

Abstract
Disruption and thrombosis of atherosclerotic plaques cause most acute cardiovascular events, but their systematic study has been hampered by the lack of suitable animal models. To assess the value of a modified rabbit model of atherothrombosis, we performed detailed histology of rabbit aortic plaques. Atherosclerosis was induced with a high cholesterol diet fed 2 weeks prior to and 6 weeks after balloon injury of the aorta, followed by 4 weeks of normal diet. We found six out of eight types of plaques cataloged by the American Heart Association in the rabbit aorta. Vulnerable plaques were defined as those with attached platelet and fibrin-rich thrombi after pharmacological triggering with Russell's viper venom and histamine. Ruptured plaques had, as also described for human plaques: i) marked medial and adventitial changes, including neovascularization and inflammation; ii) cholesterol monohydrate crystals and liquid crystalline cholesterol esters in the intima and the fibrous cap; and iii) inflamed, thin fibrous caps. Increased cholesterol monohydrate area, internal elastic lamina area, positive remodeling, fibrous cap inflammation, adventitia breakdown, and inflammation were independent predictors of plaque disruption. Our findings reveal novel insights into plaque vulnerability and could guide the design of noninvasive imaging approaches for detecting and treating high-risk plaques.
Keywords: thrombosis, lipids, vulnerable plaques
Histological studies have shown that acute coronary syndromes are caused by sudden rupture/erosion of atherosclerotic plaques that result in thrombosis (1, 2). These plaques have been termed “vulnerable” or “high-risk.” In contrast, stable plaques may remain asymptomatic for years. Ruptured human plaques (postmortem) typically exhibit a thin (2), inflamed (3, 4) fibrous cap overlaying a lipid core, increased neovascularization (5), medial and adventitial changes (6), intraplaque hemorrhage (7), and positive remodeling (8). Conversely, in eroded plaques, the thrombus forms over an intima without endothelial cells and a fibrous cap rich in smooth muscle cells (SMCs) and proteoglycans. These plaques have variable degrees of inflammation (9) and rarely contain a necrotic core.
The inability to study atherosclerosis and sudden thrombosis in a controlled manner in humans necessitates the use of animal models (10–12), such as fat-fed New Zealand White (NZW) rabbits. These rabbits developed foam-cell-rich (fatty steaks) plaques when short-term (6–10 weeks) high-fat diets were the only stimulus used to induce atherosclerosis (13, 14). However, intermittent cycles of fat feeding with periods of normal diet (15–17) induced plaques at more advanced stages that resembled human atheroma. Moreover, with the combination of arterial wall injury and hyperlipidemia, advanced lesions formed in shorter periods (18, 19).
The addition of pharmacological triggering at the end of the atherogenic diet (16) provided the first evidence of thrombosis associated with plaques in an experimental animal model, the Constantinides New Zealand white (CNZW) rabbit model, with similarities to thrombosis seen in human coronary arteries. The first in vivo magnetic resonance images of thrombus associated with plaque disruption were obtained in a CNZW rabbit model (20), and the detection of the thrombus was enhanced by molecular-targeted imaging using a fibrin binding peptide conjugated to gadolinium (21).
Although the CNZW rabbit model has great potential for studying atherothrombosis, its similarity to human atherothrombosis has been questioned (22–24) because of reports that rabbits frequently form foam-cell-rich rather than complicated plaques and concerns about the triggering method used to induce plaque disruption and thrombosis.
The goal of this study was to evaluate the applicability of a modified CNZW rabbit model that included intermittent feeding to study the human disease by performing a systematic histological analysis of aortic plaques after triggering for plaque disruption.
METHODS
Animal model
Twenty one, adult (3 months old, 2.8 kg), male, New Zealand white rabbits were purchased from Charles River Laboratories (Wilmington, MA). All rabbits were fed a 1% cholesterol diet (PharmaServe, Framingham, MA) 2 weeks prior to and 6 weeks after balloon injury of the abdominal aorta, followed by 4 weeks of normal chow diet. Under general anesthesia (acepromazine [0.75 mg/kg], ketamine [35 mg/kg], and xylazine [2.5 mg/kg], administered intramuscular), balloon injury of the abdominal aortic wall was performed using a 3F Fogarty catheter introduced through a right femoral artery cutdown. After the catheter was advanced to the diaphragm, the balloon was inflated and the catheter was gently retracted toward the iliofemoral artery. This was repeated three times. One age- and gender-matched uninjured rabbit was fed a normal diet and used as control to compare the normal histological appearance of the media and adventitia to that of disease rabbits.
At the end of the 12 weeks of preparatory period, plaque disruption was pharmacologically triggered with intraperitoneal injection of Russell's viper venom (0.15 mg/kg; Enzyme Research, South Bend, IN) followed 30 min later by intravenous injection of histamine (0.02 mg/kg; Sigma-Aldrich, St. Louis, MO). This procedure was performed twice within 48 h on two consecutive days (at 1PM of each day). Russell's viper venom is a procoagulant factor (25) and endothelial toxin (26), and histamine acts as a vasopressor in rabbits (27). Heparin (1,000 USP units; Sigma-Aldrich) was administered intravenously prior to euthanasia to prevent postmortem blood clotting.
Seven rabbits did not complete the study. Three died before the pharmacological triggering due to respiratory distress, chronic ischemic heart disease, and liver failure. Two became anorexic before the completion of the cholesterol diet. Two more became paralyzed from the waist down after the first pharmacological triggering due to occlusive thrombosis in the subrenal aorta. All experiments were done in accordance with Public Health Service Policy on Humane Care and Use of Laboratory Animals as well as guidelines approved by the Institutional Animal Care and Use Committee of Boston University.
In vivo magnetic resonance imaging experiments
See supplementary data.
Matching the in vivo magnetic resonance imaging slices with histology
The distances from the renal branches and the iliac bifurcation were used as internal reference points to match the in vivo magnetic resonance imaging (MRI) slices with histology. During extraction, the aortas were marked with suture ligature at distances above and below the left renal branch that matched the total length covered by the in vivo MRI slices. After extraction, the ligatures were used to reextend the aortas to their physiological length, at which time they were fixed with 10% formalin solution (Thermo Scientific, Waltham, MA) and cut in 1.5–2.0 cm segments. The proximal end of each segment was marked with ink on its anterior side. This procedure was used to avoid potential errors in the calculation of the percentage of stenosis, as previously reported (28). The segments where cryoprotected in 30% sucrose, embedded in tissue freezing medium (Thermo Scientific), and frozen in −80°C. Serial, 10 μm-thick cross sections were collected throughout the length of each segment.
The sections were stained with Masson's trichrome (Sigma-Aldrich) for cellular components (SMCs: pink, and red blood cells: red), fibrous tissue (blue), fibrin, and platelet-rich thrombus (purple). Picrosirius red (Electron Microscopy Sciences, Hatfield, PA) staining was used to identify the type of collagen fibers (type I: orange, and type III: green, under polarized light). Hot-stage polarized light microscopy using unstained sections was used to detect cholesteryl esters (CEs) and cholesterol monohydrate (CholM) crystals based on their birefringence (29, 30) (Figs. 4, 5; and Methods in supplementary data).
Fig. 4.

Histological examples of ruptured (A–D) and eroded (E–H) rabbit plaques associated with thrombosis. All slices were obtained from the same rabbit. A: A cross section of a ruptured plaque. B: Higher magnification of the ruptured site shows a discontinuous fibrous cap (arrow) and an attached thrombus (*). C: Corresponding polarized light micrograph shows that lipids (bright) leaked into the thrombus at the site of rupture (arrow). D: Higher-power view of the rupture site (white arrow) shows that the center of the thrombus (*) is rich in platelets and the periphery in fibrin (yellow arrows). The detachment of the thrombus from the plaque is a cutting artifact. E: Cross section of an eroded plaque located 4.5 cm distally to the ruptured site shown in A–D. F: The luminal side adjacent to the thrombus is covered by endothelial cells (black arrow), whereas the thrombus (blue arrow) is attached to the fibrous cap. G: Higher magnification of the erosion site shows no endothelial cells underneath the thrombus and a fibrous cap rich in SMCs and proteoglycans (double arrow). H: Corresponding polarized light micrograph shows no contact between the lipid core (bright) and the thrombus. Masson's trichrome (D–G), Picrosirius red under polarized light (A, B), and polarized light microscopy (C, H). Bars = 1mm in A and E, 114 μm in B, C, and F, 40 μm in D, and 62 μm in G and H. FC, fibrous cap; LC, lipid core.
Fig. 5.

Infiltration of lipids (A–J) and inflammatory (K–M) cells within the fibrous cap of ruptured rabbit plaques. A: Cross section of a ruptured plaque. B: Picrosirius stain (under bright light) of the region identified by the black box in A shows the ruptured site (red arrow) and the attachment of the thrombus (black arrow). C: Corresponding polarized light micrograph at 25°C shows lipids (bright spots). D: Overlay of B and C viewed under polarized light shows that the fibrous cap was heavily infiltrated by lipids at the site of rupture. Lipids leaked into the lumen and the thrombus (white arrow). E: Polarized light micrograph at 60°C of the section shown in C. The loss of birefringence identified the lipids as CE. F: Cross section of another ruptured plaque. G: Organized (bracket) and degraded (arrow) fibrous cap. H: Corresponding polarized light micrograph identifies lipids (bright spots). I: A higher-magnification overlay of G and H revealed lipids in the intima (*) and the fibrous cap (double arrow). J: High-power view obtained at 60°C from the region indicated by the black rectangle in I, verified the presence of cholesterol monohydrate crystals within the fibrous cap. K: Photomicrograph of the fibrous cap at the site of plaque rupture indicated by the arrow in F shows an inflamed fibrous cap. The fibrous cap appears porous and is infiltrated by lymphocytes and monocytes (L, taken from the white box in K) and eosinophils (M, taken from the black box in K). Masson's trichrome (A, F, K, L, and M), Picrosirius red (B and G), polarized light microscopy (C, E, H, and J), overlays (D and I). Bars = 1 mm in A and F, 40 μm in B–E, 114 μm in G and H, 50 μm in I, 25 μm in J and K, and 10 μm in L and M.
Atherosclerotic plaques (n = 58) were sectioned into 388 sections and categorized into early (type II, fatty streak, and type III, preatheroma) and advanced (type IV, atheroma; typeVa, fibroatheroma; typeVc, fibrotic; and typeVI, complicated) plaques according to the American Heart Association (AHA) criteria (31, 32) (Fig. 1). Type VI plaques were subdivided into VI, ruptured, and VI, eroded. Rupture was defined as a discontinuity of the fibrous cap with a contact between the thrombus and the underlying lipid core. Erosion was identified when the thrombus was attached to an intima without endothelium and a fibrous cap rich in SMCs and proteoglycans.
Fig. 1.
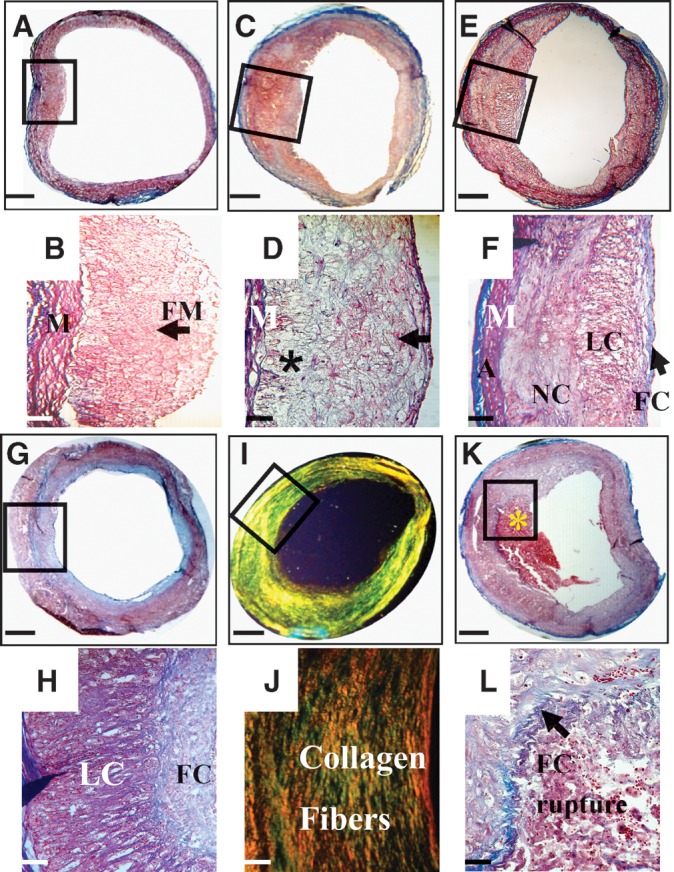
Histological classification of rabbit plaques based on the AHA criteria. A: Type II plaque. B: Higher magnification taken from the black box in A identifies foamy macrophages in the intima (arrow). C: Type III plaque. D: Higher magnification taken from the black box in C reveals foamy macrophages, foamy SMCs (arrow), and degraded tissue (*). E: Type IV plaque. F: Higher magnification taken from the black box in E shows a forming fibrous cap over a lipid core. The necrotic core is located deeper in the intima. G: Type Va plaque. H: Higher magnification of the region taken with the black box in G shows a thick fibrous cap overlaying a lipid core. I: Type Vc plaque. J: Higher magnification of the region taken with the black box identified in I demonstrates deposition of collagen fibers (type I, orange, and type III, green). K: Type VI ruptured plaque with an attached thrombus (*). L: Higher-magnification view of the ruptured site (outlined by the black box in K) revealed a discontinued fibrous cap (arrow) at which a platelet and fibrin-rich thrombus is attached. Masson's trichrome (A–H, K, and L) and Picrosirius Red viewed under polarized light (I and J). Bars = 1 mm in A, C, E, G, I, and K, 114 μm in B, D, F, H, and J, and 25 μm in L. M, media; FM, foamy macrophages; A, adventitia; FC, fibrous cap; LC, lipid core; NC, necrotic core.
Morphometry
Structural changes of the vessel wall as seen in sections stained with Masson's trichrome and/or unstained sections viewed under polarized light were recorded as categorical variables (present or absent). They included the following: i) rupture of the internal elastic lamina (IEL) (intimomedial change), ii) medial fibrosis, iii) media atrophy, iv) media breakdown, v) medial inflammation, vi) media neovascularization, vii) adventitial breakdown, viii) adventitial inflammation, and ix) adventitial neovascularization. Some of these changes have been previously described in an autopsy study of human aortic plaques (6). Rupture of the IEL was identified as a fracture of the IEL resulting in contact between the intima and media. Medial fibrosis was defined as the presence of increased collagen deposition in parts of the media. Since the media of the abdominal aorta of nondisease rabbits usually consisted of 16 ± 1.5 lamellar units, medial atrophy in disease rabbits was defined as the loss of more than 4 units and/or lamellar collapse as previously described for human plaques (6). Medial breakdown was identified when the elastic lamellae appeared fragmented and intracellular or extracellular lipid deposits were present. Adventitial breakdown was assigned when the overlying media was destroyed and lipid infiltration progressed into the adventitia. Inflammation was assigned when at least 25 or more round mononuclear cells were present within a field viewed with a ×40 magnification (33). Neovascularization was identified when red blood cells containing channels spanning the media and the adventitia were present.
The thicknesses of the fibrous cap, plaque area (PA), lipid area (LA), and cholesterol monohydrate crystals area were quantified in μm and mm2, respectively, by computerized planimetry using ImageJ (National Institutes of Health). The fibrous cap thickness was measured by drawing lines perpendicular to the lumen at five different locations of the fibrous cap on sections stained with Masson's trichrome, and the mean value was calculated. At the lesion site, the area of the lumen without plaque (lumen area) and the area circumscribed by the IEL (the potential lumen area in the absence of atherosclerosis) were traced on sections stained with Masson's trichrome. The PA was calculated by subtracting the lumen area from the IEL area. However, in plaques with discontinued IEL, the portion of the plaque that invaded the media was also included in the calculation of the PA. The cross-sectional narrowing (CSN) was defined as the extent to which the PA occupied the potential lumen area and was calculated as %CSN = (PA/IEL area)*100. The percentage of luminal stenosis was calculated by comparing the lumen area of every cross section to that of a reference site as follows: % stenosis = [1−(lumen arealesion/lumen areareference)*100]. A positive value indicates luminal narrowing, whereas a negative value indicates luminal dilation. The arterial remodeling ratio (RR) was calculated by comparing the IEL area at the lesion site to that of a reference site RR = IELlesion/IELreference. Two remodeling categories were defined: positive RR > 1.05 and negative RR < 0.95 as previously described (34). Calculations of percentage of luminal stenosis and arterial remodeling were performed after correcting for arterial tapering and interindividual variability of arterial size as previously described (35, 36). In each rabbit, the cross section with the least amount of plaque (PA = 0.76 ± 0.33mm2, IEL area = 11.5 ± 0.2 mm2, lumen area = 10.95 ± 0.6mm2, and %CSN = 5.47 ± 02.3) was chosen as a reference site, assuming that it was least affected by the disease. The percentage of total LA (cholesteryl esters and cholesterol monohydrate crystals) was calculated using polarized light photomicrographs taken at 25°C as: % lipids = (LA/PA)*100. The area occupied by CholM crystals was calculated using polarized light photomicrographs taken at 60°C.
Statistical analysis
The Statistical Package for the Social Sciences (SPSS) 11.0 was used for the analysis. Data are presented as mean ± SD. Probability values of P < 0.05 were considered significant. For two-group comparisons, continuous variables were compared using a two-tailed, Student's t-test or by the Mann-Whitney test after the variables were ranked. Multiple group comparisons were performed by one-way ANOVA test. Categorical variables were compared using the χ2 test. Qualitative data are presented as frequencies. Independent predictors of plaque disruption were identified using logistic regression analysis after the plaques where categorized as vulnerable and stable. Vulnerable plaques (rupture and eroded) were defined as those with attached platelet and fibrin-rich thrombi after pharmacological triggering for plaque disruption, whereas stable plaques had no overlying thrombus.
RESULTS
Histological features and topographic distribution of rabbit plaques
We used in vivo MRI to localize the plaques in the rabbit aorta and preselect regions for histological analysis. Six to eight plaques were detected by MRI in each aorta, all of which were validated by histology. After pharmacological triggering, 8/14 (57%) rabbits and 37% of the plaques formed thrombi. We found that each rabbit developed six out of eight types of plaques cataloged by the AHA criteria for human plaques. Examples and detailed histological characteristics of plaques classified based on the AHA criteria are shown in Fig. 1 and Table 1.
TABLE 1.
Characteristics of plaques according to the AHA classification
| AHA type | No of Sections | PA (mm2) | LA (mm2) | Lipids (%) | Luminal Stenosis (%) | CSN (%) | CholM n (%) | CholM area (mm2) | FC thickness (μm) | FC inflammation n (%) | FC Macrophage n (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fatty Streak II | 27 | 1.3 ± 0.5 | 0.5 ± 0.1 | 41.6 ± 14.6 | 14.5 ± 6.6 | 12 ± 3.8 | 0 | – | – | – | – |
| Preatheroma III | 54 | 1.6 ± 1.2 | 0.6 ± 0.2 | 41.3 ± 25 | 29 ± 22.7 | 20.7 ± 13 | 0 | – | – | – | – |
| Atheroma IV | 90 | 3.2 ± 0.8 | 1.5 ± 0.6 | 49 ± 17.7 | 39 ± 16 | 60 ± 15 | 20 (22) | 0.06 ± 0.05 | 85 ± 74 | 10 (11) | 18 (20) |
| Fibroatheroma Va | 81 | 5.4 ± 0.8 | 3.4 ± 1 | 62 ± 15.8 | 35 ± 12.4 | 57.6 ± 8.6 | 81 (100) | 0.2 ± 0.07 | 207 ± 30 | 15 (18) | 44 (54) |
| Fibrotic Vc | 36 | 4.6 ± 0.7 | 0.6 ± 0.3 | 13.8 ± 5.7 | 38 ± 15.8 | 45 ± 5.5 | 10 (28) | 0.02 ± 0.01 | 507 ± 57 | 1 (3) | 27 (75) |
| Complicated VI ruptured | 78 | 4.2 ± 1 | 2.5 ± 1 | 46 ± 12.6 | 17 ± 11.6 | 43.2 ± 11 | 78 (100) | 0.5 ± 0.2 | 57 ± 15 | 71 (91) | 69 (89) |
| Complicated VI eroded | 22 | 4.8 ± 0.8 | 1.7 ± 1.2 | 27 ± 20 | 30 ± 11.5 | 50.8 ± 7.4 | 8 (38) | 0.04 ± 0.01 | 230 ± 60 | 7 (31) | 9 (40) |
| Total and P value | 388 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| P value ruptured - eroded | 100 | 0.02 | 0.03 | <0.001 | 0.01 | 0.007 | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
FC, Fibrous cap.
The topographic distribution of each plaque type, classified based on the AHA criteria, along the aorta is illustrated in Fig. 2. Figure 2A shows an in vivo angiogram of the abdominal aorta and the corresponding excised aorta (Fig. 2B). Using the left renal branch as a reference, we found that the topological distribution of plaques along the aorta was similar between rabbits (P = 0.96); 74.8% of the plaques were located below the left renal branch, 6.3% were located at the site of the left renal branch, and 18.9% were located above the left renal branch. The distribution of plaque types tabulated based on the AHA criteria relative to the left renal branch is shown in Fig. 2C. Early plaques (types II and III) were more frequently found above and at the site of the left renal branch. In contrast, advanced plaque types (IV, Va, and Vc) were predominately found below the left renal branch. All plaque ruptures and the majority of plaque erosions occurred 1–6.5 cm below the left renal branch. The consistency between animals regarding the types of plaques found in the aorta is shown in Table 2. The frequency of each plaque type (ranging from II–Vc) was similar between rabbits with and without thrombosis.
Fig. 2.

Topographic distribution of plaque types classified based on the AHA criteria. A: In vivo magnetic resonance angiogram identifies the abdominal aorta and its renal branches. The vena cava runs parallel to the aorta. B: Corresponding excised segment of the abdominal aorta. The distribution of plaques in the aorta is described with reference to the left renal branch. C: Topographic distribution of plaques classified based on the AHA criteria. Early plaque types (II and III) are predominantly located above and at the region of the left renal branch, whereas advanced plaques (IV, Va, and Vc), including all ruptured and most of the eroded plaques, are located in the subrenal aorta. MRA, magnetic resonance angiogram.
TABLE 2.
Frequencies of plaque types classified by the AHA criteria found in rabbits with and without thrombosis
| AHA type | Percentage of Plaque Types in Rabbits with Thrombosis | Percentage of Plaque Types in Rabbits without Thrombosis | P |
|---|---|---|---|
| Fatty Streak, II | 9.20 | 14.3 | 0.50 |
| Preatheroma, III | 11.00 | 28.5 | 0.05 |
| Atheroma, IV | 19.50 | 29.0 | 0.40 |
| Fibroatheroma, Va | 9.30 | 14.5 | 0.50 |
| Fibrotic, Vc | 14.00 | 13.7 | 0.65 |
| Complicated, VI ruptured | 22.60 | – | – |
| Complicated, VI eroded | 14.40 | – | – |
| P total | 0.23 | 0.9 | – |
Medial and adventitial changes found in advanced plaques
The development of advanced rabbit plaques in this model involved not only structural and compositional changes in the intima, as described in Fig. 1, but concurrent changes in the media and adventitia. Figure 3 shows medial and adventitial changes observed in rabbit plaques compared with an aorta extracted from a control rabbit. Plaques categorized as types II and III did not show changes in the media and adventitia. In contrast, advanced plaques showed marked changes that are illustrated in detail in Fig. 3 and supplementary Tables I and II.
Fig. 3.
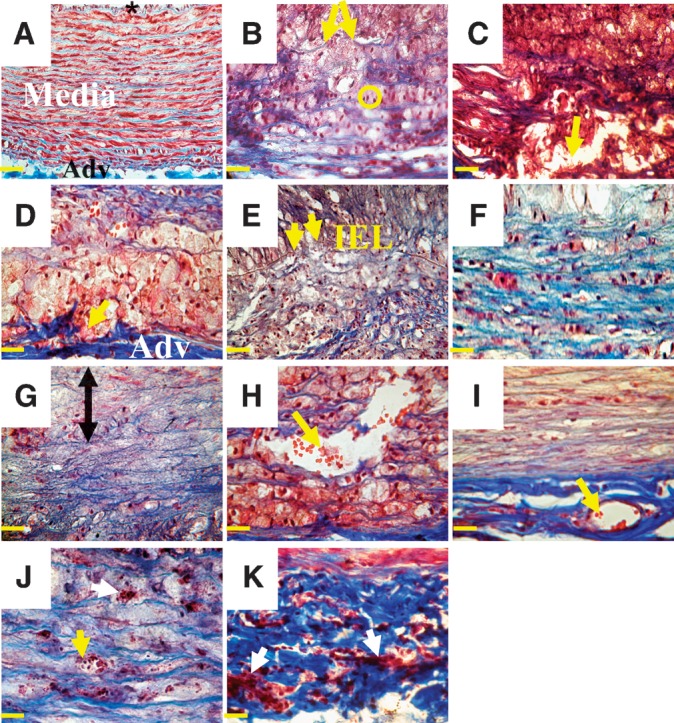
Histological examples of medial and adventitial changes in advanced rabbit plaques stained with Masson's trichrome. A: Control aorta. The intima (*) is covered by endothelial cells. Medial SMCs (red) form ordered units surrounded by collagen fibers (blue). The adventitia contains collagen and elastic fibers. B: Type IV plaque with fragmentation of medial elastic lamellae (arrows). The SMCs are reoriented longitudinally and appear as dots (circle). C: Type IV plaque with medial breakdown (arrow) and deposition of extracellular lipids and cholesterol monohydrate crystals (cholesterol clefts). D: Type Va plaque with adventitial breakdown (arrow). Foamy macrophages infiltrate the adventitia. E: Type VI eroded plaque with rupture of the IEL (arrows). F: Type VI rupture plaque with medial fibrosis (blue). G: Type VI eroded plaque with medial atrophy. Loss of SMCs starts in the upper part of the media (double arrow). H and I: Medial and adventitial neovascularization in type VI rupture plaque. The neovessels run longitudinally and contain red blood cells (red dots; arrows). J and K: Medial and adventitial inflammation in a type VI rupture plaque. Mononuclear cells are seen within neovessels (yellow arrow in J) and between the extracellular matrix (white arrows in J and K). Bars = 40 μm in A–I and 25 μm in J and K. Adv, adventitia.
Ruptured and eroded plaques
Figure 4A shows a cross section of a ruptured plaque. At the site where the fibrous cap became discontinuous, a thrombus is attached (Fig. 4B). The underlying plaque was rich in lipids that appear bright under polarized light (Fig. 4C). Some lipid leaked into the thrombus at the site of rupture. Higher-magnification view shows that the central region of the thrombus was rich in platelets, whereas the periphery was enriched in fibrin (Fig. 4D).
Figure 4E shows a cross section of an eroded plaque. A higher magnification (Fig. 4F) shows a platelet and fibrin-rich thrombus attached on a fibrous cap rich in SMCs and proteoglycans. The region of the fibrous cap without thrombus is covered by endothelial cells (Fig. 4F). In contrast, a higher-magnification view of the eroded site (Fig. 4G) revealed the absence of endothelial cells underneath the thrombus. The corresponding polarized light micrograph shows no contact between the thrombus and the lipids (Fig. 4H).
Although intraplaque hemorrhage was rarely observed in the rabbit plaques, one example is illustrated in supplementary Figure II.
Infiltration of CEs, CholM crystals, and inflammatory cells into the fibrous cap of ruptured plaques
Figure 5A shows a cross section of a ruptured plaque. Higher-magnification view (Fig. 5B) of the rupture site shows a loose fibrous cap (red) at which a thrombus is attached (brown). The corresponding polarized light micrograph at 25°C identified lipids as bright spots (Fig. 5C). An overlay of 5B and 5C under polarized light (Fig. 5D) demonstrated extensive collagen degradation and heavy infiltration by lipids at the site of fibrous cap rupture. The lipids also leaked into the thrombus. At 60°C the birefringence disappeared, indicating that the lipids infiltrating the fibrous cap at the site of rupture were crystalline cholesteryl esters that melted to an isotropic liquid (Fig. 5E).
Figure 5F shows a cross section of another plaque with a ruptured fibrous cap (arrow). Higher magnification shows a degraded fibrous cap (Fig. 5G, arrow) and abundant lipids (Fig. 5H). A higher-magnification overlay of 5G and 5H (Fig. 5I) revealed not only cholesteryl esters but also CholM crystals within the fibrous cap at 25°C. The plate-like birefringent that remained at 60°C (Fig. 5J) verified the presence of CholM crystals within the fibrous cap. Figure 5K shows that at the site of rupture (Fig. 5F, arrow) the fibrous cap is heavily inflamed. At a higher magnification the fibrous cap appeared porous and was infiltrated by lymphocytes and monocytes (Fig. 5L) as well as eosinophils (Fig. 5M). Fibrous cap inflammation was a prominent characteristic of ruptured plaques, as discussed below.
Comparison of ruptured, eroded, and advanced nonthrombosed (types IV, Va, and Vc) plaques
Ruptured plaques had a similar percentage of luminal stenosis (P = 0.08) and lower percentage of CSN (P < 0.001) compared with nonthrombosed plaques (Table 3). The LA and percentage of lipids were similar in ruptured and nonthrombosed plaques (P = 0.23 and 0.07, respectively). However, ruptured plaques more frequently had CholM crystals (P < 0.001), which occupied a larger area (P = 0.009), thinner fibrous cap (P < 0.001) with greater infiltration by foam cells (P < 0.001) and inflammatory cells (P < 0.001), higher frequencies of rupture of the IEL (P < 0.001), as well as medial and adventitial changes.
TABLE 3.
Comparison between ruptured, eroded, and advanced nonthrombosed plaques
| Ruptured (n = 78) | Eroded (n = 22) | Nonthrombosed (types IV, Va, and Vc) (n = 207) | P1 | P2 | |
|---|---|---|---|---|---|
| Intima | |||||
| PA mm2 | 4.2 ± 1 | 4.8 ± 0.8 | 4.3 ± 1.3 | 0.39 | 0.12 |
| Luminal stenosis, % | 17 ± 11.6 | 30 ± 11.5 | 37 ± 16.8 | 0.08 | 0.05 |
| CSN, % | 43 ± 11.2 | 50.8 ± 7.4 | 57 ± 12.7 | <0.001 | 0.08 |
| LA, mm2 | 2.5 ± 1 | 1.7 ± 1.2 | 2 ± 1 | 0.23 | 0.3 |
| Lipids, % | 46 ± 12.6 | 27 ± 20 | 48 ± 23 | 0.07 | <0.001 |
| ChloM crystals | 78 (100) | 8 (36) | 111 (54) | <0.001 | 0.12 |
| ChloM crystals area (mm2) | 0.5 ± 0.2 | 0.04 ± 0.01 | 0.08 ± 0.1 | 0.009 | 0.3 |
| Intimomedial change | |||||
| Rupture of the IEL, % | 70 (90) | 13 (59) | 120 (58) | <0.001 | 0.9 |
| Fibrous Cap | |||||
| FC thickness, μm | 57 ± 15 | 230 ± 60 | 204 ± 162 | <0.001 | 0.67 |
| FC macrophages, %; FC inflammation, % | 69 (89), 71 (91) | 9 (40), 7 (31) | 89 (43), 26 (12) | <0.001, <0.001 | 0.8, 0.01 |
| Media | |||||
| Breakdown, % | 78 (100) | 12 (54) | 130 (63) | <0.001 | 0.45 |
| Fibrosis, % | 66 (85) | 12 (54) | 57 (27) | <0.001 | 0.006 |
| Atrophy, % | 61 (78) | 13 (59) | 75 (36) | <0.001 | 0.03 |
| Inflammation, % | 63 (81) | 1 (4) | 18 (9) | <0.001 | 0.5 |
| Neovascularization, % | 65 (83) | 15 (68) | 100 (48) | <0.001 | 0.07 |
| Adventitia | |||||
| Breakdown, % | 70 (90) | 5 (23) | 4 (36) | <0.001 | 0.22 |
| Inflammation, % | 72 (92) | 12 (54) | 80 (39) | <0.001 | 0.14 |
| Neovascularization, % | 69 (88) | 15 (68) | 141 (68) | 0.001 | 0.9 |
P1 and P2 compare the ruptured and eroded versus nonthrombosed plaques, respectively. FC, fibrous cap.
Eroded plaques had a similar percentage of luminal stenosis (P = 0.05), percentage of CSN (0.08), LA (P = 0.3), CholM area (P = 0.3), frequency of CholM crystals (P = 0.12), and less percentage of lipid (P < 0.001) compared with nonthrombosed plaques. Fibrous cap inflammation (P = 0.01), medial fibrosis (P = 0.006), and atrophy (P = 0.03) were more prevalent in eroded plaques compared with nonthrombosed plaques.
Comparison of stable and vulnerable (ruptured and eroded) plaques
The plaque area (P < 0.001) and IEL area (P < 0.001) were significantly larger in vulnerable plaques compared to stable plaques whereas the luminal area (P = 0.1), percentage of luminal stenosis (P = 0.1), and percentage of CSN (P = 0.3) were similar between the two groups. The RR was significantly higher in vulnerable compared to stable plaques (1.05 ± 0.1 versus 0.85 ± 0.2, P < 0.001) (Table 4).
TABLE 4.
Histological measurements for stable and vulnerable (rupture and eroded) plaques
| Stable (n = 288) | Vulnerable (n = 100) | P | |
|---|---|---|---|
| PA, mm2 | 3.5 ± 1.7 | 4.3 ± 1.5 | <0.001 |
| IEL area, mm2 | 8 ± 2.3 | 9.6 ± 1.2 | <0.001 |
| Luminal area, mm2 | 4.4 ± 2.3 | 5.3 ± 1.4 | 0.100 |
| Luminal stenosis, % | 23 ± 14 | 20 ± 13 | 0.100 |
| CSN, % | 45.8 ± 21.4 | 44.8 ± 11 | 0.300 |
| RR | 0.85 ± 0.2 | 1.05 ± 0.1 | <0.001 |
Regression analysis identified increased CholM area [P < 0.001, odds ratio (OR)=1.14], IEL area (P < 0.001, OR = 3.5), positive remodeling (P < 0.001, OR = 1.12), the presence of fibrous cap inflammation (P = 0.001, OR = 14.2) CholM crystals (P = 0.1, OR = 12.3), breakdown of the adventitia (P = 0.002, OR = 18.4), and adventitial inflammation (P = 0.008, OR = 16.6) as independent predictors of thrombosis.
Effects of PA on IEL area and lumen area and effects of plaque composition on remodeling
There was a strong linear correlation between plaque area and IEL area (P < 0.001; see supplementary Figure III), indicating a change of the arterial area in response to plaque size. There was also a linear correlation between the IEL area and the lumen area (P < 0.001), suggesting that vessel wall remodeling influences the lumen area and could explain the lack of association between plaque size and lumen area (P = 0.15).
We also found that several plaque components were linearly associated with positive remodeling, such as the cholesterol monohydrate area (P < 0.001), lipid area (P = 0.02), media breakdown (P < 0.001), inflammation (P < 0.001) and fibrosis (inverse relation, P < 0.001), adventitial breakdown (P < 0.001), inflammation (P = 0.002), and neovascularization (P = 0.005).
DISCUSSION
In this study, we developed a modified protocol of the CNZW rabbit that reproduces features of atherothrombosis observed in humans. We retained the procedure of balloon injury because it accelerates atherosclerosis (18, 19) and the rabbits do not have to be kept on the cholesterol diet for longer durations, which leads to lipid toxicity (37). Furthermore, balloon injury mimics one of the postulated mechanisms for atherogenesis, i.e., injury of the endothelial cells lining the luminal site of the vessel wall (38, 39). In the rabbit aorta, we observed regeneration of endothelial cells (except in regions of plaque erosion) at 10 weeks after balloon injury, although we did not assess their functionality. In all the cases, we observed that the plaque with its constituents, including lipid, proliferating SMCs, collagen fibers, inflammatory cells, etc., accumulated under the regenerated endothelium.
Initially, we used a protocol of aortic injury followed by 6 weeks of cholesterol diet prior to pharmacological triggering. No thrombi were found, and the majority of plaques were early, while only a few were advanced (type IV). To induce plaques at more advanced stages, we included two cycles of cholesterol diet followed by normal diet prior to pharmacological triggering. We chose to perform balloon injury at 2 weeks after initiating the high-cholesterol diet so that endothelial damage occurred in a setting of increased blood lipid levels, which could further accelerate plaque formation. Subsequently, we returned the rabbits to normal chow diet for 4 weeks to extend the time interval for plaque progression and development of more advanced features without continuous feeding of cholesterol. With this protocol the rabbits developed both early (types II and III) and advanced (IV, Va, Vc, and VI) plaques. Our observations suggested that there is a limit to the severity of the lesions that rabbits develop within a short time interval. This limit could not be exceeded by simply extending the time on the cholesterol diet because the rabbits could be safely feed 1% cholesterol for only 8 weeks. Beyond that point animal mortality increased because of other adverse effects of lipid toxicity. However, the effects of the high plasma cholesterol on plaque development could be extended for longer periods by switching from the cholesterol diet to the normal diet for 4 weeks. We found that during this period the plaques developed more advanced features even though the total plasma cholesterol levels slightly decreased.
Some of the key findings of our study were that the rabbit and human plaques had remarkable similarities. In addition, several novel findings enhance our understanding of the mechanisms that promote plaque vulnerability and could guide the design of noninvasive imaging approaches for the detection of vulnerable plaque. We found that the fibrous cap is a critical factor in plaque stability, and rabbit ruptured plaques had a discontinuous fibrous cap with an attached thrombus. The fibrous cap of ruptured plaques was the thinnest (57 ± 15 μm) and had the highest frequency of inflammation (89%) and infiltration by macrophages (91%), consistent with studies of human plaques (40).
Ruptured plaques had abundant liquid CE and CholM crystals in the fibrous cap. Using polarized light microscopy and without sample exposure to organic solvents, we identified the chemical composition and phases of the lipids. In contrast, previous studies concluded that CholM crystals were present in ruptured human (41) and rabbit plaques (42) by the indirect observation of “cholesterol clefts.” Abela and Aziz (43) showed that during cholesterol crystallization, the expansion of sharp-edged crystals could damage the fibrous cap.
Together with other medial and adventitial changes, neovascularization was increased in ruptured plaques, especially in the adventitia compared with the media, as shown in human lesions (44, 45). Although neovascularization is rare in the absence of atherosclerosis, it increases concurrently with intimal thickening (5, 44) and in our study it was first seen in rabbit type IV plaques with intimal thickness ∼560 μm. Since neovessels may facilitate the delivery of inflammatory cells, this could explain the increased inflammation observed in the adventitia.
Positive remodeling was associated with vulnerable plaques and that explains why although vulnerable plaques had larger plaque areas they caused similar percentage of luminal stenosis as stable plaques. These findings are consistent with reports that human vulnerable plaques are frequently mildly stenotic (46) and emphasize that the percentage of luminal stenosis that is frequently used to triage patients in different intervention groups may not reflect the severity of the disease.
Positive remodeling was linearly associated with plaque components, such as CholM and lipid area, as well as structural changes in the media and adventitia (media breakdown and inflammation, adventitial breakdown, inflammation, and neovascularization). However, it was inversely associated with medial fibrosis. Our data are consistent with previous histological studies of coronary arteries from humans who died of acute cardiovascular events (33) and intravascular studies of patients with unstable angina (47). These findings emphasize the role of lipid-laden macrophages that secrete matrix metalloproteinases and inflammation of the media and adventitia on the disorganization of the vessel wall that might promote the expansion of the IEL.
The frequency of each plaque type was consistent between animals and plaques at more advanced stages frequently developed in the subrenal aorta. The topological distribution of rabbit plaques was similar to that reported in human aortas (48, 49). To understand the preferential accumulation of lipids in certain parts of the aorta, Barakat, Uhthoff, and Colton (50) studied the permeability of the endothelium to horseradish peroxidase in the aorta of normal rabbits. They found that the highest uptake of horseradish peroxidase was in the aortic arch; it decreased distally and reached a minimum in the lower descending aorta, and then it gradually increased toward the distal segments of the abdominal aorta. Interestingly, the region of highest uptake in the arch followed a helical turn in a clockwise direction, from the posterior to the anterior surface. These data could explain our observations that more advanced rabbit plaques were located in the subrenal aorta and also why rabbit plaques were mostly eccentric.
Conclusions and limitations of the rabbit model
The short-term feeding produced a low incidence of intraplaque hemorrhage and no calcification, unlike advanced human plaques (7). In the original CNZW model with longer preparatory periods (8.5 months) and higher exposure to dietary cholesterol (15, 16), both components were present, but animal mortality was increased. The modified CNZW rabbit model as presented here, which develops a range of human-like plaques and provides a functional rather than a histological definition of vulnerable plaque, is suitable for developing noninvasive diagnostic methods and testing of possible dietary and pharmacological interventions.
A number of animal (51–53) and human (54, 55) studies have shown that alterations in diet, including switching from a high to low lipid diet or consumption of ω3-fatty acids (56) as well as administration of drugs such as statins, can promote plaque stability. However, these studies did not establish the effect of such interventions on plaque disruption. We have previously demonstrated that an angiotensin receptor blocker attenuated atherosclerosis and reduced plaque disruption in a rabbit model of endothelial denudation followed by cholesterol feeding and pharmacological triggering for thrombosis (57). In this study, the rabbits received candesartan beginning 2 days before aortic balloon injury and continued for the 8 weeks of the cholesterol diet. However, other studies without pharmacological triggering employed different experimental protocols in which the timing of the balloon injury, the duration of cholesterol feeding, and treatment varied (52, 53). Therefore, the experimental design should be tailored to the aims of each study.
Our modified rabbit model might also offer a unique opportunity to study the effects of treatments prior to or after thrombus formation by administrating a drug before or after pharmacological triggering. For example, an antiplatelet or antithrombotic drug could be administered prior to triggering to determine its effect on reducing the size of the thrombus or even prevent its formation. Furthermore, the effect of thrombolytic drugs to dissolve the thrombus could be tested by administrating the drug after triggering. Finally, since these rabbits form nonocclusive thrombi after triggering, they could allow investigations of how to manage disrupted but nonlethal plaques such as those reported in human studies (58, 59).
The rabbit aorta offers several advantages for in vivo imaging techniques, such as MRI, computed tomography, positron emission tomography, and ultrasound: i) it is about the size (∼5 mm diameter) of a human coronary artery; ii) it has a less circuitous course; and iii) it is less prone to motion artifacts. All the rabbit aortas included in this study were also studied in detail by MRI, both in the live animal at 3 Tesla and ex vivo at 11.7 Tesla. Although we have not studied the histological characteristics of plaques prior to pharmacological triggering, our current in vivo MRI experiments, which commenced before and 48 h after pharmacological triggering, showed that the image features of the nondisrupted plaques did not change between the two imaging sessions. However, the triggering protocol makes it possible to time plaque disruption and observe the disrupted plaques with newly formed thrombi. Image features of disrupted and nondisrupted plaques can be compared, and subsequently histology can be used to interpret the images and provide additional details of plaque morphology, as illustrated herein. We will show that in vivo MRI at 3 Tesla can differentiate rabbit plaques that disrupt from plaques that do not disrupt (A. Phinikaridou et al., unpublished observations). In vivo identification of high-risk plaques in humans would have major clinical implications because current treatments rely on the degree of stenosis, which frequently fails to predict cardiac events.
Supplementary Material
Acknowledgments
The authors thank Dr. Caroline Genco, Dr. Stephen Anderson, and Dr. Alexandros Tzatsos for their comments on the manuscript and Dr. Michael LaValley for assistance with the statistical analysis.
Abbreviations
AHA, American Heart Association
CE, cholesteryl ester
CholM, cholesterol monohydrate crystals
CNZW, Constantinides New Zealand white
CSN, cross-sectional narrowing
IEL, internal elastic lamina
LA, lipid area
MRI, magnetic resonance imaging
OR, odds ratio
PA, plaque area
RR, remodeling ratio
SMC, smooth muscle cell
This work was supported by National Institutes of Health Grant P50 HL-083801 (J.A.H.) and partially funded by the Wallace H. Coulter Translational Partnership Award.
Published, JLR Papers in Press, January 12, 2009.
Footnotes
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two tables and three figures.
References
- 1.Davies M. J. 1992. Anatomic features in victims of sudden coronary death. Coronary artery pathology. Circulation. 85 I19–I24. [PubMed] [Google Scholar]
- 2.Burke A. P., A. Farb, G. T. Malcom, Y. H. Liang, J. Smialek, and R. Virmani. 1997. Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 336 1276–1282. [DOI] [PubMed] [Google Scholar]
- 3.Boyle J. J. 1997. Association of coronary plaque rupture and atherosclerotic inflammation. J. Pathol. 181 93–99. [DOI] [PubMed] [Google Scholar]
- 4.van der Wal A. C., A. E. Becker, C. van der Loos, and P. Das. 1994. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 89 36–44. [DOI] [PubMed] [Google Scholar]
- 5.Moreno P. R., K. R. Purushothaman, M. Sirol, A. P. Levy, and V. Fuster. 2006. Neovascularization in human atherosclerosis. Circulation. 113 2245–2252. [DOI] [PubMed] [Google Scholar]
- 6.Moreno P. R., K. R. Purushothaman, V. Fuster, and W. N. O'Connor. 2002. Intimomedial interface damage and adventitial inflammation is increased beneath disrupted atherosclerosis in the aorta: implications for plaque vulnerability. Circulation. 105 2504–2511. [DOI] [PubMed] [Google Scholar]
- 7.Kolodgie F. D., H. K. Gold, A. P. Burke, D. R. Fowler, H. S. Kruth, D. K. Weber, A. Farb, L. J. Guerrero, M. Hayase, R. Kutys, et al. 2003. Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 349 2316–2325. [DOI] [PubMed] [Google Scholar]
- 8.Pasterkamp G., A. H. Schoneveld, D. J. Hijnen, D. P. de Kleijn, H. Teepen, A. C. van der Wal, and C. Borst. 2000. Atherosclerotic arterial remodeling and the localization of macrophages and matrix metalloproteases 1, 2 and 9 in the human coronary artery. Atherosclerosis. 150 245–253. [DOI] [PubMed] [Google Scholar]
- 9.Farb A., A. P. Burke, A. L. Tang, T. Y. Liang, P. Mannan, J. Smialek, and R. Virmani. 1996. Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 93 1354–1363. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong M. L., and D. D. Heistad. 1990. Animal models of atherosclerosis. Atherosclerosis. 85 15–23. [DOI] [PubMed] [Google Scholar]
- 11.Drew, A. F. 2000. Atherosclerosis: Experimental Methods and Protocols. Humana Press, Totowa, NJ.
- 12.Lutgens E., R. J. van Suylen, B. C. Faber, M. J. Gijbels, P. M. Eurlings, A. P. Bijnens, K. B. Cleutjens, S. Heeneman, and M. J. Daemen. 2003. Atherosclerotic plaque rupture: local or systemic process? Arterioscler. Thromb. Vasc. Biol. 23 2123–2130. [DOI] [PubMed] [Google Scholar]
- 13.Anitschkow N., and S. Chalatow. 1913. Ueber experimentelle cholesterinsteatose und ihre bedeutung für die entstehung einiger pathologischer prozesse. Centrbl. Allg. Pathol. Pathol. Anat. 24 1–9. [Google Scholar]
- 14.Constantinides P., N. Gutmann-Auersperg, and D. Hospes. 1958. Acceleration of intimal atherogenesis through prior medial injury. AMA Arch. Pathol. 66 247–254. [PubMed] [Google Scholar]
- 15.Constantinides P., J. Booth, and G. Carlson. 1960. Production of advanced cholesterol atherosclerosis in the rabbit. Arch. Pathol. 70 80–92. [PubMed] [Google Scholar]
- 16.Constantinides P., and R. N. Chakravarti. 1961. Rabbit arterial thrombosis production by systemic procedures. Arch. Pathol. 72 197–208. [PubMed] [Google Scholar]
- 17.Vesselinovitch D., R. W. Wissler, K. Fisher-Dzoga, R. Hughes, and L. Dubien. 1974. Regression of atherosclerosis in rabbits. I. Treatment with low-fat diet, hyperoxia and hypolipidemic agents. Atherosclerosis. 19 259–275. [DOI] [PubMed] [Google Scholar]
- 18.Weidinger F. F., J. M. McLenachan, M. I. Cybulsky, J. T. Fallon, N. K. Hollenberg, J. P. Cooke, and P. Ganz. 1991. Hypercholesterolemia enhances macrophage recruitment and dysfunction of regenerated endothelium after balloon injury of the rabbit iliac artery. Circulation. 84 755–767. [DOI] [PubMed] [Google Scholar]
- 19.Abela G. S., P. D. Picon, S. E. Friedl, O. C. Gebara, A. Miyamoto, M. Federman, G. H. Tofler, and J. E. Muller. 1995. Triggering of plaque disruption and arterial thrombosis in an atherosclerotic rabbit model. Circulation. 91 776–784. [DOI] [PubMed] [Google Scholar]
- 20.Johnstone M. T., R. M. Botnar, A. S. Perez, R. Stewart, W. C. Quist, J. A. Hamilton, and W. J. Manning. 2001. In vivo magnetic resonance imaging of experimental thrombosis in a rabbit model. Arterioscler. Thromb. Vasc. Biol. 21 1556–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Botnar R. M., A. Buecker, A. J. Wiethoff, E. C. Parsons, Jr., M. Katoh, G. Katsimaglis, R. M. Weisskoff, R. B. Lauffer, P. B. Graham, R. W. Gunther, et al. 2004. In vivo magnetic resonance imaging of coronary thrombosis using a fibrin-binding molecular magnetic resonance contrast agent. Circulation. 110 1463–1466. [DOI] [PubMed] [Google Scholar]
- 22.Wissler, R. W., and D. Vesselinovitch. 1974. Differences between Human and Animal Atherosclerosis. In Atherosclerosis III. G. Schettler and A. Weizel, editors. Springer, New York. 319–325.
- 23.Badimon L. 2001. Atherosclerosis and thrombosis: lessons from animal models. Thromb. Haemost. 86 356–365. [PubMed] [Google Scholar]
- 24.Rekhter M. D., G. W. Hicks, D. W. Brammer, C. W. Work, J-S. Kim, D. Gordon, J. A. Keiser, and M. J. Ryan. 1998. Animal model that mimics atherosclerotic plaque rupture. Circ. Res. 83 705–713. [DOI] [PubMed] [Google Scholar]
- 25.Davey M. G., and E. F. Luscher. 1965. Actions of some coagulant snake venoms on blood platelets. Nature. 207 730–732. [DOI] [PubMed] [Google Scholar]
- 26.Constantinides P., and M. Robinson. 1969. Ultrastructural injury of arterial endothelium. Arch. Pathol. 88 99–117. [PubMed] [Google Scholar]
- 27.Awano K., M. Yokoyama, and H. Fukuzaki. 1989. Role of serotonin, histamine, and thromboxane A2 in platelet-induced contractions of coronary arteries and aortae from rabbits. J. Cardiovasc. Pharmacol. 13 781–792. [PubMed] [Google Scholar]
- 28.Stary H. C. 1989. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis. 9 I19–I32. [PubMed] [Google Scholar]
- 29.Small, D. M. 1986. Handbook of Lipid Research: The Physical Chemistry of Lipids. From Alkanes to Phospholipids. D.J. Hanahan, editor. Plenum Press, New York and London.
- 30.Waugh D. A., and D. M. Small. 1984. Identification and detection of in situ cellular and regional differences of lipid composition and class in lipid-rich tissue using hot stage polarizing light microscopy. Lab. Invest. 51 702–714. [PubMed] [Google Scholar]
- 31.Stary H. C., A. B. Chandler, R. E. Dinsmore, V. Fuster, S. Glagov, W. Insull, Jr., M. E. Rosenfeld, C. J. Schwartz, W. D. Wagner, and R. W. Wissler. 1995. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 15 1512–1531. [DOI] [PubMed] [Google Scholar]
- 32.Stary H. C., A. B. Chandler, R. E. Dinsmore, V. Fuster, S. Glagov, W. Insull, Jr., M. E. Rosenfeld, C. J. Schwartz, W. D. Wagner, and R. W. Wissler. 1995. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 92 1355–1374. [DOI] [PubMed] [Google Scholar]
- 33.Burke A. P., F. D. Kolodgie, A. Farb, D. Weber, and R. Virmani. 2002. Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation. 105 297–303. [DOI] [PubMed] [Google Scholar]
- 34.Pasterkamp G., C. Borst, E. J. Gussenhoven, W. P. Mali, M. J. Post, S. H. The, J. A. Reekers, and F. G. van den Berg. 1995. Remodeling of de novo atherosclerotic lesions in femoral arteries: impact on mechanism of balloon angioplasty. J. Am. Coll. Cardiol. 26 422–428. [DOI] [PubMed] [Google Scholar]
- 35.Pasterkamp G., A. H. Schoneveld, W. van Wolferen, B. Hillen, R. J. Clarijs, C. C. Haudenschild, and C. Borst. 1997. The impact of atherosclerotic arterial remodeling on percentage of luminal stenosis varies widely within the arterial system. A postmortem study. Arterioscler. Thromb. Vasc. Biol. 17 3057–3063. [DOI] [PubMed] [Google Scholar]
- 36.Shiomi M., T. Ito, M. Hasegawa, K. Yoshida, and K. L. Gould. 2004. Novel insights into coronary lumen preservation during progression of coronary atherosclerosis in coronary atherosclerosis-prone rabbits. Coron. Artery Dis. 15 419–426. [DOI] [PubMed] [Google Scholar]
- 37.Constantinides, P. 1965. Experimental Atherosclerosis. Elsevier Publishing, New York.
- 38.Ross R., and J. A. Glomset. 1976. The pathogenesis of atherosclerosis (first of two parts). N. Engl. J. Med. 295 369–377. [DOI] [PubMed] [Google Scholar]
- 39.Ross R., and J. A. Glomset. 1976. The pathogenesis of atherosclerosis (second of two parts). N. Engl. J. Med. 295 420–425. [DOI] [PubMed] [Google Scholar]
- 40.Galis Z. S., G. K. Sukhova, M. W. Lark, and P. Libby. 1994. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest. 94 2493–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Virmani R., A. P. Burke, A. Farb, and F. D. Kolodgie. 2006. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 47 C13–C18. [DOI] [PubMed] [Google Scholar]
- 42.Ma H., K. S. Aziz, R. Huang, and G. S. Abela. 2006. Arterial wall cholesterol content is a predictor of development and severity of arterial thrombosis. J. Thromb. Thrombolysis. 22 5–11. [DOI] [PubMed] [Google Scholar]
- 43.Abela G. S., and K. Aziz. 2006. Cholesterol crystals rupture biological membranes and human plaques during acute cardiovascular events–a novel insight into plaque rupture by scanning electron microscopy. Scanning. 28 1–10. [DOI] [PubMed] [Google Scholar]
- 44.Kumamoto M., Y. Nakashima, and K. Sueishi. 1995. Intimal neovascularization in human coronary atherosclerosis: Its origin and pathophysiological significance. Hum. Pathol. 26 450–456. [DOI] [PubMed] [Google Scholar]
- 45.Moreno P. R., K. R. Purushothaman, V. Fuster, D. Echeverri, H. Truszczynska, S. K. Sharma, J. J. Badimon, and W. N. O'Connor. 2004. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation. 110 2032–2038. [DOI] [PubMed] [Google Scholar]
- 46.Glagov S., E. Weisenberg, C. K. Zarins, R. Stankunavicius, and G. J. Kolettis. 1987. Compensatory enlargement of human atherosclerotic coronary arteries. N. Engl. J. Med. 316 1371–1375. [DOI] [PubMed] [Google Scholar]
- 47.Varnava A. M., P. G. Mills, and M. J. Davies. 2002. Relationship between coronary artery remodeling and plaque vulnerability. Circulation. 105 939–943. [DOI] [PubMed] [Google Scholar]
- 48.Roberts J. C. J., C. Moses, and R. H. Wilkins. 1959. Autopsy studies in atherosclerosis. I. Distribution and severity of atherosclerosis in patients dying without morphologic evidence of atherosclerotic catastrophe. Circulation. 20 511–519. [Google Scholar]
- 49.Glagov S., D. A. Rowley, and R. I. Kohut. 1961. Atherosclerosis of human aorta and its coronary and renal arteries. A consideration of some hemodynamic factors which may be related to the marked differences in atherosclerotic involvement of the coronary and renal arteries. Arch. Pathol. 72 558–571. [PubMed] [Google Scholar]
- 50.Barakat A. I., P. A. Uhthoff, and C. K. Colton. 1992. Topographical mapping of sites of enhanced HRP permeability in the normal rabbit aorta. J. Biomech. Eng. 114 283–292. [DOI] [PubMed] [Google Scholar]
- 51.Williams J. K., G. K. Sukhova, D. M. Herrington, and P. Libby. 1998. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. J. Am. Coll. Cardiol. 31 684–691. [DOI] [PubMed] [Google Scholar]
- 52.Aikawa M., E. Rabkin, Y. Okada, S. J. Voglic, S. K. Clinton, C. E. Brinckerhoff, G. K. Sukhova, and P. Libby. 1998. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 97 2433–2444. [DOI] [PubMed] [Google Scholar]
- 53.Corti R., J. I. Osende, J. T. Fallon, V. Fuster, G. Mizsei, H. Jneid, S. D. Wright, W. F. Chaplin, and J. J. Badimon. 2004. The selective peroxisomal proliferator-activated receptor-gamma agonist has an additive effect on plaque regression in combination with simvastatin in experimental atherosclerosis: in vivo study by high-resolution magnetic resonance imaging. J. Am. Coll. Cardiol. 43 464–473. [DOI] [PubMed] [Google Scholar]
- 54.Crisby M., G. Nordin-Fredriksson, P. K. Shah, J. Yano, J. Zhu, and J. Nilsson. 2001. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 103 926–933. [DOI] [PubMed] [Google Scholar]
- 55.Smilde T. J., S. van Wissen, H. Wollersheim, M. D. Trip, J. J. Kastelein, and A. F. Stalenhoef. 2001. Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolaemia (ASAP): a prospective, randomised, double-blind trial. Lancet. 357 577–581. [DOI] [PubMed] [Google Scholar]
- 56.von Schacky C., P. Angerer, W. Kothny, K. Theisen, and H. Mudra. 1999. The effect of dietary {omega}-3 fatty acids on coronary atherosclerosis: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 130 554–562. [DOI] [PubMed] [Google Scholar]
- 57.Johnstone M. T., A. S. Perez, I. Nasser, R. Stewart, A. Vaidya, F. Al Ammary, B. Schmidt, G. Horowitz, J. Dolgoff, J. Hamilton, et al. 2004. Angiotensin receptor blockade with candesartan attenuates atherosclerosis, plaque disruption, and macrophage accumulation within the plaque in a rabbit model. Circulation. 110 2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burke A. P., F. D. Kolodgie, A. Farb, D. K. Weber, G. T. Malcom, J. Smialek, and R. Virmani. 2001. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 103 934–940. [DOI] [PubMed] [Google Scholar]
- 59.Qiao Y., A. Farber, E. Semaan, and J. A. Hamilton. 2008. Images in cardiovascular medicine. Healing of an asymptomatic carotid plaque ulceration. Circulation. 118 e147–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.