

Abstract
The 3′untranslated region (UTR) of human LDL receptor (LDLR) mRNA contains three AU-rich elements (AREs) responsible for rapid mRNA turnover and mediates the stabilization induced by berberine (BBR). However, the identities of the specific RNA binding proteins involved in the regulation of LDLR mRNA stability at the steady state level or upon BBR treatment are unknown. By conducting small interfering RNA library screenings, biotinylated RNA pull-down, mass spectrometry analysis, and functional assays, we now identify heterogeneous nuclear ribonucleoprotein D (hnRNP D), hnRNP I, and KH-type splicing regulatory protein (KSRP) as key modulators of LDLR mRNA stability in liver cells. We show that hnRNP D, I, and KSRP interact with AREs of the LDLR 3′UTR with sequence specificity. Silencing the expression of these proteins increased LDLR mRNA and protein levels. We further demonstrate that BBR-induced mRNA stabilization involves hnRNP I and KSRP, as their cellular depletions abolished the BBR effect and BBR treatment reduced the binding of hnRNP I and KSRP to the LDLR mRNA 3′UTR. These new findings demonstrate that LDLR mRNA stability is controlled by a group of ARE binding proteins, including hnRNP D, hnRNP I, and KSRP. Our results suggest that interference with the ability of destabilizing ARE binding proteins to interact with LDLR-ARE motifs is likely a mechanism for regulating LDLR expression by compounds such as BBR and perhaps others.
Keywords: 3′untranslated region, berberine, mRNA stability, hypercholesterolemia
Regulation of human hepatic LDL receptor (LDLR) expression occurs at the transcriptional, posttranscriptional, and translational levels. The transcriptional regulation of the LDLR gene through a cholesterol-governed negative feedback mechanism has been well characterized and is primarily mediated by family members of the sterol-regulatory element binding proteins (SREBPs) (1, 2). The identification of proprotein convertase subtilisin/kexin type 9 (PCSK9) defined a cellular mechanism for controlling LDLR expression at the protein level (3–7). In contrast to transcriptional and translational control, molecular mechanisms underlying posttranscriptional regulation of the LDLR remain largely unknown.
One aspect of posttranscriptional regulation is the modulation of mRNA stability through cis-regulatory elements residing in the mRNA 3′untranslated region (UTR) and RNA binding proteins (RBPs) that interact with their cognate sequences within the 3′UTR (8). LDLR mRNA has a 2.5 kb long stretch of 3′UTR (9) in which three AU-rich elements (AREs) have been identified based on their sequence similarity to the classic motif UUAUUUAUU (10). AREs are the best characterized sequence determinants of messenger stability among known RNA cis-regulatory elements (11). AREs are present in 3′UTRs of many short-lived mRNA species. The destabilizing functions of ARE sequences are mediated through their interaction with ARE binding proteins (ARE-BPs) (12). Some ARE-BPs are decay promoting factors, such as KH-type splicing regulatory proteins (KSRPs), which interact with AREs and recruit RNA degradation machinery to the mRNA (13, 14). Others are functional stabilizing trans-acting factors, such as embryonic lethal abnormal vision Drosophila-like 1 (ELAVL1), also known as HuR, that bind to ARE motifs and stabilize the ARE-mRNAs (12, 15, 16). Heterogeneous nuclear ribonucleoprotein D/AU-rich element RNA binding protein 1 (hnRNP D/AUF1) functions either as a destabilizer or a stabilizer, and this seems to vary depending on cellular context or its isoform expression profiles (15–18).
The ability of AREs of LDLR mRNA to target host mRNA toward degradation was demonstrated in a heterologous system. It was shown that inclusion of the most 5′ ARE (ARE1) of the LDLR 3′UTR into a β-globin-fusion mRNA resulted in a 3-fold increase in its turnover rate, while inclusion of all three AREs to the coding region of β-globin mRNA resulted in further destabilization of the β-globin-fusion transcripts (10). However, the critical question of which ARE-BPs participate in degrading LDLR mRNA via these destabilizing sequences has not been answered.
The importance of the 3′UTR in control of liver LDLR expression is further highlighted by our recent demonstration that the natural cholesterol-lowering drug berberine (BBR) increases LDLR mRNA half-life nearly 3-fold without affecting gene transcription (19–22). The action of BBR is mediated through the LDLR mRNA 3′UTR. Interestingly, the BBR-responsive region is confined to the 1 kb of 5′ proximal ARE-containing section of the 3′UTR (19). This suggests that these ARE sequences are possibly involved in the mRNA stabilizing process as well as in the rapid degradation process.
The identification of RBPs that play critical roles in control of LDLR mRNA stability in unstimulated and in BBR-stimulated liver cells is of clinical relevance and could provide new molecular targets for therapeutic intervention. To accomplish this goal, we took a systematic approach that integrated different lines of investigation, including small interfering RNA (siRNA) library construction and screening, reporter gene transfection, biotinylated RNA pull down, mass spectrometry (MS) analysis, and functional assays. Through these comprehensive studies, we now demonstrate that LDLR mRNA stability is regulated by a complex network of RBPs involving ARE-BPs that destabilize the mRNA as well as RBPs that are required for maintaining mRNA stability. Interestingly, our results suggest that BBR-mediated mRNA half-life prolongation is achieved through interfering with the ability of specific ARE binding proteins KSRP and hnRNP I to interact with ARE sequences of the LDLR mRNA 3′UTR.
To the best of our knowledge, this is the first study of the identification of LDLR mRNA binding proteins.
MATERIALS AND METHODS
Generation of a luciferase-UTR reporter cell line and siRNA library screening
The plasmid pLuc-UTR-1 contains LDLR mRNA 3′UTR sequence from nucleotides 2677 to 5100 at the end of the luciferase coding region (20). This plasmid was transfected into HepG2 cells, and stable clones were selected by Zeocin (Invitrogen) at a concentration of 40 μg/ml. The clone LDLR-Luc6 that expressed a high level of luciferase activity and was responsive to BBR stimulation was used to screen the siRNA library. To construct a siRNA library targeting a sufficient number of known human RBPs, we conducted a Medline-based literature search. Based upon search results, 46 RBPs were selected as our initial targets for siRNA-mediated knockdown. Three siRNAs were designed to target different sequences of each transcript with exceptions of three RBPs to which a single validated siRNA was available and was used in the screening. Thus, the siRNA library was comprised of 132 siRNAs and was manufactured by Applied Biosystems. For conducting the siRNA library screening, LDLR-Luc6 cells (1 × 104 cells/well) in suspension were mixed with 12 nM siRNA in siPORT™ NeoFX™ transfection reagent and were plated in 96 well plates. Two days after transfection, cells were cultured in MEM medium containing 0.5% FBS overnight, and BBR at a concentration of 15 μg/ml was added for 8 h before cell lysis. Luciferase activity was measured in an Lmax luminomitor (Molecular Devices). Three separate transfections were conducted for each siRNA in which triplicate wells were used for each condition. Silencer negative control siRNA (Cat. No. 4618G) with scrambled nucleotide sequence was included in the screening as nonspecific control.
Analysis of knockdown effects of siRNA on mRNA and protein expressions of targeted RBPs
Total RNA was isolated from HepG2 cells untreated or treated with BBR (15 μg/ml) for 8 h. The reverse transcription was conducted with random primers using M-MLV (Promega) at 37°C for 1 h in a volume of 20 μl containing 2 μg of total RNA. RT-PCR assays were performed to measure mRNA levels of siRNA-targeted RBPs using primers and conditions listed in supplementary Table I. Western blot analysis was performed to examine the protein expression in siRNA-transfected cells as previously described (21). The antibodies directed to hnRNP D (sc-22368), E1 (sc-16503), F (sc-10045), I (sc-56701), K (sc-25373), L (sc-16550), M (sc-20002), cleavage and polyadenylation specific factor (CPSF1), and IGF-II mRNA binding protein 3 (IMP3) (sc-47893) were obtained from Santa Cruz Biotechnology. The mouse anti-KSRP antibody was generously provided by Dr. Ching-Yi Chen from the University of Alabama at Birmingham. Chicken polyclonal antibody to LDLR was purchased from Abcam (ab14056).
Quantitation of LDLR mRNA expression by Northern blot analysis and real-time PCR
Northern blot analyses of LDLR and GAPDH mRNA using 32P-labeled DNA probes were conducted as previously described (19). Real-time PCR was performed on the cDNA using an ABI Prism 7900-HT Sequence Detection System and Universal MasterMix. Human LDLR and GAPDH PreDeveloped TaqMan Assay Reagents (Applied Biosystems) were used to assess mRNA expression in HepG2 cells with or without BBR treatment.
Preparation of cytoplasmic and nuclear fractions
Cytoplasmic and nuclear fractions were prepared from HepG2 cells. Briefly, cells were trypsinized and collected by centrifugation. After two washes in PBS, cells were resuspended in 5× packed cell volume of cold buffer A (10 mM HEPES-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCL, 1 mM DTT, and 1 mM PMSF) and incubated on ice for 10 min. Swelled cells were centrifuged at 4,500 g for 5 min at 4°C, and the cell pellets were resuspended in two original packed cell volume of buffer A and disrupted by applying 20 strokes of a tight pestle of a Dounce homogenizer (Wheaton). The cell lysates were centrifuged at 4,500 g for 2 min at 4°C to pellet nuclei,and the supernatant was collected as the cytoplasmic fraction. The pelleted nuclei were resuspended in 1/2 packed nuclear volume of Low Salt Buffer (20 mM HEPES, pH 7.9, 25% glycerol, 20 mM KCl, 1.5 mM MgCl2, 20 mM EDTA, 0.5 mM DTT, and 0.2 mM PMSF) before addition of 1/2 packed nuclear volume of High Salt Buffer (1.2 M KCl) under agitation for 30 min at 4°C then centrifuged for 15 min. The supernatant was used as nuclear extract.
Plasmid construction and in vitro transcription
pLDLR2 plasmid was used as the template to PCR amplify the LDLR coding sequence or the 3′UTR using 5′ KpnI and 3′ XbaI-tailed primers. The LDLR primer sequences for in vitro transcription and luciferase reporter constructions are listed in supplementary Table I online. The plasmid pcDNA3.1(+) contains a T7 promoter upstream of the multiple cloning sites. The individual PCR fragments were cut with KpnI and XbaI and cloned into the KpnI and XbaI sites of pcDNA3.1(+) to yield pcDNA-LDLR-CDS, pcDNA-UTR1, or pcDNA-UTR2. All constructs were sequenced, and clones with correct sequence were further propagated to isolate plasmid DNA. These plasmids served as templates for the in vitro synthesis of various biotinylated LDLR transcripts by using the AmpliScribe™ T7-Flash™ transcription Kit and Biotin-16-UTP (EPICENTRE® Biotechnologies) according to the manufacturer's instructions. The reaction was conducted at 42°C for 3 h in a volume of 20 μl. Products were loaded onto a 1% agarose gel containing 1% formaldehyde. The RNA band was visualized by ethidium bromide staining. pLuc-UTR-2 was used as the template to generate luciferase-UTR reporters with ARE sites individually mutated.
Biotinylated RNA pull-down assay
Thirty micrograms of purified biotinylated transcripts were incubated with 500 μg of cytosolic protein in binding buffer (10 mM HEPES, pH 7.4, 3 mM MgCl2, 5% glycerol, and 1 mM DTT) for 30 min at room temperature. Yeast transfer RNA (50 ng/ml) and heparin (5 mg/ml) were added to mixtures, and binding reactions were allowed to continue for 10 more min. Afterwards, NeutrAvidin™ Agarose Resin (Pierce Biotechnology) was added, and the reaction was carried out at 4°C overnight under constant mixing. After a brief centrifugation, bound proteins in the pull-down material were separated by 4–20% SDS-PAGE gel. Afterwards, proteins were either subjected to direct staining using GelCode® Blue Stain Reagent (Pierce Biotechnology) or transferred to nitrocellulose membrane for Western blot analysis. In these assays, avidin-coated agarose resin was mixed with cytoplasmic proteins in the absence of biotinylated transcript as a control for nonspecific binding.
MS
Protein bands that were prominently present in the LDLR 3′UTR binding lanes, in comparison with those in control and LDLR coding sequence lanes, were excised from SDS-PAGE gels stained with GelCode® Blue and digested using the In-Gel Tryptic Digestion Kit (Pierce Biotechnology) according to the manufacturer's instructions. Tryptic samples were analyzed by an Agilent XCT Plus ion trap mass spectrometer equipped with an 1100 series liquid chromatography unit, nanoflow pump, and orthogonal nanospray source. The resulting data were analyzed with Mascot (Matrix Science; http://www.matrixscience.com). Search parameters are as follows: type of search: MS/MS Ion Search; database: SwissProt; enzyme: trypin; taxonomy: Homo sapiens; fixed modification: carbamidomethyl (C); variable modifications: oxidation (M), phospho (ST), and phospho (Y); mass values: monoisotopic: peptide mass tolerance: ± 1.2 Da; fragment mass tolerance: ± 0.6 Da; max missed cleavages: 1; peptide charge: +1, +2, and +3; protein mass: unrestricted; and instrument: ESI-TRAP. As an individual peptide may belong to different proteins, only proteins identified by MS/MS Ion Search that matched their molecular weight estimated from the SDS-PAGE gels were listed in Table 3.
TABLE 3.
List of proteins binding to LDLR mRNA identified by MS
| Characteristics of LDLR mRNA 3′UTR Binding Proteins | Accession Number | MW (Da) | Peptides Identified (% Sequence Coverage) | Protein Score | |
|---|---|---|---|---|---|
| ARE-BPs | |||||
| KSRP/FUBP2 | KH-type splicing regulatory protein, ARE-BP | Q92945 | 73063 | 19 (27.18%) | 280 |
| hnRNP D/AUF1 | AU-rich element RNA binding protein | Q14103 | 38581 | 4 (9.58%) | 67 |
| hnRNP-DL | hnRNP D-like protein | O14979 | 46580 | 2 (6.98%) | 58 |
| mRNA stabilizing factors | |||||
| HuR/ELAVL1 | Hu-antigen R, ELAV-like protein 1 | Q15717 | 36240 | 1 (3.37%) | 55 |
| Splicing factors | |||||
| hnRNP M | Splicing regulatory factor | P52272 | 77749 | 1 (1.37%) | 56 |
| HnRNP A/B | Heterogeneous nuclear ribonucleoprotein A/B | Q99729 | 36704 | 1 (3.01%) | 43 |
| ROA1 | hnRNP core protein A1, single-strand RNA binding protein | P09651 | 38936 | 2 (1.3%) | 46 |
| SF3B3 | Splicing factor 3B subunit 3 | Q15393 | 136575 | 2 (2.14%) | 115 |
| SFR16 | Splicing factor, arginine/serine-rich | Q8N2M8 | 75340 | 1 (1.74%) | 45 |
| STRAP | Serine-threonine kinase receptor-associated protein, involving in assembly of spliceosomal snRNP | Q9Y3F4 | 38756 | 3 (10%) | 40 |
| IGF-II mRNA BP | |||||
| IMP-1/IF2B1 | Insulin-like growth factor 2 mRNA binding protein 1, coding region determinant binding protein | Q9NZI8 | 63759 | 16 (18.37%) | 201 |
| IMP-2/IF2B2 | Insulin-like growth factor 2 mRNA binding protein 2 | Q9Y6M1 | 61918 | 8 (10.25%) | 122 |
| IMP-3/IF2B3 | Insulin-like growth factor 2 mRNA binding protein 3 | O00425 | 64023 | 6 (9.33%) | 83 |
| Elongation factors | |||||
| EF1A1 | Elongation factor 1-α 1 | P68104 | 50451 | 2 (4.98%) | 49 |
| EF2 | Elongation factor 2 (EF-2) | P13639 | 96246 | 7 (7.11%) | 109 |
| EF1A2 | Elongation factor 1-α 2 | Q05639 | 50780 | 1 (2.38%) | 52 |
| Poly(rC) binding protein | |||||
| PCBP1/hnRNP E1 | Poly(rC) binding protein 1, hnRNP-E1 | Q15365 | 37987 | 14 (39.89%) | 131 |
| PCBP2 | Poly(rC) binding protein 2 | Q15366 | 38955 | 8 (21.10%) | 109 |
| PCBP3 | Poly(rC) binding protein 3 | P57721 | 36201 | 8 (13.57%) | 228 |
| Poly(A) binding protein | |||||
| PABP1 | Poly(A) binding protein 1 | P11940 | 70854 | 2 (3.14%) | 62 |
| RNA helicase | |||||
| DDX3X | ATP-dependent RNA helicase | O00571 | 73597 | 2 (3.47%) | 45 |
| DDX3Y | ATP-dependent RNA helicase, DEAD box protein 3, Y-chromosomal | O15523 | 73564 | 2 (3.48%) | 45 |
| DDX17 | RNA-dependent helicase p72 | Q92841 | 72953 | 5 (8.92%) | 94 |
| DDX47 | Probable ATP-dependent RNA helicase | Q9H0S4 | 50900 | 1 (2.42%) | 85 |
| DHX36 | NA helicase associated with AU-rich element ARE | Q9H2U1 | 115673 | 2 (2.48%) | 82 |
| ExoRNase | |||||
| XRN2 | 5′-3′ exoRNase 2 | Q9H0D6 | 109426 | 3 (4.95%) | 111 |
| Others | |||||
| ACF | APOBEC1 complementation factor, a family member of hnRNP | Q9NQ94 | 65446 | 3 (5.89%) | 43 |
| CAPR1 | Cytoplasmic activation- and proliferation-associated protein 1, bound to c-myc mRNA | Q14444 | 72935 | 4 (4.80%) | 136 |
| CSDE1 | Cold shock domain-containing protein E1, cytoplasmic RNA PB, N-ras upstream gene protein | O75534 | 89684 | 4 (4.14%) | 127 |
| FUBP1/HDH V | Far upstream element binding protein 1, DNA helicase V | Q96AE4 | 67690 | 13 (24.53%) | 119 |
| FUBP3 | Far upstream element binding protein 3 | Q96I24 | 61944 | 3 (4.90%) | 56 |
| hnRPUL1 | Heterogeneous nuclear ribonucleoprotein U-like protein 1, RNA transport | Q9BUJ2 | 96250 | 2 (2.34%) | 88 |
| U5S1 | 116 kDa U5 small nuclear ribonucleoprotein component | Q15029 | 110336 | 1 (1.52%) | 41 |
| HS90A | Heat shock protein HSP 90-α (HSP 86) | P07900 | 85006 | 4 (6.56%) | 152 |
| HS90B | Heat shock protein HSP 90-β (HSP 84) | P08238 | 83554 | 7 (11.19%) | 169 |
Statistical analysis
Significant differences between control and treatment groups or between scrambled and gene-specific siRNAs were assessed by two-tailed Student's t-test. P < 0.05 was considered statistically significant.
RESULTS
Construction and screening of a human RBP siRNA library
Since it was unknown which mRNA binding proteins could interact with LDLR mRNA, we constructed an siRNA library with a capacity to silence expression of 46 known human RBPs. To screen this library, we established a clone of HepG2 cells (LDLR-Luc6) that stably express a luciferase-LDLR 3′UTR chimeric transcript. We also set up a control for the siRNA transfection with an siRNA of scrambled sequence that does not match any known gene sequence. Transfection of this control siRNA did not change cell growth, the expression level of endogenous LDLR mRNA, and the luciferase activity compared with untransfected cells. Thus, scrambled siRNA was included in the subsequent library screening assays and other functional assays as a negative control for transfection conditions.
Individual siRNA was transfected into LDL-Luc6 cells, and luciferase activity was measured in control and BBR-treated cells. Effects of siRNA on luciferease activity in unstimulated and BBR-stimulated cells were compared with the scrambled siRNA control. Analysis of summarized results of three independent screenings revealed that transfection of 23 siRNAs either did not alter luciferase activities at all or only caused marginal differences (<30% of control siRNA). The remaining 23 siRNAs affected luciferase activity and were categorized into four functional groups in Table 1.
TABLE 1.
siRNAs targeted to 23 human mRNA binding proteins showing significant effects on LDLR mRNA 3′UTR luciferase reporter activity
| Group 1: Lowered Basal Activity (Fold Change of Basal Activity) | Group 2: Increased Basal Activity (Fold Change of Basal Activity) | Group 3: Lost BBR Stimulation (BBR Fold Stimulation) | Group 4: Lost BBR Stimulation and Affected Basal Activity (Fold Basal; BBR Fold Stimulation) |
|---|---|---|---|
| Scrambled siRNA, Control (1.0) | Scrambled siRNA, Control (1.0) | Scrambled siRNA, BBR (2.18 ± 0.29; P = 0.00) | Scrambled siRNA (Control, 1.0; BBR, 2.18) |
| ACF (0.56 ± 0.02, P = 0.00) | hnRNP D (2.54 ± 0.46, P = 0.01) | hnRNP L (1.36 ± 0.15, P = 0.83) | ELAVL1 (1.46 ± 0.23, P = 0.02; 1.62 ± 0.31, P = 0.20) |
| AUH (0.39 ± 0.19, P = 0.01) | PABPC1 (3.44 ± 1.0, P = 0.01) | hnRNP M (1.32 ± 0.17, P = 0.56) | ELAVL3 (1.83 ± 0.39, P = 0.02; 1.29 ± 0.08; P = 0.08) |
| CPSF1 (0.27 ± 0.19, P = 0.00) | hnRNP U (1.43 ± 0.14, P = 0.59) | hnRNP I (1.44 ± 0.19, P = 0.01; 1.36 ± 0.09, P = 0.59) | |
| CUGBP2 (0.38 ± 0.20, P = 0.01) | PCBP3 (1.53 ± 0.33, P = 0.23) | IMP-3 (1.59 ± 0.23; P = 0.01; 1.30 ± 0.07, P = 0.10) | |
| ELAVL4 (0.57 ± 0.06, P = 0.00) | SF1 (1.58 ± 0.26, P = 0.01; 1.40 ± 0.08, P = 0.31) | ||
| GRSF1 (0.66 ± 0.16, P = 0.03) | KSRPa (1.62 ± 0.02, P = 0.00; 1.13 ± 0.08, P = 0.15) | ||
| hnRNP A3 (0.33 ± 0.08, P = 0.00) | |||
| hnRNP E1 (0.70 ± 0.06, P = 0.00) | |||
| RNPS1 (0.65 ± 0.01, P = 0.00) | |||
| SYNCRIP (0.65 ± 0.13, P = 0.01) | |||
| TIA1 (0.35 ± 0.01, P = 0.00) |
Data are mean ± SD of luciferase activities of three independent transfections. The Luc activity in scrambled siRNA-transfected control cells was defined as 1. The fold change in basal Luc activity was calculated by dividing Luc activity in gene-targeted siRNA-transfected control cells with Luc activity in scrambled siRNA-transfected cells without BBR treatment. BBR fold changes were calculated by dividing Luc activity of BBR-treated cells over control cells transfected with the same siRNA. Student's two-tailed t-test was used to determine statistical differences of basal luciferase activity between scrambled siRNA and gene-targeted siRNA transfection and between control and BBR-treated cells. P < 0.05 was considered to be statistically significant.
Effects of KSRP siRNA were demonstrated in transient cotransfection with pLuc-UTR-1 and pRL-SV40 as a normalization vector.
Eleven siRNAs (Group 1) reduced basal luciferase activities by 30–73% as compared with scrambled siRNA (P < 0.05) and did not affect BBR inducibility. Some of these RBPs, such as Apolipoprotein-1 complementation factor (23) and CPSF1 (24), are known to be involved in general RNA processing, suggesting that these factors participate in different processing events of the LDLR transcript.
Group 2 contains two siRNAs (PABPC1 and hnRNP D) that increased basal luciferase activity without significant effects on BBR stimulation. PABPC1 is a poly(A) binding protein involved in the general process of mRNA decay (25, 26) of various mRNA species, whereas hnRNP D is known to recognize specific sequence motifs to regulate mRNA decay (15).
Group 3 contains four siRNAs (hnRNP L, hnRNP M, hnRNP U, and PCBP3) that did not affect basal luciferase activity but abrogated the stimulation observed with BBR. BBR treatment resulted in a 2.2-fold increase in luciferase activity (P < 0.001) in control cells transfected with scrambled siRNA.
Group 4 contains five siRNAs that appeared to have dual effects. Depletion of their target RBPs increased basal luciferase activity by 44–83% of control (P < 0.05) and abolished the BBR stimulatory effects (P > 0.05). Although siRNA to KSRP did not change luciferase activity in the stable clone during the initial screening, it consistently increased basal luciferase activity and blocked stimulation by BBR when assayed in transient transfections with pLuc-UTR-1 and a renilla luciferase reporter (pRL-SV40) (see supplementary Fig. I); therefore, it was included in Group 4.
RT-PCR assays confirmed the effective knockdown of endogenous RBP mRNAs in 11 siRNA transfected cells without or with BBR treatment (Fig. 1). Therefore, we elected to evaluate these 11 siRNAs in more detail. The other 12 siRNAs in Table 1 that had shown alterations in the luciferase screen were not pursued further due either to difficulty in detecting the target gene by RT-PCR (nine genes) or to a lack of evidence of a significant knockdown of the target mRNA by the siRNA (three genes). Using quantitative real-time RT-PCR assays, we examined endogenous LDLR mRNA levels in untreated or BBR-treated HepG2 cells that were individually transfected with these siRNAs. Table 2 summarizes the results of three independent transfections. Cellular depletions of AUH, CPSF1, and GRSF1 reduced LDLR mRNA expression levels in the range of 36–44%. Of importance, we found that elimination of hnRNP D, I, and KSRP increased LDLR mRNA expression by >50% of control. To ensure the effects of siRNA transfections specifically altered LDLR mRNA, we examined mRNA levels of ApoB, HMG-CoA reductase (HMGCR), SREBP2, and PCSK9 in hnRNP D siRNA-transfected cells (see supplementary Fig. II). Depletion of hnRNP D increased LDLR mRNA abundance without significant effects on the other genes tested. Changes in gene expression of SREBP2 or HMGCR could indirectly alter LDLR mRNA levels through SRE-1-mediated transcriptional regulation. The fact that siRNA to hnRNP D did not alter the expression of these mRNAs supports the direct involvement of hnRNP D in LDLR mRNA decay. Similarly, we did not observe consistent changes in mRNA levels of SREBP2 or HMGCR in hnRNP I or KSRP siRNA-transfected cells.
Fig. 1.
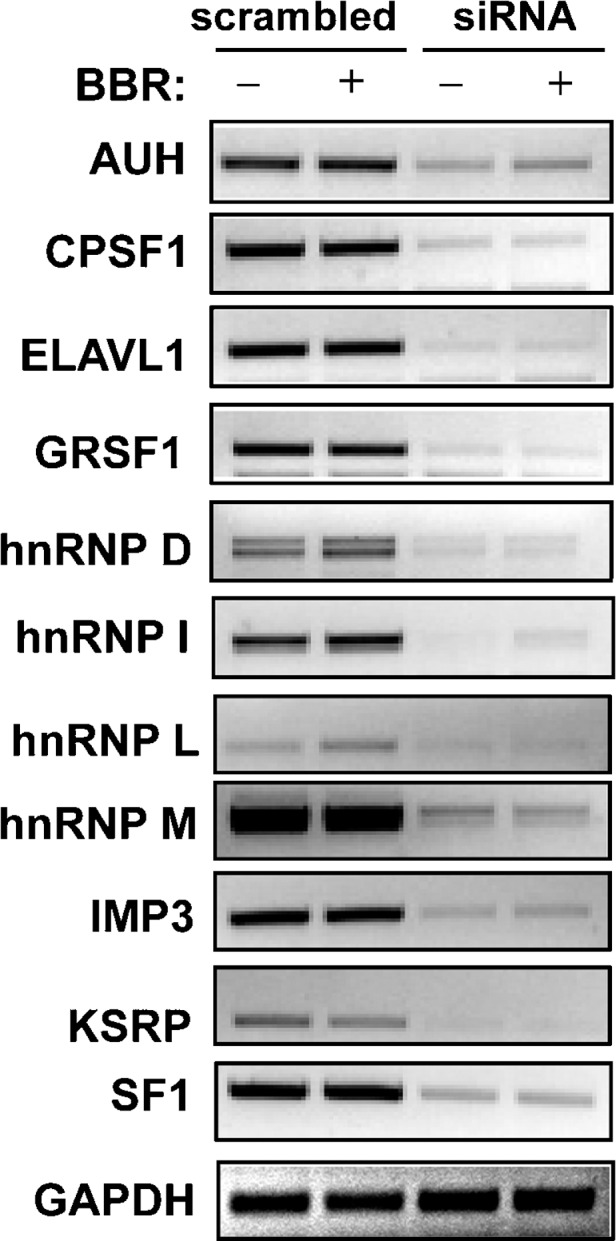
RT-PCR analysis of target gene expression in siRNA-transfected cells. HepG2 cells were plated into six-well plates and were transfected with 12 nM concentration of individual siRNAs against genes encoding RBPs or scrambled siRNA using siPORT™ NeoFX™ transfection reagent. Forty-eight hours after transfection, cells were incubated in 0.5% FBS MEM medium overnight prior to treatment with BBR (15 μg/ml) for 8 h, and total RNA was isolated. RT-PCR was carried out using specific primers. RT-PCR analyses showed that GAPDH mRNA levels were not affected by different siRNA transfection. Thus, one representative RT-PCR product of GAPDH was presented in this figure.
TABLE 2.
LDLR mRNA expression in siRNA transfected cells
| siRNA | Basal Change (Mean ± SD) | P | BBR Fold Increase (Mean ± SD) | P |
|---|---|---|---|---|
| Scrambled | 1.00 ± 0.00 | 1.82 ± 0.13 | ||
| AUHa | 0.56 ± 0.13 | 0.009 | 2.70 ± 0.58 | 0.104 |
| CPSF1a,b | 0.63 ± 0.25 | 0.028 | 2.27 ± 0.30 | 0.030 |
| ELAVL1/HuRa,c | 1.11 ± 0.02 | 0.001 | 1.26 ± 0.18 | 0.028 |
| GRSF1a,b | 0.64 ± 0.06 | 0.001 | 2.46 ± 0.11 | 0.006 |
| hnRNP Da | 1.55 ± 0.21 | 0.020 | 1.74 ± 0.21 | 0.704 |
| hnRNP Ia,c | 1.81 ± 0.22 | 0.006 | 1.20 ± 0.10 | 0.012 |
| hnRNP Lc | 1.03 ± 0.32 | 0.988 | 1.37 ± 0.56 | 0.040 |
| hnRNP Ma | 1.11 ± 0.05 | 0.037 | 1.42 ± 0.29 | 0.163 |
| IMP3a,c | 1.32 ± 0.22 | 0.028 | 1.32 ± 0.17 | 0.004 |
| KSRPa,c | 1.56 ± 0.21 | 0.021 | 1.08 ± 0.10 | 0.003 |
| SF1c | 1.17 ± 0.38 | 0.604 | 1.46 ± 0.24 | 0.040 |
HepG2 cells were transfected with individual siRNA for 2 d. Cells then were cultured in MEM containing 0.5% FBS overnight prior to the treatment of vehicle or BBR (15 μg/ml) for 8 h. LDLR mRNA levels were determined by quantitative real-time RT-PCR. The data are mean ± SD of three independent transfection assays. For changes in basal expression, two-tailed Student's t-test was used to compare the mean values of normalized LDLR mRNA levels between scrambled siRNA and a gene-specific siRNA in cells without BBR treatment. For changes in BBR activity, two-tailed Student's t-test was used to compare the mean values of BBR fold activity between scrambled siRNA and a gene-specific siRNA.
siRNA transfection statistically altered the basal LDLR mRNA abundance (P < 0.05) in control cells.
siRNA transfection statistically increased cell response to BBR stimulation compared with BBR effect in scrambled siRNA transfected cells (P < 0.05).
siRNA transfection statistically reduced cell response to BBR stimulation compared with BBR effect in scrambled siRNA-transfected cells (P < 0.05).
Table 2 also shows that BBR treatment increased endogenous LDLR mRNA level by 1.8-fold compared with untreated cells. This stimulatory effect was significantly diminished by siRNA transfections of ELAVL1, hnRNP I, hnRNP L, IMP3, KSRP, and G-rich RNA sequence binding factor 1 (GRSF1) (P < 0.05). Transfection of siRNAs of CPSF1 and G-rich RNA sequence binding factor 1 (GRSF1) caused a modest increase in BBR activity on LDLR mRNA expression. Thus, the results of the luciferase reporter assay used in the library screening are consistent with the examination of endogenous mRNA expression for 11 RBPs. This suggests that, as a minimum, these 11 RBPs are involved in control of LDLR mRNA stability, and a subset of these might participate in BBR-mediated mRNA half-life prolongation.
Biochemical characterizations of LDLR mRNA binding proteins
In parallel with the siRNA library screening, we conducted RNA pull-down assays followed by MS to identify RBPs that bind to the LDLR 3′UTR. Biotinylated transcripts corresponding to the coding sequence (CDS) or to the 3′UTR (UTR1) (see supplementary Fig. IIIA, B) were incubated with cytosolic proteins isolated from untreated or BBR-treated HepG2 cells. Avidin-coated agarose resin was subsequently added to the reactions, and ribonucleoprotein complexes were pulled down by centrifugation. Bound proteins were separated by SDS-PAGE gel and visualized by blue stain. Multiple proteins were readily detected in UTR-containing complexes (see supplementary Fig. IIIC, lanes 5 and 6) but were either not present or showed weak staining signals in CDS-containing complexes (lanes 3 and 4), whereas a few proteins bound to the resin nonspecifically (lane 2). The bands that were prominently present in UTR complexes were excised and analyzed by MS.
MS analyses (Table 3) revealed that LDLR 3′UTR binding proteins include three sequence-specific and decay-promoting ARE-BPs (KSRP, hnRNP D, and hnRNP-DL) (13, 17, 27, 28), one known stabilizing factor (ELAV1/HuR) (18), and three poly(rC) binding proteins (PCBP1/hnRNP E1, PCBP2, and PCBP3) (29, 30). In addition, a number of factors that are involved in different steps of RNA processing, such as RNA helicases, exoRNase XRN2 (31), and poly(A) binding protein PABP1 (32), were found. Notably, six hnRNPs involved in RNA splicing were identified. MS identified a total of 35 proteins from the pull-down assay.
Based upon the availability of specific antibodies, we were able to perform Western blot analyses to verify the identity of five proteins found by MS (hnRNP D, KSRP, CPSF1, hnRNP E1, and IMP3), which had also previously been observed through siRNA screening. Despite the fact that hnRNP I and hnRNP L were not found by MS, we also examined their presence in the pulled down materials by Western blotting. This decision was based upon the screening results that showed a marked increase of LDLR mRNA expression in cells transfected with hnRNP I siRNA and an ablation of BBR effect in hnRNP L transfected cells. Fig. 2 shows that three proteins, including hnRNP D, I, and KSRP, specifically bind to the LDLR mRNA 3′UTR without binding to the CDS, whereas four proteins (CPSF1, hnRNP E1, hnRNP L, and IMP3) bind to the 3′UTR and to the CDS.
Fig. 2.
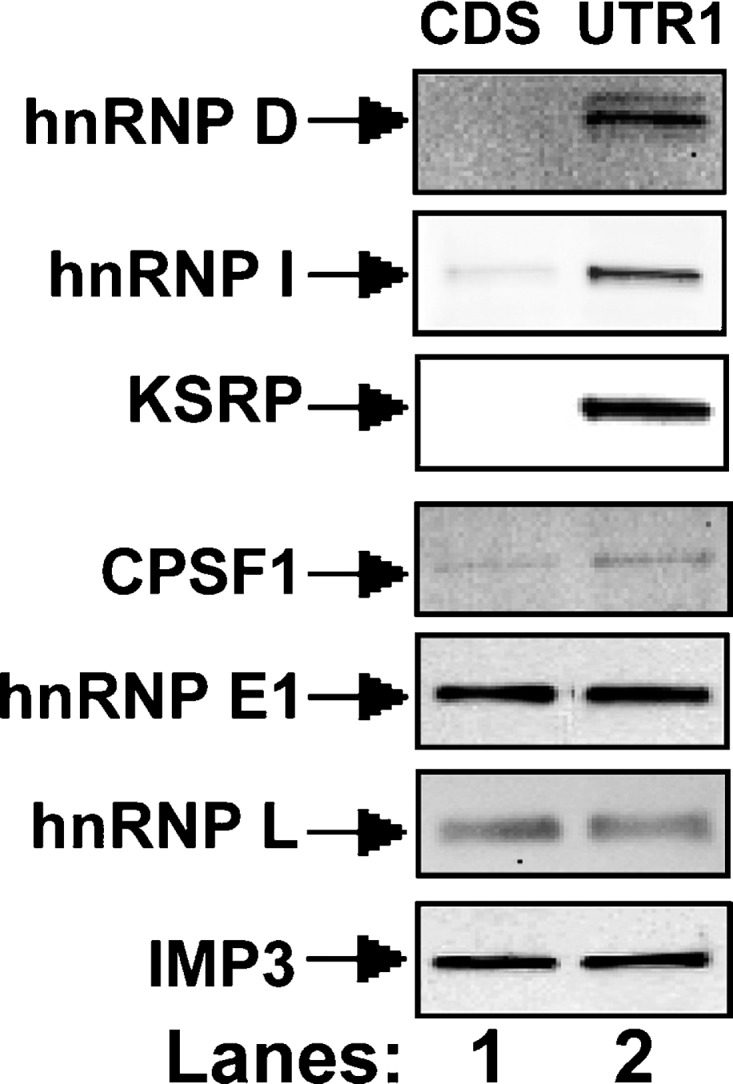
Western blot analysis following RNA pull-down assays. Pull-down assays were carried out by incubating 50 μg of cytosolic protein with 5 μg of biotinylated RNA fragments corresponding to the CDS or to the UTR1. Bound proteins in the pull-down material were analyzed by Western blotting using indicated antibodies.
Identification of hnRNP D, I, and KSRP as key regulators of LDLR mRNA stability through AREs
Since the results of the siRNA library screening and the biotinylated RNA pull-down assays both suggested that hnRNP D, I, and KSRP are functionally involved in the regulation of LDLR mRNA stability through the 3′UTR, our further investigations were focused on these three RBPs. Western blotting using specific antibodies demonstrated substantially reduced expressions of these RBPs in siRNA-transfected HepG2 cells, thereby establishing their target-specific depletion (Fig. 3A).
Fig. 3.

Effects of siRNA knockdown on LDLR mRNA and protein expressions. A: Western blot analysis of hnRNP D (four isoforms), hnRNP I, and KSRP in cells transfected with targeted siRNA or control siRNA. The figure shown is representative of three separate transfection experiments. B: Northern blot and real-time RT-PCR analyses of LDLR mRNA in HepG2 cells transfected with scrambled siRNA or gene-specific siRNAs. In the left panel, 5 μg of total RNA was used per sample. The membrane was first hybridized to a 32P-labeled LDLR probe and reprobed with a 32P-labeled GAPDH probe to demonstrate equal RNA loading. The figure shown is representative of two separate experiments with similar results. In the right panel, real-time RT-PCR assays show the fold difference of LDLR mRNA levels after normalization with GAPDH mRNA. The data represent the means ± SD derived from six independent transfection experiments. ***P < 0.001 compared with scrambled siRNA. C: Effects of siRNA knockdown on LDLR protein expression. siRNAs were transfected into HepG2 cells in a six-well plate, and protein lysates were extracted after 2 d of transfection. Western blot analysis was performed using chicken anti-LDLR polyclonal antibody at 1:5,000 dilution. Detection of β-actin is shown as a loading control. The immunoblot shown is representative of two independent experiments with similar results.
Northern blot analysis of LDLR mRNA showed that depletion of hnRNP D, I, and KSRP increased mRNA abundance (Fig. 3B, left panel). These data were corroborated by results of quantitative real-time RT-PCR assays derived from a total of six independent siRNA transfections (Fig. 3B, right panel). Western blot analysis using specific anti-LDLR antibody further illustrated increased LDLR protein expression in hnRNP D, I, and KSRP depleted cells (Fig. 3C). In addition, we measured LDLR-mediated uptake of fluorescently labeled LDL (DiI-LDL) particles in siRNA-transfected cells by fluorescence microscopy and by fluorescence-activated cell sorting. Quantitative analysis of fluorescence-activated cell sorting indicated that LDL uptake was increased by 35, 50, and 41% in hnRNP D, I, and KSRP siRNA-transfected cells, respectively (see supplementary Fig. IV), consistent with the increases in LDLR mRNA and LDLR protein levels observed. Simultaneous transfection of two siRNAs further elevated LDLR expression as measured by DiI-LDL uptake assays (see supplementary Fig. V). Together, these results demonstrate that hnRNP D, I, and KSRP are negative regulators of LDLR mRNA expression.
It is possible that LDLR gene transcription could be suppressed by these RBPs, and release of that suppression might result in higher levels of LDLR expression in siRNA-transfected cells. To address this possibility, we used a stable HepG2-derived cell line (B11) that expresses an LDLR promoter luciferase reporter, pLDLR234Luc (33, 34). B11 cells were transfected with siRNAs for 2 d, followed by an 8-h treatment with vehicle, BBR, or the cytokine oncostatin M. The results showed that LDLR promoter activity was not increased by siRNAs directed to hnRNP D or KSRP and was only slightly enhanced by hnRNP I siRNA. BBR had no effect on LDLR gene transcription. By contrast, promoter activity was increased 1.8-fold by oncostatin M, a known activator of LDLR transcription through the sterol-independent regulatory element motif of the LDLR promoter (34) (Fig. 4). These results exclude the possibility of a transcriptional effect and further support the functional roles of these RBPs in the regulation of LDLR mRNA stability through the 3′UTR.
Fig. 4.

Knockdown of LDLR mRNA binding proteins does not affect LDLR gene transcription. The plasmid pLDLR234Luc contains a 177 bp fragment of the LDLR promoter sequence that includes SP1 binding sites, sterol-regulatory element-1 (SRE-1), and the sterol-independent regulatory element (SIRE) placed in the 5′ of the luciferase CDS. This plasmid was stably expressed in a HepG2 cell line B11 (44). B11 cells were seeded in 96-well plates and transfected with individual siRNAs for 48 h. Afterwards, cells were incubated in MEM containing 0.5% FBS overnight prior to the addition of BBR (15 μg/ml) or the cytokine oncostatin M (50 ng/ml) for 8 h. After cell lysis, luciferase activities were measured. The data shown (mean ± SD) are representative of two separate experiments in which triplicate wells were assayed for each siRNA.
While hnRNP I is mostly known for its activity in regulation of RNA splicing (35), hnRNP D and KSRP have been shown to act as destabilizing trans-factors (14, 16). To determine whether their destabilizing effects on LDLR mRNA are mediated through ARE sequences, we first conducted site-directed mutagenesis on the pLuc-UTR2 (19) construct to mutate the core ARE consensus sequence individually (Fig. 5A). To validate the loss of the destabilizing function of these ARE mutant reporters, wild-type (wt) and mutated reporters were transfected into HepG2 cells, and luciferase activities were measured. The renilla luciferase expression vector was cotransfected in these experiments to normalize differences in transfection efficiency. The normalized luciferase activity of each ARE mutant was statistically higher than the wt reporter (Fig. 5B). Thus, we used these mutant vectors as templates to generate biotinylated UTR transcripts with each ARE site individually mutated (Fig. 5C). The wt and ARE-mutated transcripts were individually incubated with HepG2 cytoplasmic proteins, and pull-down assays were performed. The upper panels of Fig. 5D show the results of representative Western blots. The amounts of proteins bound to each transcript were quantitated by densitometry analysis, and the data are presented as mean ± SEM from results of four separate pull-down assays (Fig. 5D, lower panel). The binding of hnRNP D to the LDLR 3′UTR was greatly diminished by each ARE mutation, suggesting that hnRNP D binds to all three ARE sites. The binding of hnRNP I was reduced to 42% of wt by ARE1 mutation (P < 0.01) and was not significantly affected by ARE2 or ARE3 mutation. The interaction of KSRP to the LDLR 3′UTR exhibited a strong reduction by ARE1 and ARE3 mutation. These results clearly demonstrate that hnRNP D, hnRNP I, and KSRP are trans-factors that bind to the destabilizing ARE sequences of LDLR mRNA with some apparent site specificity or preference.
Fig. 5.


hnRNP D, I, and KSRP are ARE-BPs of LDLR mRNA. A: Schematic presentation of the chimeric Luc-LDLR 3′UTR fusion constructs that contain either the wt or individually mutated AREs. B: Effects of ARE mutations on luciferase-LDLR 3′UTR reporter activity. HepG2 cells were transiently transfected with individual luciferase-UTR reporter plasmids along with pRL-SV40 as the normalizing transfection vector. Cells were harvested 2 d after transfection, and dual reporter assays were performed. The normalized firefly luciferase activity in cells transfected with the wt vector was defined as 100%, and luciferase activities in cells transfected with mutated vectors were plotted relative to that value. The data represent the means ± SD derived from three independent transfection experiments. ***P < 0.001 compared with UTR-wt. C: Agarose gel stained with ethidium bromide shows the in vitro synthesized, biotin-labeled mRNA fragments of the LDLR transcript. D: Pull-down assays were carried out by incubating 50 μg of cytosolic protein with 5 μg of each biotinylated transcript. In the upper panels, bound proteins in the pull-down material were analyzed by Western blotting using antibodies recognizing hnRNP D, I, and KSRP. The membrane was reprobed with anti-IMP3 antibody to show the equal binding of IMP3 to CDS and UTR transcripts. In the lower panel, after Western blotting, amounts of proteins bound to each transcript were quantitated by densitometry analysis, and the data are presented as mean ± SEM from four separate pull-down assays. **P < 0.01 and ***P < 0.001 compared with UTR-wt.
We further tested various combinational mutations among the three AREs in direct binding and functional assays. While combining mutations of two AREs were not substantially different than mutating a single ARE, changing the AU-rich sequences within all three AREs nearly eliminated the bindings of these ARE-BPs to the LDLR 3′UTR mRNA (see supplementary Fig. VIA). Reporters in which two or three ARE mutations were combined further increased luciferase activity as compared with single ARE mutants (see supplementary Fig. VIB).
BBR regulates LDLR mRNA stability through KSRP and hnRNP I
We further investigated the functional involvement of these three regulators of LDLR mRNA decay in BBR-induced mRNA stabilization. Additional experiments of siRNA transfection and quantitative real-time RT-PCR confirmed the results in Table 2 and clearly showed that the ability of BBR to upregulate LDLR mRNA expression in siRNA-transfected cells was not affected by siRNA to hnRNP D but was abolished by siRNAs targeted to hnRNP I and KSRP (Fig. 6A).
Fig. 6.

BBR-induced mRNA stabilization is mediated by KSRP and hnRNP I through 3′UTR ARE motifs. A: HepG2 cells were transfected with different siRNAs for 48 h. The medium was changed to 0.5% FBS overnight prior to BBR stimulation for 8 h. The LDLR and GAPDH mRNA levels were quantitated by real-time RT-PCR. The normalized LDLR mRNA abundance in scrambled siRNA-transfected cells without BBR treatment is expressed as 1, and the amounts of LDLR transcripts in gene-specific siRNA-transfected cells with or without BBR treatment are plotted relative to that value. The graph represents the means ± SD from five independent experiments. **P < 0.01 and ***P < 0.001 compared with untreated control cells. B: Nuclear and cytoplasmic distributions of hnRNP I and KSRP. HepG2 cells were untreated or treated with BBR for 8 h. Cytoplasmic and nuclear fractions were extracted, and 50 μg of each sample were separated by SDS-PAGE. hnRNP I and KSRP were examined by specific antibodies. Anti-γ-tubulin and anti-HDAC1 were used as indicators for cytoplasmic and nuclear fractions, respectively. C: BBR reduced the binding of hnRNP I and KSRP to the LDLR mRNA 3′UTR. Cytosolic proteins (100 μg) isolated from control or BBR-treated cells were incubated with equal amounts of biotinylated UTR1 or biotinylated CDS, and pull-down assays were performed. The amounts of hnRNP I and KSRP in the reaction mixtures (input, lanes 1 and 2) and in the pulled down materials (lanes 4 to 7) were analyzed by Western blotting. Lane 3 was a control sample without biotinylated transcripts. The membrane was reprobed with anti-IMP3 antibody to show the lack of BBR effect on IMP3 binding. D: Amounts of proteins bound to each transcript were quantitated by densitometry analysis, and the data are presented as mean ± SD from results of three separate pull-down assays. ***P < 0.001 compared with control.
To determine whether BBR treatment alters the protein abundance or subcellular distributions of hnRNP I and KSRP, nuclear and cytoplasmic fractions were isolated from control or BBR-treated HepG2 cells after cell lysis. Western blot analysis showed that hnRNP I and KSRP were clearly detected in both the cytoplasmic and the nuclear fractions. BBR treatment did not alter their protein levels or the nuclear/cytoplasmic distribution (Fig. 6B). Next we examined the binding of hnRNP I and KSRP to the LDLR 3′UTR in the presence or absence of BBR. Equal amounts of cytoplasmic proteins from control and BBR-treated HepG2 cells were incubated with biotinylated UTR1 transcript or biotinylated CDS. Western blotting with anti-hnRNP I and KSRP antibodies detected the same abundance of hnRNP I and KSRP in the reaction mixtures of control and BBR treated cells before the pull-down assay (Fig. 6C, lanes 1 and 2) but showed reduced amounts in the UTR1-bound complexes from BBR-treated cells (lane 5 versus lane 4). In contrast, the binding of IMP3 to the LDLR transcript was not affected by BBR treatment. Results from three independent pull-down assays show that BBR treatment lowered bound KSRP to 59% of control (P < 0.001) and reduced hnRNP I binding to 61% of control (P < 0.001) (Fig. 6D).
DISCUSSION
It has been known that liver LDLR mRNA is labile, having a half-life of 45–60 min, but it is readily stabilized with 2- to 3-fold increases of its t1/2 in response to extracellular stimuli, including PMA (36), bile acids (37), and the cholesterol lowering compound BBR (19–21). However, which cellular factors are involved in LDLR mRNA degradation and how the process of mRNA stabilization occurs is unknown.
In this study using a luciferase reporter assay and RT-PCR analysis of endogenous transcripts, our RNA interference experiments identified 11 RBPs whose cellular depletion by siRNA transfection affected the expression levels of endogenous LDLR mRNA as well as a luciferase-LDLR 3′UTR chimeric transcript in steady state and/or in BBR stimulated cells (Table 2). Independently, our MS analyses identified a total of 35 proteins that bound to biotinylated LDLR transcripts, binding either to the 3′UTR alone or to both the 3′UTR and the coding region (Table 3). From these two lines of investigation, our studies uncovered a network of RBPs, at least 11 of which are likely involved in control of LDLR mRNA stability or processing. The complexity of this network can be inferred from the fact that these proteins belong to different classes of hnRNPs, including decay-promoting ARE-BPs, mRNA stabilizing proteins, splicing factors, RNA helicase, IGF-II mRNA binding proteins, and hnRNPs that function in general mRNA processing without sequence specificity. While additional studies are needed to fully characterize the functions of all of these proteins in LDLR mRNA processing, our current in-depth investigations have focused on three ARE-BPs and demonstrated that hnRNP D, hnRNP I, and KSRP act as negative regulators of LDLR mRNA stability. Further examination of their functions in BBR-induced mRNA stabilization showed the involvement of KSRP and hnRNP I in the response of cells to BBR stimulation.
hnRNP D comprises four isoforms of 37, 40, 42, and 45 kDa. In some cases, the expression profile of its isoforms dictates whether hnRNP D functions as a destabilizer or a stabilizer of mRNA (15, 38). It has been shown that isoforms p37 and p42 have the highest ARE binding affinity and exert the most profound effect on ARE-mRNA degradation (15, 39). In our experiments, p37 and p42 were strongly reduced in siRNA transfected cells as compared with p40 and p45, suggesting that p37 and p42 are the major isoforms involved in LDLR mRNA degradation. It has been shown that hnRNP D isoforms associate with heat shock proteins hsc70-hsp70, translation initiation factor, and PABP to promote mRNA degradation (40, 41). It is noteworthy that PABP and two subunits of hsp90 were identified in our biotinylated RNA pull-down materials (Table 3). It is possible that these proteins are components of an hnRNP D-containing degradation complex that participates in the basal turnover of LDLR mRNA in liver cells.
KSRP is a well-documented mRNA destabilizer that promotes decay of ARE-mRNAs through its association with the exosome, a large multiprotein complex (13, 14). In this study, we show that the depletion of KSRP by siRNA transfection resulted in increases in both LDLR mRNA and protein levels. Although we did not observe a significant increase in luciferase activity in LDLR-Luc6 cells that stably express the 3′UTR luciferase reporter construct, we consistently observed an increase in pLuc-UTR-1 activity in HepG2 cells that were transiently transfected with pLuc-UTR-1 and pRL-SV40. It is possible that siRNA to KSRP somehow inhibited cell proliferation and lowered luciferase activity, thereby masking its induction of LDLR 3′UTR reporter gene expression. The facts that KSRP has a strong binding affinity to the LDLR 3′UTR region and that this binding exhibits specificity toward destabilizing elements ARE1 and ARE3 argue that its primary involvement in LDLR expression is to affect mRNA decay through the 3′UTR.
Unlike the well-established roles of hnRNP D and KSRP in mRNA degradation, hnRNP I is known as a factor primarily involved in premRNA splicing (35, 42). It has been shown to repress the inclusion of alternative exons in numerous systems. Recently, hnRNP I was shown to bind to the 3′UTR of inducible nitric oxide synthase mRNA, and the binding was correlated with mRNA destabilization (43). In this study, we show that hnRNP I is a decay-promoting factor for the LDLR transcript through its binding to ARE motifs. Mutation in ARE1 produced the most inhibition of binding of this protein to the LDLR 3′UTR, suggesting that hnRNP I preferably binds to the ARE1 site. Depletion of hnRNP I increased mRNA levels of the endogenous LDLR transcript detected by Northern blots as well as the luciferase-LDLR 3′UTR chimeric transcript. These results, combined with the direct binding of hnRNP I to ARE sequences of the LDLR 3′UTR, strongly support the functional role of this protein as an mRNA decay promoting factor in the posttranscriptional regulation of LDLR expression.
Among the three LDLR ARE-BPs, our data suggest that KSRP and hnRNP I have dual functions in the decay process of LDLR mRNA. While depletion of hnRNP D increased LDLR mRNA levels regardless of BBR treatment, depletion of KSRP and hnRNP I only increased LDLR mRNA levels in resting cells but failed to show the BBR induction. Using the biotinylated RNA probe, we showed that the binding of these two proteins to the 3′UTR was reduced by BBR treatment. This reduced binding was not caused by decreased mRNA or protein expression. We also detected similar patterns of subcellular distribution of these proteins between nuclear and cytoplasmic fractions before and after BBR treatment, which ruled out a regulatory effect of BBR in the shuttling process of these proteins between the nuclear and cytoplasmic compartments. Further studies to address how BBR affects the binding affinity of RBPs to ARE-LDLR mRNA are currently ongoing in our laboratory.
In summary, our studies have revealed that LDLR mRNA stability is controlled by a network of hnRNPs functioning in different processes of mRNA decay. We further identified three ARE-BPs that are negative regulators of LDLR mRNA stability. Our results suggest that interference with the interaction of decay-promoting RBPs with ARE motifs of LDLR mRNA is one of the underlying mechanisms for BBR-induced LDLR mRNA stabilization and perhaps other compounds that influence LDLR mRNA half-life. This work sheds new light on the regulation of LDLR gene expression at the posttranscriptional level.
Supplementary Material
Acknowledgments
We thank Dr. Ching-Yi Chen for providing the anti-KSRP antibody.
Abbreviations
ARE, AU-rich element
ARE-BP, ARE binding protein
BBR, berberine
CDS, coding sequence
CPSF1, cleavage and polyadenylation specific factor
ELAVL1, embryonic lethal abnormal vision Drosophila-like 1
HMGCR, HMG-CoA reductase
hnRNP, heterogeneous nuclear ribonucleoprotein
IMP3, IGF-II mRNA binding protein 3
KSRP, KH-type splicing regulatory protein
LDLR, LDL receptor
MS, mass spectrometry
RBP, RNA binding protein
siRNA, small interfering RNA
UTR, untranslated region
wt, wild type
This study was supported by the Department of Veterans Affairs (Office of Research and Development, Medical Research Service) and by grants (1RO1 AT002543-01A1 and 1R21 AT003195-01A2) from the National Center for Complementary and Alternative Medicine.
Published, JLR Papers in Press, January 13, 2009.
Footnotes
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of a six figures and one table.
References
- 1.Brown M. S., and J. L. Goldstein. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of the membrane bound transcription factor. Cell. 89 331–340. [DOI] [PubMed] [Google Scholar]
- 2.Brown M. S., and J. L. Goldstein. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA. 96 11041–11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seidah N. G. 2003. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1):liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA. 100 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abifadel M., M. Varret, J. P. Rabès, D. Allard, K. Ouguerram, M. Devillers, C. Cruaud, S. Benjannet, L. Wickham, D. Erlich, et al. 2003. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34 154–156. [DOI] [PubMed] [Google Scholar]
- 5.Maxwell K. N., R. E. Soccio, E. M. Duncan, E. Sehayek, and J. L. Breslow. 2003. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J. Lipid Res. 44 2109–2119. [DOI] [PubMed] [Google Scholar]
- 6.Horton J. D., N. A. Shah, J. A. Warrington, N. N. Anderson, S. W. Park, M. S. Brown, and J. L. Goldstein. 2003. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA. 100 12027–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton J. D., J. C. Cohen, and H. H. Hobbs. 2007. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem. Sci. 32 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross J. 1995. mRNA stability in mammalian cells. Microbiol. Rev. 59 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto T., C. G. Davis, M. S. Brown, W. J. Schneider, M. L. Casey, J. L. Goldstein, and D. W. Russell. 1984. The human LDL receptor: a cystein-rich protein with multiple Alu sequence in its mRNA. Cell. 39 27–38. [DOI] [PubMed] [Google Scholar]
- 10.Wilson G. M., M. Z. Vasa, and R. G. Deeley. 1998. Stabilization and cytoskeletal-association of LDL receptor mRNA are mediated by distinct domains in its 3′ untranslated region. J. Lipid Res. 39 1025–1032. [PubMed] [Google Scholar]
- 11.Zhang T., V. Kruys, G. Huez, and C. Gueydan. 2002. AU-rich element-mediated translational control: complexity and multiple activity of trans-activating factors. Biochem. Soc. Trans. 30 952–958. [DOI] [PubMed] [Google Scholar]
- 12.Barreau C., L. Paillard, and B. H. Osborne. 2005. Survey and summary: AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33 7138–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gherzi R., M. Trabucchi, M. Ponassi, T. Ruggiero, G. Corte, C. Moroni, C-Y. Chen, K. S. Khabar, J. S. Andersen, and P. Briata. 2006. The RNA-binding protein KSRP promotes decay of b-catenin mRNA and is inactivated by PI3K-AKT signaling. PLoS Biol. 5 e5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Ruggiero T., M. Trabucchi, M. Ponassi, G. Corte, C. Y. Chen, L. al-Haj, K. S. Khabar, P. Briata, and R. Gherzi. 2007. Identification of a set of KSRP target transcripts upregulated by PI3K-AKT signaling. BMC Mol. Biol. 8 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raineri I., D. Wegmueller, B. Gross, U. Certa, and C. Moroni. 2004. Roles of AUF1 isoforms, HuR, and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res. 32 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pullmann R., Jr., H. H. Kim, K. Abdelmohsen, A. Lal, J. L. Martindale, X. Yang, and M. Gorospe. 2007. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol. Cell. Biol. 27 6265–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao B., Y. Hu, and G. Brewer. 2007. Competitive binding of AUF1 and TIAR to MYC mRNA controls its translation. Nat. Struct. Mol. Biol. 14 511–518. [DOI] [PubMed] [Google Scholar]
- 18.David P. S., R. Tanveer, and J. D. Port. 2007. FRET-detectable interactions between the ARE binding proteins, HuR and p37AUF1. RNA. 13 1453–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong W., J. Wei, P. Abidi, M. Lin, S. Inaba, C. Li, Y. Wang, Z. Wang, S. Si, H. Pan, et al. 2004. Berberine is a promising novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat. Med. 10 1344–1352. [DOI] [PubMed] [Google Scholar]
- 20.Abidi P., Y. Zhou, J. D. Jiang, and J. Liu. 2005. ERK-dependent regulation of hepatic LDL receptor expression by herbal medicine berberine. Arterioscler. Thromb. Vasc. Biol. 25 2170–2176. [DOI] [PubMed] [Google Scholar]
- 21.Abidi P., W. Chen, F. B. Kraemer, H. Li, and J. Liu. 2006. The medicinal plant goldenseal is a natural LDL-lowering agent with multiple active components and new action mechanisms. J. Lipid Res. 47 2134–2147. [DOI] [PubMed] [Google Scholar]
- 22.Chen W., S. Wang, Y. Ma, Y. Zhou, H. Liu, P. Strnad, F. B. Kraemer, R. M. Krauss, and J. Liu. 2008. Analysis of polymorphisms in 3′ untranslated region of LDL receptor gene and their effect on plasma cholesterol levels and drug response. Int. J. Mol. Med. 21 345–353. [PubMed] [Google Scholar]
- 23.Lehmann D. M., C. A. Galloway, M. P. Sowden, and H. C. Smith. 2006. Metabolic regulation of ApoB mRNA editing is associated with phosphorylation of APOBEC-1 complementation factor. Nucleic Acids Res. 34 3299–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufmann I., G. Martin, A. Friedlein, H. Langen, and W. Keller. 2004. Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 23 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosoda N., F. Lejeune, and L. E. Maquat. 2006. Evidence that poly(A) binding protein C1 binds nuclear pre-mRNA poly(A) tails. Mol. Cell. Biol. 26 3085–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behm-Ansmant I., D. Gatfield, J. Rehwinkel, V. Hilgers, and E. Izaurralde. 2007. A conversed role for cytoplasmic poly(A)-binding protein 1 (PABPC1) in nonsense-mediated mRNA decay. EMBO J. 26 1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kajita Y., J. Nakayama, M. Aizawa, and F. Ishikawa. 1995. The UUAG-specific RNA binding protein, heterogeneous nuclear ribonucleotprotein D0. J. Biol. Chem. 270 22167–22175. [DOI] [PubMed] [Google Scholar]
- 28.Kawamura H., Y. Tomozoe, T. Akaqi, D. Kamei, M. Ochiai, and M. Yamada. 2002. Identification of the nucleocytoplasmic shuttling sequence of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its interaction with mRNA. J. Biol. Chem. 277 2732–2739. [DOI] [PubMed] [Google Scholar]
- 29.Ostareck-Lederer A., D. H. Ostareck, and M. W. Hentze. 1998. Cytoplasmic regulatory functions of the KH-domain proteins hnRNPs K and E1/E2. Trends Biochem. Sci. 23 409–411. [DOI] [PubMed] [Google Scholar]
- 30.Thiele B. J., A. Doller, T. Kähne, R. Pregla, R. Hetzer, and V. Regitz-Zagrosek. 2004. RNA-binding proteins heterogeneous nuclear ribonucleoprotein A1, E1, and K are involved in post-transcriptional control of collagen I and III synthesis. Circ. Res. 95 1058–1066. [DOI] [PubMed] [Google Scholar]
- 31.Kaneko S., O. Rozenblatt-Rosen, M. Meyerson, and J. L. Manley. 2007. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 21 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bollig F., R. Winzen, M. Gaestel, S. Kostka, K. Resch, and H. Holtmann. 2003. Affinity purification of ARE-binding proteins identifies polyA-binding protein 1 as a potential substrate in MK2-induced mRNA stabilization. Biochem. Biophys. Res. Commun. 301 665–670. [DOI] [PubMed] [Google Scholar]
- 33.Liu J., R. Streiff, Y. L. Zhang, R. E. Vestal, M. J. Spence, and M. R. Briggs. 1997. Novel mechanism of transcriptional activation of hepatic LDL receptor by oncostatin M. J. Lipid Res. 38 2035–2048. [PubMed] [Google Scholar]
- 34.Liu J., T. E. Ahlborn, M. R. Briggs, and F. B. Kraemer. 2000. Identification of a novel sterol-independent regulatory element (SIRE) in the human LDL receptor promoter. J. Biol. Chem. 275 5214–5221. [DOI] [PubMed] [Google Scholar]
- 35.Paradis C., P. Cloutier, L. Shkreta, J. Toutant, K. Klarskov, and B. Chabot. 2007. hnRNP I/PTB can antagonize the splicing repressor activity of SRp30c. RNA. 13 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson G. M., E. A. Roberts, and R. G. Deeley. 1997. Modulation of LDL receptor mRNA stability by phorbol esters in human liver cell culture models. J. Lipid Res. 38 437–446. [PubMed] [Google Scholar]
- 37.Nakahara M., H. Fujii, P. R. Maloney, M. Shimizu, and R. Sato. 2002. Bile acids enhance low density lipoprotein receptor gene expression via a MAPK cascade-mediated stabilization of mRNA. J. Biol. Chem. 277 37229–37234. [DOI] [PubMed] [Google Scholar]
- 38.Sarkar B., Q. Xi, C. He, and R. J. Schneider. 2003. Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol. Cell. Biol. 23 6685–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner B. J., C. T. DeMaria, Y. Sun, G. M. Wilson, and G. Brewer. 1998. Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics. 48 195–202. [DOI] [PubMed] [Google Scholar]
- 40.Lu J. Y., N. Bergman, N. Sadri, and R. J. Schneider. 2007. Assembly of AUF1 with eIF4G-poly(A) binding protein complex suggests a translation function in AU-rich mRNA decay. RNA. 12 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laroia G., R. Cuesta, G. Brewer, and R. J. Schneider. 1999. Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science. 284 499–502. [DOI] [PubMed] [Google Scholar]
- 42.Wagner E. J., and M. A. Garcia-Blanco. 2001. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 21 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Söderberg M., F. Raffalli-Mathieu, and M. A. Lang. 2007. Regulation of the murine inducible nitric oxide synthase gene by dexamethasone involves a heterogenous nuclear ribonucleoprotein I (hnRNP I) dependent pathway. Mol. Immunol. 44 3204–3210. [DOI] [PubMed] [Google Scholar]
- 44.Liu H., C. Fenollar-Ferrer, A. Cao, C. Anselmi, P. Carloni, and J. Liu. 2009. Molecular dissection of human oncostatin M-mediated signal transductions through site-directed mutagenesis. Int. J. Mol. Med. 23 161–172. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.