andreas Rett and benign familial neonatal convulsions revisited. Neurology. 2006;67:864–866. doi: 10.1212/01.wnl.0000234066.46806.90. [DOI] [PubMed] [Google Scholar]
- 46.Leppert M, Anderson VE, Quattlebaum T, et al. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nature. 1989;337:647–648. doi: 10.1038/337647a0. [DOI] [PubMed] [Google Scholar]
- 47.Lewis TB, Leach RJ, Ward K, et al. Genetic heterogeneity in benign familial neonatal convulsions: identification of a new locus on chromosome 8q. Am J Hum Genet. 1993;53:670–675. [PMC free article] [PubMed] [Google Scholar]
- 48.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 49.Cooper EC, Harrington E, Jan YN, Jan LY. M-channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008 doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- 52.Schenzer A, Friedrich T, Pusch M, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brooks-Kayal AR, Shumate MD, Jin H, et al. gamma-Aminobutyric acid(A) receptor subunit expression predicts functional changes in hippocampal dentate granule cells during postnatal development. J Neurochem. 2001;77:1266–1278. doi: 10.1046/j.1471-4159.2001.00329.x. [DOI] [PubMed] [Google Scholar]
- 54.Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- 55.Glier C, Dzietko M, Bittigau P, et al. Therapeutic doses of topiramate are not toxic to the developing rat brain. Exp Neurol. 2004;187:403–409. doi: 10.1016/j.expneurol.2004.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.