

Abstract
The role of erythropoietin receptor (EpoR) expression in tumor cells and the potential of EpoR-mediated signaling to contribute to cellular proliferation and invasiveness require further characterization. To determine whether EpoR expression and activation in tumor cells modulates intracellular signal transduction to promote cellular proliferation and migration, we employed a novel experimental model using human breast cancer cells engineered to stably express a constitutively active EpoR-R129C variant. EpoR-R129C expression resulted in increased cellular proliferation and migration of breast cancer cells and these effects were associated with significantly increased Epo-induced phosphorylation of ERK1/2, AKT and c-Jun-NH2-kinase (SAPK/JNK) proteins. Expression of the constitutively active EpoR-R129C receptor promoted the proliferation and migration of breast cancer cells via activation of ERK- and SAPK/JNK-dependent signaling pathways, respectively. These findings suggest that EpoR over-expression and activation in breast cancer cells has the potential to contribute to tumor progression by promoting the proliferation and invasiveness of the neoplastic cells.
Keywords: Erythropoietin, erythropoietin receptor, extracellular regulated kinase, c-Jun-NH2 kinases, breast cancer, signal transduction
INTRODUCTION
The recombinant forms of human erythropoietin (Epo)– the principal cytokine that regulates red blood cell production– have constituted an important component of the supportive therapy of cancer patients with chemotherapy-induced anemia to reduce red blood cell transfusions and to improve quality of life. The use of erythropoiesis-stimulating agents in some cancer patients has been associated with adverse clinical outcomes resulting in enhanced tumor progression and impaired survival, the mechanisms of which are not completely understood. A series of recent clinical trials have reported adverse outcomes associated with Epo therapy leading to impaired progression-free and overall survival[1,2]. Ubiquitous EpoR expression in non-hematopoietic tissues and in tumor cells has raised the possibility that Epo may directly modulate tumor growth via activation of functional EpoR signaling in tumor cells, however, the mechanisms of the detrimental effects of Epo in some cancer patients require further characterization[3]. The expression of EpoR mRNA transcripts and protein in tumor cell lines and in primary tumors has not been associated with predictable effects of exogenous Epo in pre-clinical experimental models[4, 5]. In some studies employing exogenous Epo treatment of tumor cells in vitro, increased cell proliferation[6-9], migration capacity[10-12], apoptosis inhibition[13-15], and chemo-radiation resistance[16-19] have been reported, whereas several other studies have not found a significant effect of Epo on cellular proliferation[20-24] or sensitivity to chemotherapeutic agents[25]. These divergent findings have led to the emergence of a controversy as to whether functional EpoR-mediated signaling may be activated in cancer cells to modulate tumor cell behavior. To investigate this question of EpoR functionality in cancer cells and to further characterize the biology of Epo signaling in cancer cells, the development of novel pre-clinical experimental models designed to examine the functionality of EpoR signaling in cancer cells may be useful.
The purpose of this study was to determine whether expression of a constitutively active EpoR variant EpoR-R129C promotes functional intracellular signaling to modulate the cellular proliferation and migration of breast cancer cells. The mutant EpoR-R129C receptor confers growth factor-independent growth and tumorigenicity when expressed in hematopoietic Ba/F3 cells that are normally dependent on interleukin-3 for survival and growth[26, 27], but EpoR-R129C expression does not transform normal fibroblasts[28]. Hematopoietic cells expressing EpoR-R129C exhibit constitutive receptor activation due to ligand-independent formation of disulfide-linked receptor homodimers[29] leading to increased cellular proliferation associated with constitutive JAK2-STAT5 activation[30-32]. Here we show that expression of EpoR-R129C in breast cancer cells promotes cellular proliferation and migration by specific activation of the MAP kinase pathway but not the JAK2-STAT5 axis.
Materials and Methods
Cell lines, tissue culture and transfections
MCF-7 and MDA-MB-231 human breast cancer cells and P769 renal carcinoma cells were obtained from American Type Culture Collection(ATCC, Rockville, MD) and cultured in RPMI 1640 medium(Invitrogen, Carlsbad, CA) and 10% fetal bovine serum(FBS, Hyclone, Logan, UT). Cells were stably transfected using Lipofectamine reagent(Invitrogen) either with empty mammalian expression vector pCR3.1(Invitrogen) or a pCR3.1 plasmid encoding EpoR-R129C[33]. Single cell clones were selected in G418(Invitrogen) at a concentration of 0.8mg/mL and mock-transfected cells incubated with G418 as controls exhibited 100% cell death. Isolated, G418-resistant single cell clones were expanded and whole cell extracts were analyzed by immunoblotting for EpoR expression. Anemic mouse spleen extracts served as a positive control for EpoR expression and P769 cell extracts were used as negative control.
Reagents
Human recombinant Epo(Procrit) was purchased from the outpatient pharmacy at Duke University Medical Center. Antibodies against total and phospho-specific AKT-Ser473, JAK2-Tyr1007/1008 (lot 6), ERK1/2-Thr202/Tyr204, SAPK/JNK-Thr183/Tyr185, STAT5-Tyr694 were from Cell Signaling Technology(Beverly, MA). Antibodies against actin, total JAK2 and EpoR(M-20) were from Santa Cruz Biotechnology(Santa Cruz, CA). Kinase inhibitors against MEK(PD 98059) and SAPK/JNK(SP600125) were from EMD Chemicals(San Diego, CA). Soluble EpoR protein as an Epo antagonist was from R&D Systems(Minneapolis, MN).
Cell proliferation and migration assays
MCF-7 cells(1.0×104/well) or MDA-MB-231 cells(1.5×104/well) were seeded in multi-well tissue culture plates and cultured in RPMI and 10% FBS for 24 hours. The medium was changed to 2ml RPMI supplemented with charcoal-dextran-treated FBS (10% MCF-7 or 5% MDA-MB-231 cells). Three single cell clones each of EpoR-R129C or empty vector-transfected cells were analyzed in triplicates by counting the cells for 5 to 9 days using trypan blue exclusion assays to generate growth curves. The effect of MEK inhibitor PD98059 on proliferation was examined by adding the kinase inhibitor(50μM) or the vehicle DMSO (0.1%) in negative control cultures.
Colony formation assays in agarose were performed with cell suspensions (2.0×103cells/60mm dish) in RPMI (without phenol red) containing 10% FBS and 0.35% agarose (Promega, Madison,WI) layered on 0.6% base agarose gel (in RPMI/10% FBS,5 ml/dish). In some cultures, Epo was added to final concentration 10units/ml in the presence or absence of MEK inhibitor PD98059(50μM) or the solvent DMSO(0.1%) as negative control mixed with the cell suspension before cells were seeded in triplicates for each condition. Colonies were defined as a cluster consisting of >30 cells and counted using a microscope at day 15.
Cell migration assays were performed using Transwell chambers(Corning Costar, Corning, NY) as described[10]. Epo was added to the cell suspension at a concentration of 1unit/ml based on results of dose-response experiments(data not shown). Cells (3.0×105/ml) in 100μl/well were transferred to the top chamber of each Transwell and allowed to migrate under normoxic conditions for 6 hours. In experiments involving kinase inhibitors, cells were pre-incubated with JNK kinase inhibitor SP600125(20μM) or DMSO vehicle(0.2%) as control for 15 minutes prior to seeding.
Western Blotting and ERK kinase assays
Cells were starved in serum-free RPMI medium for 1 hour and either left unstimulated or incubated with vehicle consisting of 100mM NaCl, 20mM sodium citrate, 0.3mM citric acid [pH6.9] and 2.5 mg/ml human albumin(Sigma, St Louis, MO), or increasing concentrations of Epo as indicated for 10 minutes at 37°C. As a control in some experiments, Epo was heat-denatured by boiling the protein for 5 minutes prior to addition to the cultures. As another control, Epo (10units) was preincubated with excess soluble EpoR protein (0.5μg) for 1 hour at room temperature prior to treatment of the cells. Whole cell extracts were resolved by SDS-PAGE and subjected to immunoblotting with phospho-specific antibodies.
In vitro kinase assays were performed using a kit(Cell Signaling Technologies) by immunoprecipitation with a monoclonal phospho-specific ERK1/2 antibody. The immunoprecipitates were incubated with an ELK-1 fusion protein in the presence of ATP and kinase buffer allowing immunoprecipitated active MAP kinase to phosphorylate its substrate ELK-1 measured by Western blotting using phospho-specific(Ser383) ELK-1 antibody. Quantification of the density of the bands was performed using NIH ImageJ software.
Statistical Analyses
Statistical analyses were performed by using GrapdPad InStat software version 3.0(San Diego, CA). Comparisons between two groups were performed by t-tests. Comparisons between multiple groups were performed by one-way ANOVA and Bonferroni post-hoc multiple comparisons test. A two-tailed P value of P<0.05 was considered statistically significant.
RESULTS AND DISCUSSION
We investigated the effect of exogenous Epo treatment and/or EpoR-R129C over-expression on cancer cell proliferation, migration and signaling. An expression plasmid encoding EpoR-R129C[33] was stably introduced into human breast cancer cells and single cell clones were isolated(Fig.1A). Cells expressing EpoR-R129C exhibited 2.2-fold increased proliferation compared to empty-vector transfected controls or untransfected cells(Fig.1B). Epo treatment did not further stimulate the proliferation of EpoR-R129C expressing cells but resulted in a significant 1.7-fold increase in proliferation of empty vector-transfected MCF-7 cell clones(Fig.1C). Expression of EpoR-R129C in MDA-MB-231 breast cancer cells was also associated with increased proliferation rate of the cells(Supplementary Fig.S1).
Fig.1. EpoR expression in breast cancer cells and effect of EpoR-R129C expression on cellular proliferation.
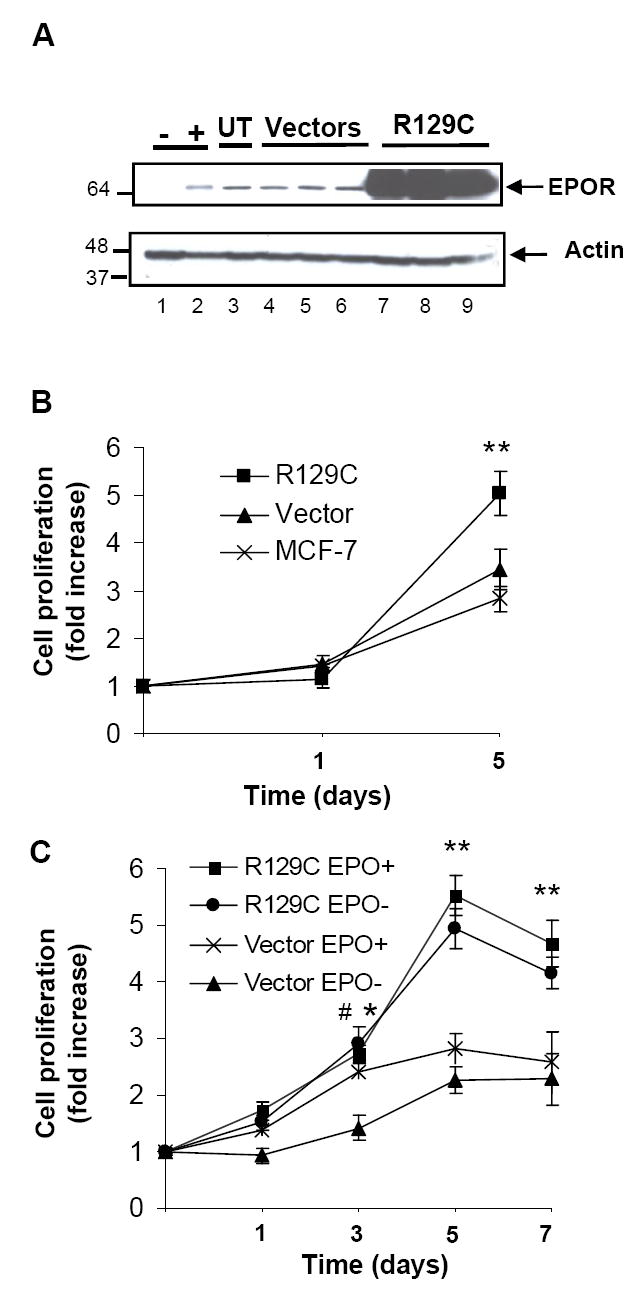
A.MCF-7 cells stably transfected with empty vector or plasmid encoding EpoR-R129C were analyzed by immunoblotting for EpoR expression. Lane 1.negative control P769 cells without EpoR expression, Lane 2.anemic mouse spleen positive control, Lane 3.untransfected MCF-7 cells (UT), Lanes 4-6.Empty expression vector-transfected single cell clones, Lanes 7–9.EpoR-R129C transfected clones. The lower portion of the blot was analyzed for actin expression demonstrating protein integrity and comparable loading. Molecular weight marker(kDa) positions are illustrated. B.EpoR-R129C expressing cells exhibit increased proliferation compared to empty vector-transfected(Vector) or untransfected MCF-7 cells. The data of three independent clones was pooled and expressed as fold-increase in cell number to generate growth curves (mean±sem, n=8 replicates/group).**P<0.01 compared to vector-transfected or untransfected cells. C.Epo treatment (10units/ml) promotes the proliferation of vector-transfected but not EpoR-R129C expressing cells. Three single cell clones each of EpoR-R129C and vector-transfected cells were analyzed in triplicates, (n=9 replicates/group).*P<0.05 Epo-treated vector cells compared to no Epo treatment, #P<0.05 and **P<0.01 EpoR-R129C-expressing cells compared to vector-transfected cells without Epo treatment.
We then investigated EpoR-mediated signaling in breast cancer cells to characterize the mechanisms of the proliferative effects of EpoR-R129C expression. In untransfected MCF-7 cells, Epo treatment induced the increased phosphorylation of p44/42 MAP kinases ERK1/2(Fig.2) but not JAK2, STAT5 or AKT(Supplementary Fig.S2 and data not shown). Following Epo treatment, there was a dose- and time-dependent increase in ERK1/2 phosphorylation with a rapid increase in ERK1/2 phosphorylation within 5-10 minutes of Epo treatment that gradually decreased to basal levels by 60 minutes in the continued presence of Epo(Fig.2A and 2B). We determined whether the rapid Epo-mediated phoshorylation of ERK1/2 is associated with increased kinase enzymatic activity using in vitro kinase assays. Epo treatment resulted in a significant 2.3 ± 0.3 fold increase in ERK kinase activity in MCF-7 cells(Fig.2C). We next examined the activation of intracellular signaling in MCF-7 cells engineered to express EpoR-R129C. There was a significant 2.6±0.4-fold increase in basal phosphorylation level of ERK1/2 and a 1.6±0.1-fold increase in basal AKT phosphorylation compared to vector-transfected controls(Fig.3A). Epo treatment of cells expressing EpoR-R129C significantly enhanced ERK1/2 phosphorylation by 2.7±0.3-fold (Fig.3B) and SAPK/JNK phosphorylation by 5.38±1.6 compared to vector controls(Fig.3C). Whereas Epo treatment did not induce the phosphorylation of AKT in untransfected or vector-transfected MCF-7 cells, in EpoR-R129C expressing cells there was a significant, dose-dependent, 2.29±0.5 fold increase in AKT phosphorylation in response to Epo(Fig.3D).
Fig.2. Epo-induced ERK pathway activation in MCF-7 breast cancer cells.
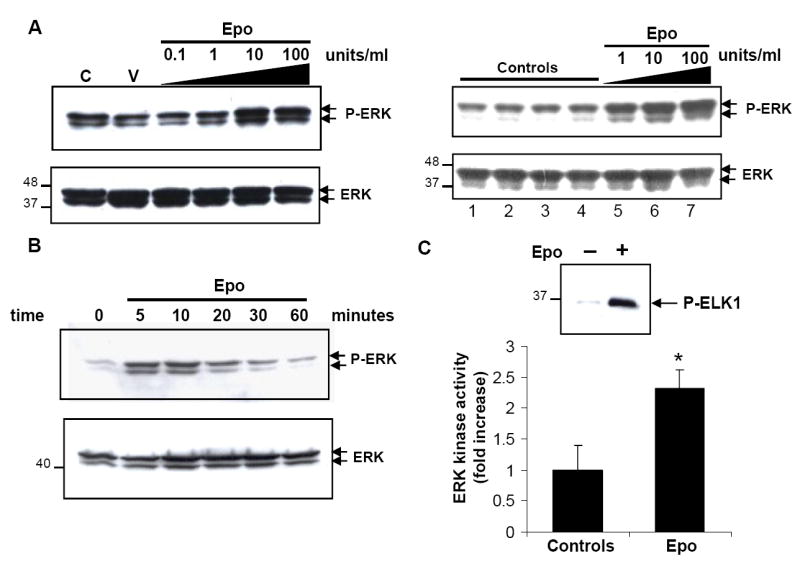
A.Epo phosphorylates ERK1/2 in a dose-dependent manner. Cells were starved for 1 hour and left unstimulated (C), incubated with vehicle (V), or increasing concentrations of Epo. Whole cell lysates were analyzed by immunoblotting using phospho-specific ERK1/2 antibody. The blots were stripped and re-probed with antibody against total ERK. Specific Epo-induced ERK phosphorylation(P-ERK) was further confirmed by additional controls (panels shown on right). Lane 1.untreated cells, Lane 2.vehicle, Lane 3.heat-denatured Epo(10units/ml), Lane 4.Epo pre-incubated with soluble EpoR antagonist prior to cell culture addition, Lanes 5-7.Epo treatment at indicated concentrations demonstrating increased ERK phosphorylation only in the presence of Epo. B.Time course of Epo-mediated phosphorylation of ERK1/2 in breast cancer cells. Cells were either left unstimulated (0) or stimulated with Epo (10units/ml) for the indicated time course. C.EPO-mediated induction of ERK kinase activity in breast cancer cells. Cells were either left untreated(−) or treated(+) with recombinant Epo (10units/ml) for 10 minutes at 37°C. Kinase assays were performed to measure ability of immunoprecipitated active ERK kinase to phosphorylate its substrate ELK-1. A representative immunoblot illustrating increased ELK-1 phosphorylation in response to Epo is shown(upper panel). Quantitative representation(mean±s.e.m) of fold-increase in ERK kinase activity(lower panel) in three independent experiments.*P< 0.05.
Fig.3. Increased phosphorylation of ERK1/2, JNK and AKT in breast cancer cells expressing EpoR-R129C.
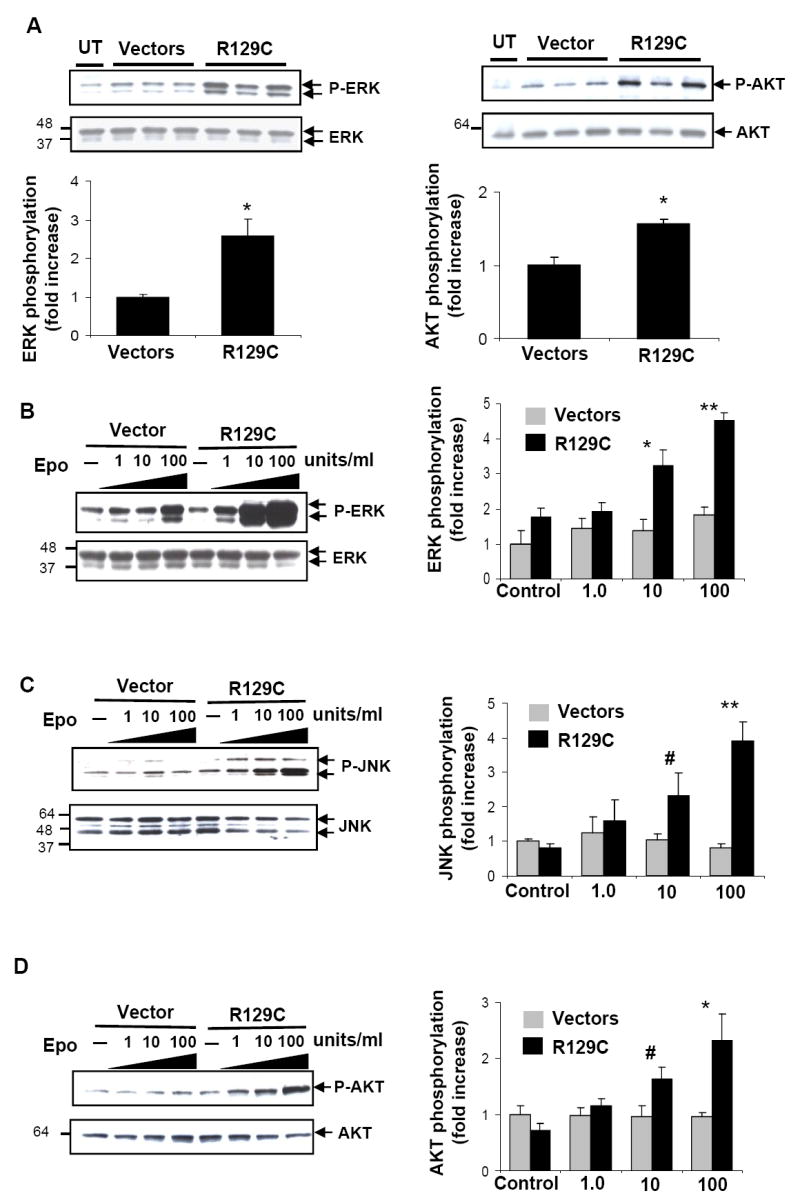
A.Increased basal phosphorylation of ERK (panels on left) and AKT (panels on right). Cells during exponential growth phase in RPMI and 10%FBS were removed from culture and whole cell lysates were analyzed for basal P-ERK and P-AKT by immunoblotting. Three independent single cell clones of empty vector and EpoR-R129C-transfected cells were analyzed. The blots were re-probed with total ERK and AKT antibodies to normalize phosphorylated protein amounts. Quantitative representation of P-ERK and P-AKT are illustrated below the blots.*P<0.05, n=3 clones/group. UT: untransfected MCF-7 cells. B-C.Epo-induced ERK1/2 (B), SAPK/JNK (C) and AKT (D) phosphorylation is increased in breast cancer cells expressing EpoR-R129C. Cells were starved for 1 hour and either left unstimulated(–) or incubated with increasing concentrations of Epo. Representative immunoblots are shown. Three independent single cell clones of empty vector or EpoR-R129C-transfected cell lines were analyzed. Epo-induced phoshorylation of the proteins was quantified by densitometry and normalized to total protein content. The data was pooled and expressed as fold-increase over control (mean±sem).#P<0.05,*P<0.01,**P<0.001 EpoR-R129C expressing cells compared to vector-transfected cells.
To determine whether enhanced ERK signaling pathway activation contributes to the increased cellular proliferation of cells expressing EpoR-R129C, we examined the effect of MEK kinase inhibitor PD98059 on cell growth. Treatment of breast cancer cells with PD98059 abolished the Epo-induced and constitutive phosphorylation of ERK1/2(Supplementary Fig.S3A). In proliferation assays, EpoR-R129C expressing cells exhibited significantly decreased growth in the presence of inhibitor(Fig.4A). We examined the effect of Epo and EpoR-R129C expression on anchorage independent growth using soft-agar colony formation assays(Fig.4B). Epo treatment or the expression of EpoR-R129C in breast cancer cells significantly enhanced colony formation and both the effect of exogenous Epo and EpoR-R129C expression were blocked by the presence of MEK inhibitor. We then examined the effect of Epo treatment or EpoR-R129C expression on the migration capacity of breast cancer cells in an in vitro assay. In empty vector-transfected cells, Epo treatment significantly increased the migration of the cells by 1.95±0.2-fold(Fig.4C). EpoR-R129C expression in the absence of Epo treatment was also associated with a significant 1.51±0.18-fold increase in cellular migration compared to control cells. Epo treatment of EpoR-R129C expressing cells led to a minor, non-significant increase of migration. To determine the role of SAPK/JNK kinase in increased cellular migration, cells were treated with the kinase inhibitor SP600125 which blocks the phosphorylation of SAPK/JNK(Supplementary Fig.S3B). In the presence of SAPK/JNK kinase inhibitor, we found significant inhibition of cellular migration in response to Epo in vector-transfected cells and in cells expressing the constitutively active EpoR-R129C(Fig.4C), consistent with the important role for SAPK/JNK activation in promoting the migration capacity of other types of cancer cells[34, 35].
Fig.4. Kinase inhibitors targeting MEK/ERK and SAPK/JNK block the proliferation and migration of breast cancer cells expressing EpoR-R129C.
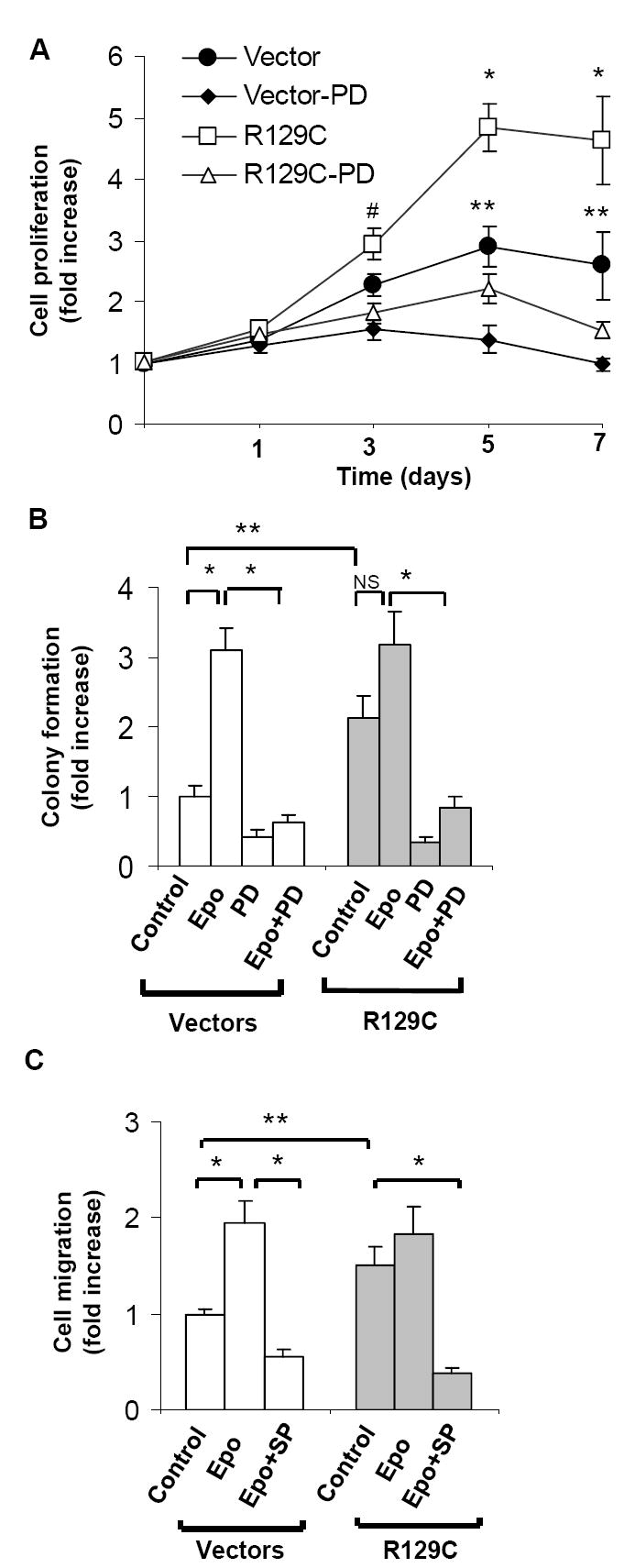
A.Growth curves of stably transfected MCF-7 cells cultured in the presence or absence of MEK inhibitor PD98059. Three independent single cell clones of empty vector- or EpoR-R129C expression plasmid-transfected cell lines were analyzed and the data was expressed as fold-increase in cell proliferation (n=9 replicates, *P<0.001 R129C compared to vector and to R129C-PD, #P<0.05 R129C compared to R129C-PD, **P<0.001 vector compared to vector-PD. B.Soft agar colony formation assays. Two independent single cell clones of empty vector or EpoR-R129C expressing cell lines were analyzed in the presence or absence of Epo (10units/ml) or MEK inhibitor for anchorage-independent growth with triplicate assays for each condition (n=11 replicates/group,**P<0.05,*P<0.001). C.Epo treatment or expression of EpoR-R129C promotes the migration of breast cancer cells. MCF-7 cells were placed in Transwell chambers in the presence or absence of JNK inhibitor SP600125 and Epo (1unit/ml). Three single cell clones in each group were analyzed for cell migration, the data was pooled and expressed as fold-increase over control(n=12/group,*P<0.001,**P<0.05).
In these studies we show that induction of EpoR signaling by either exogenous Epo treatment or over-expression of EpoR-R129C leads to the activation of MAP kinase pathway in MCF-7 breast cancer cells. We found that unlike ERK1/2, the constitutive phosphorylation of JAK2 protein in MCF-7 cells was not increased further by either Epo treatment or over-expression of EpoR-R129C. The mechanism of the increased cellular proliferation as a result of constitutively active EpoR-R129C expression in breast cancer cells primarily involves increased activation of the ERK1/2 pathway and is not associated with increased JAK2-STAT5 phosphorylation. This finding contrasts with the predominant signaling mechanism in hematopoietic cells where increased proliferation as a result of EpoR-R129C expression is associated with constitutive activation of JAK2-STAT5 pathway and not ERK1/2 activation[31,32,36]. Our studies also indicate that in human breast cancer cells, EpoR-R129C expression enhances Epo responsiveness as manifested by increased Epo-induced phosphorylation of ERK1/2 consistent with the results of our previous in vivo studies in which EpoR-R129C expression in rodent mammary carcinoma cells was associated with increased intra-tumor phospho-ERK content and stimulation of tumor xenograft growth[33]. The present studies also demonstrate that the expression of EpoR-R129C is associated with enhanced Epo-mediated phosphorylation of SAPK/JNK, a pathway that is involved in increased migration capacity of breast cancer cells in vitro. In conclusion, using a novel cell culture model to investigate Epo biology in tumor cells, these findings suggest that EpoR expression and activation in breast cancer cells has the potential to contribute to tumor progression by promoting cellular proliferation and invasiveness.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health CA100844 and Department of Defense DAMD-17-03-1-0968. The authors thank Dr. Harvey F. Lodish for generously providing the EpoR-R129C cDNA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khuri FR. Weighing the hazards of erythropoiesis stimulation in patients with cancer. N Engl J Med. 2007;356:2445–2448. doi: 10.1056/NEJMp078101. [DOI] [PubMed] [Google Scholar]
- 2.Bennett CL, Silver SM, Djulbegovic B, Samaras AT, Blau CA, Gleason KJ, Barnato SE, Elverman KM, Courtney DM, McKoy JM, Edwards BJ, Tigue CC, Raisch DW, Yarnold PR, Dorr DA, Kuzel TM, Tallman MS, Trifilio SM, West DP, Lai SY, Henke M. Venous thromboembolism and mortality associated with recombinant erythropoietin and darbepoetin administration for the treatment of cancer-associated anemia. Jama. 2008;299:914–924. doi: 10.1001/jama.299.8.914. [DOI] [PubMed] [Google Scholar]
- 3.Arcasoy MO. The non-haematopoietic biological effects of erythropoietin. Br J Haematol. 2008;141:14–31. doi: 10.1111/j.1365-2141.2008.07014.x. [DOI] [PubMed] [Google Scholar]
- 4.Hardee ME, Arcasoy MO, Blackwell KL, Kirkpatrick JP, Dewhirst MW. Erythropoietin biology in cancer. Clin Cancer Res. 2006;12:332–339. doi: 10.1158/1078-0432.CCR-05-1771. [DOI] [PubMed] [Google Scholar]
- 5.Arcasoy MO. Erythropoiesis-stimulating agent use in cancer: preclinical and clinical perspectives. Clin Cancer Res. 2008;14:4685–4690. doi: 10.1158/1078-0432.CCR-08-0264. [DOI] [PubMed] [Google Scholar]
- 6.Westenfelder C, Baranowski RL. Erythropoietin stimulates proliferation of human renal carcinoma cells. Kidney Int. 2000;58:647–657. doi: 10.1046/j.1523-1755.2000.00211.x. [DOI] [PubMed] [Google Scholar]
- 7.Acs G, Acs P, Beckwith SM, Pitts RL, Clements E, Wong K, Verma A. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res. 2001;61:3561–3565. [PubMed] [Google Scholar]
- 8.Pajonk F, Weil A, Sommer A, Suwinski R, Henke M. The erythropoietin-receptor pathway modulates survival of cancer cells. Oncogene. 2004;23:8987–8991. doi: 10.1038/sj.onc.1208140. [DOI] [PubMed] [Google Scholar]
- 9.Feldman L, Wang Y, Rhim JS, Bhattacharya N, Loda M, Sytkowski AJ. Erythropoietin stimulates growth and STAT5 phosphorylation in human prostate epithelial and prostate cancer cells. Prostate. 2006;66:135–145. doi: 10.1002/pros.20310. [DOI] [PubMed] [Google Scholar]
- 10.Lester RD, Jo M, Campana WM, Gonias SL. Erythropoietin promotes MCF-7 breast cancer cell migration by an ERK/mitogen-activated protein kinase-dependent pathway and is primarily responsible for the increase in migration observed in hypoxia. J Biol Chem. 2005;280:39273–39277. doi: 10.1074/jbc.M509446200. [DOI] [PubMed] [Google Scholar]
- 11.Mohyeldin A, Lu H, Dalgard C, Lai SY, Cohen N, Acs G, Verma A. Erythropoietin signaling promotes invasiveness of human head and neck squamous cell carcinoma. Neoplasia. 2005;7:537–543. doi: 10.1593/neo.04685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai SY, Childs EE, Xi S, Coppelli FM, Gooding WE, Wells A, Ferris RL, Grandis JR. Erythropoietin-mediated activation of JAK-STAT signaling contributes to cellular invasion in head and neck squamous cell carcinoma. Oncogene. 2005;24:4442–4449. doi: 10.1038/sj.onc.1208635. [DOI] [PubMed] [Google Scholar]
- 13.Acs G, Chen M, Xu X, Acs P, Verma A, Koch CJ. Autocrine erythropoietin signaling inhibits hypoxia-induced apoptosis in human breast carcinoma cells. Cancer Lett. 2004;214:243–251. doi: 10.1016/j.canlet.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 14.Hardee ME, Rabbani ZN, Arcasoy MO, Kirkpatrick JP, Vujaskovic Z, Dewhirst MW, Blackwell KL. Erythropoietin inhibits apoptosis in breast cancer cells via an AKT-dependent pathway without modulating in vivo chemosensitivity. Mol Cancer Ther. 2006;5:356–361. doi: 10.1158/1535-7163.MCT-05-0196. [DOI] [PubMed] [Google Scholar]
- 15.Um M, Lodish HF. Antiapoptotic effects of erythropoietin in differentiated neuroblastoma SH-SY5Y cells require activation of both the STAT5 and AKT signaling pathways. J Biol Chem. 2006;281:5648–5656. doi: 10.1074/jbc.M510943200. [DOI] [PubMed] [Google Scholar]
- 16.Acs G, Zhang PJ, McGrath CM, Acs P, McBroom J, Mohyeldin A, Liu S, Lu H, Verma A. Hypoxia-inducible erythropoietin signaling in squamous dysplasia and squamous cell carcinoma of the uterine cervix and its potential role in cervical carcinogenesis and tumor progression. Am J Pathol. 2003;162:1789–1806. doi: 10.1016/S0002-9440(10)64314-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belenkov AI, Shenouda G, Rizhevskaya E, Cournoyer D, Belzile JP, Souhami L, Devic S, Chow TY. Erythropoietin induces cancer cell resistance to ionizing radiation and to cisplatin. Mol Cancer Ther. 2004;3:1525–1532. [PubMed] [Google Scholar]
- 18.Kumar SM, Acs G, Fang D, Herlyn M, Elder DE, Xu X. Functional erythropoietin autocrine loop in melanoma. Am J Pathol. 2005;166:823–830. doi: 10.1016/S0002-9440(10)62303-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solar P, Feldman L, Jeong JY, Busingye JR, Sytkowski AJ. Erythropoietin treatment of human ovarian cancer cells results in enhanced signaling and a paclitaxel-resistant phenotype. Int J Cancer. 2007;122:281–288. doi: 10.1002/ijc.23071. [DOI] [PubMed] [Google Scholar]
- 20.Berdel WE, Oberberg D, Reufi B, Thiel E. Studies on the role of recombinant human erythropoietin in the growth regulation of human nonhematopoietic tumor cells in vitro. Ann Hematol. 1991;63:5–8. doi: 10.1007/BF01714953. [DOI] [PubMed] [Google Scholar]
- 21.Rosti V, Pedrazzoli P, Ponchio L, Zibera C, Novella A, Lucotti C, Della Cuna GR, Cazzola M. Effect of recombinant human erythropoietin on hematopoietic and non-hematopoietic malignant cell growth in vitro. Haematologica. 1993;78:208–212. [PubMed] [Google Scholar]
- 22.Westphal G, Niederberger E, Blum C, Wollman Y, Knoch TA, Rebel W, Debus J, Friedrich E. Erythropoietin and G-CSF receptors in human tumor cells: expression and aspects regarding functionality. Tumori. 2002;88:150–159. doi: 10.1177/030089160208800214. [DOI] [PubMed] [Google Scholar]
- 23.Rossler J, Stolze I, Frede S, Freitag P, Schweigerer L, Havers W, Fandrey J. Hypoxia-induced erythropoietin expression in human neuroblastoma requires a methylation free HIF-1 binding site. J Cell Biochem. 2004;93:153–161. doi: 10.1002/jcb.20133. [DOI] [PubMed] [Google Scholar]
- 24.Liu WM, Powles T, Shamash J, Propper D, Oliver T, Joel S. Effect of haemopoietic growth factors on cancer cell lines and their role in chemosensitivity. Oncogene. 2004;23:981–990. doi: 10.1038/sj.onc.1207294. [DOI] [PubMed] [Google Scholar]
- 25.Gewirtz DA, Di X, Walker TD, Sawyer ST. Erythropoietin fails to interfere with the antiproliferative and cytotoxic effects of antitumor drugs. Clin Cancer Res. 2006;12:2232–2238. doi: 10.1158/1078-0432.CCR-05-2287. [DOI] [PubMed] [Google Scholar]
- 26.Yoshimura A, Longmore G, Lodish HF. Point mutation in the exoplasmic domain of the erythropoietin receptor resulting in hormone-independent activation and tumorigenicity. Nature. 1990;348:647–649. doi: 10.1038/348647a0. [DOI] [PubMed] [Google Scholar]
- 27.Longmore GD, Pharr PN, Lodish HF. A constitutively activated erythropoietin receptor stimulates proliferation and contributes to transformation of multipotent, committed nonerythroid and erythroid progenitor cells. Mol Cell Biol. 1994;14:2266–2277. doi: 10.1128/mcb.14.4.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longmore GD, Lodish HF. An activating mutation in the murine erythropoietin receptor induces erythroleukemia in mice: a cytokine receptor superfamily oncogene. Cell. 1991;67:1089–1102. doi: 10.1016/0092-8674(91)90286-8. [DOI] [PubMed] [Google Scholar]
- 29.Watowich SS, Yoshimura A, Longmore GD, Hilton DJ, Yoshimura Y, Lodish HF. Homodimerization and constitutive activation of the erythropoietin receptor. Proc Natl Acad Sci U S A. 1992;89:2140–2144. doi: 10.1073/pnas.89.6.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shikama Y, Barber DL, D’Andrea AD, Sieff CA. A constitutively activated chimeric cytokine receptor confers factor-independent growth in hematopoietic cell lines. Blood. 1996;88:455–464. [PubMed] [Google Scholar]
- 31.Longmore GD, You Y, Molden J, Liu KD, Mikami A, Lai SY, Pharr P, Goldsmith MA. Redundant and selective roles for erythropoietin receptor tyrosines in erythropoiesis in vivo. Blood. 1998;91:870–878. [PubMed] [Google Scholar]
- 32.Moucadel V, Constantinescu SN. Differential STAT5 signaling by ligand-dependent and constitutively active cytokine receptors. J Biol Chem. 2005;280:13364–13373. doi: 10.1074/jbc.M407326200. [DOI] [PubMed] [Google Scholar]
- 33.Hardee ME, Cao Y, Fu P, Jiang X, Zhao Y, Rabbani ZN, Vujaskovic Z, Dewhirst MW, Arcasoy MO. Erythropoietin blockade inhibits the induction of tumor angiogenesis and progression. PLoS ONE. 2007;2:e549. doi: 10.1371/journal.pone.0000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia BP, Schlaepfer DD. Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res. 2001;61:7079–7090. [PubMed] [Google Scholar]
- 35.Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219–223. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- 36.Miura Y, Miura O, Ihle JN, Aoki N. Activation of the mitogen-activated protein kinase pathway by the erythropoietin receptor. J Biol Chem. 1994;269:29962–29969. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.