

Abstract
Oxidized LDL (oxLDL) increase in patients affected by type-2 diabetes, obesity, and metabolic syndrome. Likewise, insulin resistance, an impaired responsiveness of target tissues to insulin, is associated with those pathological conditions. To investigate a possible causal relationship between oxLDL and the onset of insulin resistance, we evaluated the response to insulin of 3T3-L1 adipocytes treated with oxLDL. We observed that oxLDL inhibited glucose uptake (−40%) through reduced glucose transporter 4 (GLUT4) recruitment to the plasma membrane (−70%), without affecting GLUT4 gene expression. These findings were associated to the impairment of insulin signaling. Specifically, in oxLDL-treated cells insulin receptor (IR) substrate-1 (IRS-1) was highly degraded likely because of the enhanced Ser307phosphorylation. This process was largely mediated by the activation of the inhibitor of κB-kinase β (IKKβ) and the c-Jun NH2-terminal kinase (JNK). Moreover, the activation of IKKβ positively regulated the nuclear content of nuclear factor κB (NF-κB), by inactivating the inhibitor of NF-κB (IκBα). The activated NF-κB further impaired per se GLUT4 functionality. Specific inhibitors of IKKβ, JNK, and NF-κB restored insulin sensitivity in adipocytes treated with oxLDL. These data provide the first evidence that oxLDL, by activating serine/threonine kinases, impaired adipocyte response to insulin affecting pathways involved in the recruitment of GLUT4 to plasma membranes (PM). This suggests that oxLDL might participate in the development of insulin resistance.
Keywords: insulin resistance, GLUT4, IRS-1, IKKβ, NF-κB
Insulin resistance, commonly considered the major risk factor for type 2 diabetes (1, 2), is characterized by a reduced ability of insulin to regulate glucose homeostasis in target tissues. In particular, the sensitivity of adipose tissue toward insulin is a crucial event for normal total body fuel metabolism (3). Fatless mouse models and various conditions of lipodystrophy in humans are associated with severe total body insulin resistance (3–5) due to the lack of responsive tissue. On the other hand, obesity is closely associated with type 2 diabetes, likely by inducing insulin resistance through different mechanisms such as high plasma level of free fatty acids, increased production of cytokines, and alteration of insulin signaling (6–9). The entry of glucose in fat and muscle cells in response to insulin is mediated by the facilitative glucose transporter 4 (GLUT4), which is mainly expressed in those insulin-sensitive cell types (10–12). GLUT4 is stored in intracellular vesicles from which, in response to acute insulin stimulation, translocates to the plasma membrane facilitating the uptake of glucose (13, 14). In the skeletal muscles of type 2 diabetes patients, the insulin-induced GLUT4 translocation is markedly reduced (15). Several signaling cascades triggered by insulin participate in this process, in particular the insulin receptor substrate (IRS)-phosphatidyl inositol 3-kinase-dependent pathway and its downstream kinases such as Akt play a prominent role (16). However, the mechanism by which the insulin-signaling pathway can induce the translocation process is still an unanswered question. The binding of insulin to insulin receptor (IR), by triggering autophosphorylation, induces, in turn, the phosphorylation of several interacting proteins such as IRS proteins. In particular, IRS-1 contains several potential phosphorylation sites such as tyrosines and serines/threonines that can be phosphorylated in response to different signals that modulate IRS-1 activity. Specifically, in response to insulin, IRS-1 is phosphorylated on specific phosphotyrosine binding domains, which can couple IRS-1 to activated IR, and recruit a number of signal transducers, including phosphatidyl inositol 3-kinase (17, 18). This leads, through a complex mechanism, to the phosphorylation and activation of Akt, which plays an essential role in GLUT4 translocation (19–24). Conversely, the serine phosphorylation of IRS-1 (in particular at ser307 in mice) has been shown as a major player in the desensitization of insulin action (18, 25, 26) because it can interrupt IRS/IR interaction or promote IRS-1 protein degradation. In both cases, serine phosphorylation of IRS-1 may affect IRS-1 functions that have been postulated to represent a unifying mechanistic link among all the factors involved in the establishment of insulin resistance (25, 27). In this regard, many authors have found a marked reduction of IRS-1 and GLUT4 content and functionality in the adipose tissue of diabetic patients (28, 29). Consequently, the decreased sensitivity to insulin of adipose tissue isolated from type 2 diabetes patients seems to be caused by impaired insulin signaling and reduced GLUT4 levels. Both, the inhibitors of kappa B kinase (IKK), in particular IKKβ and the c-Jun NH2-terminal kinase (JNK), have been shown to phosphorylate IRS-1 at Ser307 in mice in response to different stimuli, among which TNFα and free fatty acids (25, 30–34). Furthermore, phosphorylation induced by the IKK complex and several other signaling pathways is a major mechanism in the activation of nuclear factor κB (NF-κB) (35, 36). The NF-κB complex, which is activated by several factors such as inflammatory cytokines (6, 37, 38) and oxidized LDL (oxLDL) (39), appears to influence the establishment of insulin resistance (40) likely by inducing inappropriate inflammatory responses, which can provoke the disruption of insulin signaling cascade. Specifically, NF-κB activation has recently been shown to affect IRS-1 phosphorylation and glucose uptake in 3T3-L1 adipocytes exposed to oxidative stress (41).
OxLDL, considered to play a major role in the formation and development of atherosclerotic plaques (42), have been found in the course of atherosclerosis, type 2 diabetes and obesity (43, 44). Interestingly, several studies have demonstrated that increased plasma oxLDL concentration is associated with the metabolic syndrome (45–48). Although the actual extent of in vivo interactions between adipose tissue and lipoproteins is still to be defined, a bulk of data suggests that adipocytes could have a role in the metabolism of circulating lipoproteins including oxLDL. This hypothesis is based on several observations. First, in adipose tissue there is the largest amount of free cholesterol in the body, which originates mainly from circulating lipoproteins because of the low degree of cholesterol biosynthesis in adipocytes (49). Second, a number of lipoprotein receptors, such as those for VLDL and several LDL receptor family members, are expressed on adipocytes supporting a role for adipose tissue in the modulation of lipid transport as recently demonstrated in mice (50). Finally, specific receptors for oxLDL, which are active in lipoprotein internalization, have been detected on the membranes of adipocyte cell lines such as 3T3-L1, as well as on human and animal adipocytes (51, 52). In particular, the presence of the fatty acid transporter/scavenger receptor CD36, and the oxLDL receptor 1 (ORL1) on adipocytes support the hypothesis that the adipose tissue could be involved in oxLDL metabolism in vivo (53, 54). We have recently demonstrated that oxLDL can be actively taken up by 3T3-L1 preadipocytes through a CD36-mediated transport and that they can induce a significant increase in membrane receptor content. This enables oxLDL to interfere with the homeostasis of adipose tissue by altering the normal balance between differentiation and proliferation in preadipocytes, thus potentially contributing to the appearance of insulin resistance (55, 56). Furthermore, oxLDL are a causative factor in lowering insulin sensitivity in cultured cells, possibly by inhibiting the signaling kinases responsible for the cellular response to insulin (57) and/or by activating the NF-κB complex, responsible for the regulation of genes involved in inflammation and cell survival (58). However, to our knowledge, no data are available regarding the effects of oxLDL on the response of adipocytes to insulin.
To elucidate a possible role of oxidized lipoproteins in the development of insulin resistance, we evaluated the effect of oxLDL on the uptake of glucose and the insulin-induced translocation of GLUT4 in 3T3-L1 adipocytes, extensively used as the golden standard in evaluating insulin-stimulated GLUT4 traffic (59). We observed that oxLDL treatment induced a marked insulin-desensitization characterized by a decrease in glucose uptake likely due to the impairment of GLUT4 translocation. Furthermore, we investigated the molecular events underlying the oxLDL-induced insulin resistance, demonstrating that the Ser307 phosphorylation of IRS-1 was associated with its degradation and with the impairment of insulin signaling. These effects were largely mediated by the activation of the two serine/threonine kinases IKKβ and JNK. A parallel activation of NF-κB was also detected, which, in turn, affected insulin signaling cascade and GLUT4 translocation. The treatment of oxLDL-treated adipocytes with specific inhibitors of IKKβ, JNK, and NF-κB, in fact, restored insulin sensitivity. Finally, we assessed whether the oxidative stress or the proinflammatory cytokines TNFα and IL-6, which have been demonstrated to promote insulin resistance (41, 60), are involved in mediating the effect of oxLDL on GLUT4 functionality and glucose uptake.
MATERIALS AND METHODS
Materials
DMEM was purchased from Flow Laboratories (Irvine, UK); fetal calf serum, glutamine, and antibiotics were from Hyclone (Cramlington, UK). 2-Deoxy-D-1[3H]glucose was purchased from Amersham Biosciences Corp. (Piscataway, NJ). Oil Red O, insulin, isobuthylmetylxanthine, dexamethasone, phloretin, and Protein G Sepharose were from Sigma (St. Louis, MO). Electrophoresis reagents were from Bio-Rad (Hercules, CA). Anti-IRS-1, anti-GAPDH, monoclonal anti-IκBα, and anti-NF-κB p65, NF-κB inhibitor SN50, anti-Lamin B, horseradish peroxidase-conjugate anti-goat, anti-mouse, and anti-rabbit antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-GLUT4 was from Chemicon International, Inc. (Temecula, CA). Monoclonal anti-phospho-IκB-α (Ser32/36), anti-IKKβ, anti-phospho-IKKα (Ser180)/IKKβ (Ser181), anti-SAPK/JNK, anti-phospho-SAPK (Tyr183/Tyr185), anti-phospho-IRS-1(Ser307) was purchased from Cell Signaling Technology (Beverly, MA). 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) SP600125 and BAY-11-7082 were from Biomol Research Laboratories (Plymouth, PA). Murine monoclonal anti -CD-36, -TNFα, and -IL-6 and nonimmune IgG antibodies were purchased from R and D Systems (Minneapolis, MN).
Plasma LDL isolation and oxidation
LDL (1.019–1.063 g/ml) were isolated by density gradient ultracentrifugation in vertical rotor from fresh pooled plasma of healthy volunteers and provided by the Centro Trasfusionale, University of Rome “La Sapienza,” as described elsewhere (61). The protein content was measured by Lowry's method using BSA as standard (62). Native LDL (nLDL) were oxidized with 20 μM CuSO4 for 18 h at 37°C. Oxidation was stopped by 1 mM EDTA. The degree of LDL oxidation was assessed by determining the thiobarbituric acid reactive substances content according to Yagi (63) and the increase in electrophoretic mobility on agarose gel (61); the thiobarbituric acid reactive substances content of oxLDL was 45 ± 7 nmol malondialdehyde equivalent/mg LDL protein (vs. 4 ± 0.3 nmol in nLDL). The electrophoretic mobility of oxLDL vs. nLDL was 1.9 ± 0.2.
3T3-L1 preadipocyte differentiation
Murine 3T3-L1 preadipocytes (American Cell Culture Collection) were cultured in DMEM supplemented with 10% fetal calf serum, 0.2 mmol/l glutamine, and 10 U/ml antibiotics. Two days after confluence, the cells were treated with the differentiation mixture containing 1 μM insulin, 0.25 μM dexamethasone, and 0.5 mM isobuthylmetylxantine for 48 h. The medium was changed by adding 1 μM insulin for 48 h. The cells were used for the experiments on day 14, when more than 90% of cells presented the adipocyte phenotype as detected by Oil Red O staining and mRNA expression of aP2 and leptin, markers of mature adipocytes (55).
Adipocytes treatment
OxLDL were sterilized by a 0.22 μm membrane (Millipore Corporation, Bedford, MA) and incubated with the cells. Different oxLDL concentrations (25–200 mg protein/l) were used to test oxLDL cytotoxicity and their effect on glucose uptake in order to choose the best concentration. The dose-response curve of the effect of oxLDL on glucose uptake (Table 1) showed that the reduction of cellular response to insulin was dependent to oxLDL concentration. The 100 mg/l concentration was chosen because it induced approximately 50% decrease in glucose uptake without showing any sign of cytotoxicity, as assessed by Neutral Red assay, or affecting the morphology or the metabolism of differentiated 3T3-L1 adipocytes, as determined by the expression of leptin and aP2 and the incorporation of H3-Uridine, which were both comparable to the controls (data not shown). Furthermore, this concentration was of the same order of magnitude as that found in patients with coronary artery diseases (approximately 50 mg/l) (64).
TABLE 1.
Dose response curve of oxLDL on glucose uptake
| OxLDL (mg/L) | 0 | 25 | 50 | 100 | 200 |
|---|---|---|---|---|---|
| 2-DG uptake (% of control) | 100 ± 0,02 | 85 ± 2,2 | 70 ± 2a | 56 ± 2,8a | 37 ± 1,2a |
2-DG, 2-deoxyglucose; oxLDL, oxidized LDL. The data are means ± SEM of three independent experiments.
P < 0.05 compared with control.
Under all the experimental conditions described below, differentiated 3T3-L1 cells, untreated and treated with nLDL (100 mg/l), were used as controls. As we obtained wholly overlapping results, the data reported for controls regard only nLDL-treated cells. 3T3-L1 adipocytes were serum starved for 18 h and incubated with 100 mg/l nLDL or oxLDL for 4 h before incubation with 20 nM insulin for 15 min. In a set of experiments addressed to evaluate the involvement of the serine kinases and the transcription factor NF-κB, specific inhibitors were added to the cell culture 30 min before the oxLDL treatment, specifically, 50 μM SP600125, (JNK inhibitor), 50 μM 15d-PGJ2 (IKKβ inhibitor), 10 μM NF-κB SN50, or 5 μM BAY11-7082 (NF-κB inhibitors) were added to the cells.
Glucose uptake assay
Mature adipocytes, plated in 24-well plates in low-glucose DMEM (1,000 mg/L D-(+)-glucose) were serum starved for 18 h and stimulated with 20 nM insulin for 15 min. [H3]2-DG (2-deoxyglucose) (1 μCi/well) was added to the cells for 45 min to allow the cells to uptake it. The reaction was stopped with ice-cold PBS. The cells were washed three times with PBS and solubilized in 0.5 ml PBS containing 0.5% TRITON X (65). The total incorporated radioactivity was determined in a liquid scintillation counter. The results were corrected for aspecific absorption by subtracting the remaining radioactivity after the addition of 0.5 mM phloretin, a GLUT4 inhibitor. Aspecific absorption was always less than 10% of total uptake. Results were normalized for protein content measured by Lowry assay (62). In the experiments addressed to test the effect of lipoproteins on glucose uptake, the cells were incubated with 100 mg/L nLDL or oxLDL for 4 h before stimulation with insulin.
Evaluation of oxidative stress occurrence and oxLDL internalization
Intracellular reactive oxygen species (ROS) levels were determined using a fluorescence probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA), which is converted to highly fluorescent dichlorofluorescein in the presence of intracellular ROS. Cells were washed with PBS and incubated with freshly diluted CM-H2DCFDA (25 μM) in PBS for 1 h and washed twice with PBS. The cell fluorescence intensity was measured on a spectrofluorometer with an excitation wavelength of 485 nm and emission wavelength of 530 nm. The level of reduced glutathione (GSH), oxidized GSH (GSSG), and the GSH/GSSG ratio were determined by the Bioxytech GSH/GSSG-412 assay kit (OXIS International, Inc., Portland, OR) according to the manufacturer's instructions. In some experiments aimed to assess whether the occurrence of oxidative stress could have a role in oxLDL effect on adipocytes, the cells were treated with the antioxidant vitamin E for 30 min before oxLDL treatment, and then glucose uptake was determined.
To determine whether the effect of oxLDL on glucose uptake was mediated by oxidized lipids or needed internalization via membrane receptors such as CD36, we measured glucose uptake in adipocytes treated with 10 μg/ml monoclonal anti-CD36 antibody for 30 min before oxLDL treatment.
Western blotting and immunoprecipitation
3T3-L1 adipocytes were lysed by cell-solubilizing buffer containing 1% TRITON X in PBS, protease inhibitor cocktail (SIGMA), 1 mM Na3VO4 and 50 mM NaF. The whole cell lysates were centrifuged at 4°C for 20 min at 16,000 g to remove insoluble materials. For immunoprecipitation the supernatants were incubated for 2 h at 4°C with specific antibodies, namely anti-IRS-1 or anti-IKKβ. Then the samples were incubated with protein G-Sepharose for 30 min and the beads washed with the same lysis buffer for three times. For NF-κB determination, nuclear protein extracts were prepared by Nuclear/Cytosol fractionation Kit (Medical and Biological Laboratories, Watertown, LA) according to the manufacturer's instructions. The whole cell lysates, the immunoprecipitates and the nuclear extracts were boiled with Laemmli sample buffer for 5 min, resolved by 7.5% (for IRS-1) or 12% SDS-PAGE (for the others), and transferred onto polyvinylidene difluoride membranes. The membranes were blocked by 5% nonfat dry milk and incubated with the appropriate antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for chemiluminescent detection. Densitometric analysis was performed by a molecular imager FX (BIORAD). Equal loading of proteins was verified by immunoblotting with a goat anti-GAPDH and goat anti-Lamin B antibodies for whole cell and nuclear extracts, respectively.
Membrane GLUT4 determination
To evaluate GLUT4 in plasma membranes (PM), the cells were fractionated according to McKeel and Jarett (66). Briefly, cultures were washed once with TES buffer (10 mM TRIS-HCl, 1 mM EDTA, 255 mM sucrose, pH 7.4) and homogenized in ice-chilled TES using a 1 ml Dounce homogenizer. The homogenate was centrifuged at 16,000 g for 20 min at 4°C and solidified surface fat was removed. The resulting pellets were resuspended in TES, layered on a 1.12M sucrose cushion in 10 mM TRIS-HCl, 1 mM EDTA and centrifuged at 200,000 g for 15 min at 4°C in a Beckman ultracentrifuge SW 60 rotor. PM were collected at the interface, diluted in TES buffer (4:1) (v/v) and centrifuged at 16,000 g for 15 min at 4°C. PM pellets were resuspended in TES and immunoblotted for GLUT4. Equal loading of proteins was verified by immunoblotting with a goat anti-GAPDH.
Total RNA extraction and RT-PCR analysis
Total RNA was extracted from cultured 3T3-L1 adipocytes by the TRIZOL isolation method (Invitrogen-Life technologies) according to the manufacturer's recommendations. One microgram of total RNA was used for RT-PCR analysis (Invitrogen). RT-PCR was done with the following couples of primers: 5′-GCA TCG CCT AAC ACA TGG TGA C-3′ and 5′-GCT GCA CCC TGG GAA AGG AGG TCT A-3′ for amplification of GLUT4; 5′-CCA CCC ATG GCA AAT TCC ATG GCA-3′ and 5′-TCT AGA CGG CAG GTC AGG TCC ACC-3′ for GAPDH used as housekeeping gene. The samples were incubated in an automated heat-block (Minicycler, MJ Research) under the following conditions: 94°C for 30 s, 60°C for 30 s, and 72°C for 40 min using 25–28 cycles. The PCR products were electrophoresed on a 1.5% agarose gel containing ethidium bromide. Densitometric analysis was performed by a molecular imager FX (BIORAD).
Assessment of PPARγ activity
PPARγ activity was determined in nuclear extracts with the TransAM ELISA Kit (Active Motif Europe Rixensart, Belgium) according to the manufacturer's instructions.
Evaluation of inflammatory cytokines
To evaluate the involvement of proinflammatory cytokines in oxLDL-induced insulin resistance, TNFα and IL-6 secretion was determined in the culture media by Elisa kit (R and D Systems Inc., Minneapolis, MN) according to the manufacturer's instructions. Furthermore we studied the effect of neutralizing monoclonal anti-TNFα (2 μg/ml) and anti-IL-6 (0.5 μg/ml) antibodies on GLUT4 translocation by adding them to the cell cultures 30 min before oxLDL treatment.
Statistical analysis
Student's t-test was used for statistical analysis. Data are expressed as mean ± SEM. A value of P < 0.05 was considered significant.
RESULTS
Insulin-induced glucose uptake and GLUT4 translocation in oxLDL-treated 3T3-L1 adipocytes
3T3-L1 mature adipocytes differentiated by hormonal mixture proved to be a suitable model for detecting the cellular response to insulin stimulation. To determine insulin sensitivity, we evaluated glucose uptake demonstrating that it was strongly increased in cells treated with insulin, with respect to untreated cells (Fig. 1). To evaluate whether lipoproteins affected cell sensitivity toward insulin, adipocytes were incubated with oxLDL or nLDL for 4 h before insulin stimulation and glucose uptake determination. The results showed that insulin-induced glucose uptake was significantly inhibited (−40%) after oxLDL treatment with respect to the untreated and nLDL treated cells (Fig. 1). We also demonstrated that the activity of oxLDL on 3T3-L1 cells required the internalization of the lipoproteins through the scavenger receptor CD36. In fact, by treating the adipocytes with 10 μg/ml of monoclonal anti-CD36 antibody before oxLDL, we completely counteracted the impairment of glucose uptake (Fig. 1).
Fig. 1.

Oxidized LDL (OxLDL) decrease glucose uptake in 3T3-L1 adipocytes after internalization by CD36 3T3-L1 adipocytes were serum starved in low-glucose medium for 18 h, preincubated with 10 μg/ml monoclonal anti-CD36 antibody for 30 min, treated with 100 mg/l native LDL (nLDL) or oxLDL for 4 h before incubation with 20 nM insulin for 15 min. The rate of glucose uptake was determined upon addition of [H3]2-DG (2-deoxyglucose) for 45 min. Basal glucose uptake in untreated cells was set at 100% to which all other values were related. The data are means ± SEM of three independent experiments. * P < 0.05 compared with the basal uptake; # P < 0.05 compared with untreated or nLDL treated cells; § P < 0.05 compared with oxLDL treated cells.
The lowered glucose uptake found after oxLDL treatment was associated with an impairment of the facilitative glucose transporter GLUT4. Specifically, by assessing GLUT4 protein level by Western blot analysis in whole cell lysates and PM, we found that the GLUT4 protein level was reduced in the plasma membrane by as much as 70% with respect to the controls (Fig. 2A), while the total level of the protein was unaffected by the treatment with oxLDL (Fig. 2B). Furthermore, oxLDL exerted only a moderate effect on GLUT4 mRNA expression (Fig. 2C). These results indicated that the insulin-induced translocation of GLUT4 was significantly reduced in oxLDL-treated 3T3-L1 adipocytes. Thus, the decreased uptake of glucose induced by oxLDL seemed to be determined by the impairment of GLUT4 translocation rather than by a reduced GLUT4 total protein level or mRNA expression.
Fig. 2.
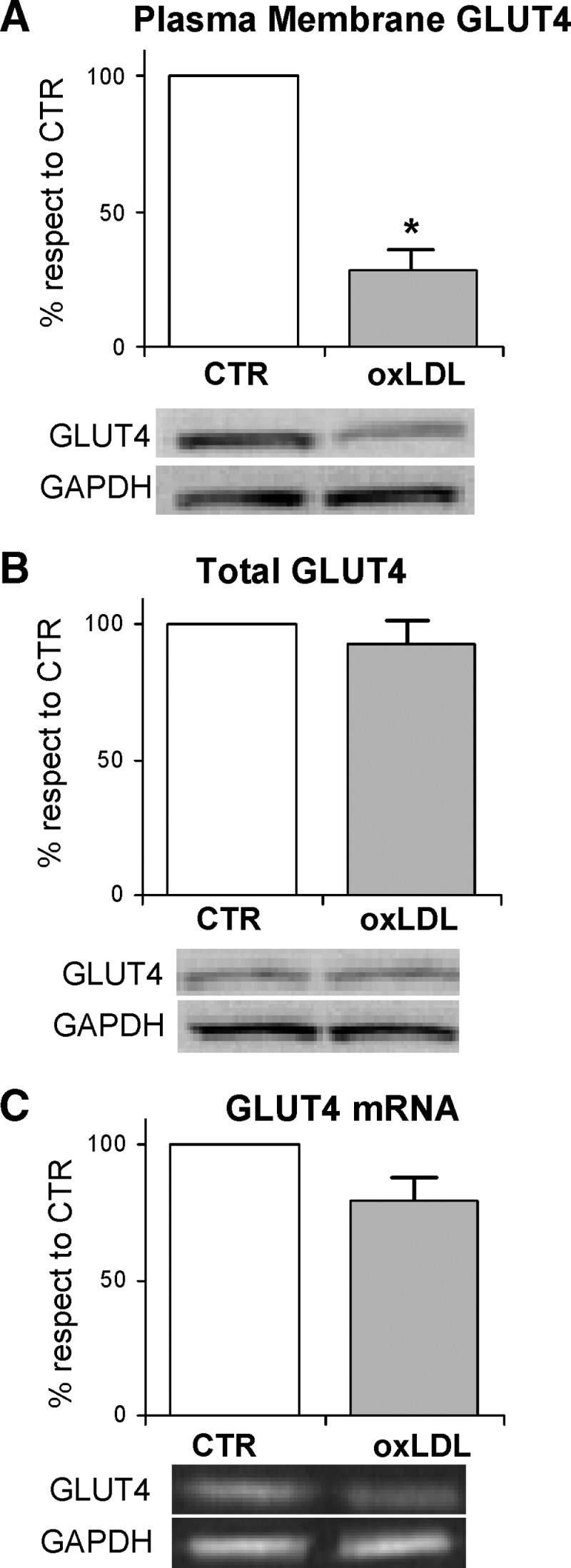
Gene and protein expressions of glucose transporter 4 (GLUT4) in oxLDL-treated adipocytes 3T3-L1 adipocytes were serum starved for 18 h and incubated with 100 mg/l nLDL or oxLDL for 4 h before incubation with 20 nM insulin for 15 min. A: Plasma membrane fractions were prepared as described in Materials and Methods. Immunoblotting of GLUT4 and GAPDH were determined. B: The cell lysates were resolved by SDS-PAGE and analyzed using antibodies against GLUT4 and GAPDH. The blots are representative of at least three independent experiments. C: Total RNA was analyzed by RT-PCR as described in Materials and Methods. The values indicate expression of target gene normalized to GAPDH RNA and expressed as percentage of nLDL control (CTR). The data are means of three independent experiments ± SEM. * P < 0.001 compared with nLDL (CTR).
The impairment of glucose uptake induced by oxLDL is not related to the occurrence of oxidative stress
Because hydrogen peroxide has been demonstrated to affect GLUT4 functionality (41, 67), and oxLDL can induce oxidative stress in several cell types, we carried out experiments to evaluate intracellular redox balance in 3T3-L1 after 4 h oxLDL exposure. To this aim, we determined the intracellular production of ROS, which was unchanged in oxLDL-treated cells with respect to the control (Table 2). Furthermore, because GSH is the major nonenzymatic regulator of intracellular redox homeostasis, we evaluated the levels of GSH, GSSG, and the GSH/GSSG ratio after oxLDL treatment: we found no difference between treated and untreated cells (Table 2). These results showed that effect of oxLDL on adipocyte glucose uptake was not dependent on oxidative stress. As further confirmation we demonstrated that the antioxidant Vitamin E could not revert the impairment of the cellular response to insulin induced by oxLDL (data not shown).
TABLE 2.
Evaluation of oxidative stress occurrence in oxLDL-treated 3T3-L1 adipocytes
| GSH μM/mg cell protein | GSSG μM/mg cell protein | GSH/GSSG RATIO | ROS (%) | |
|---|---|---|---|---|
| CTR | 169,33 ± 15 | 7,60 ± 1,86 | 19,96 ± 2 | 100 ± 0,03 |
| oxLDL | 158,27 ± 13a | 7,97 ± 2,06a | 17,59 ± 2a | 112 ± 3a |
CTR, control; GSH, glutathione; GSSG, oxidized glutathione; ROS, reactive oxygen species. The data are means ± SEM of three independent experiments.
P < 0.05 compared with control.
OxLDL effects on IRS-1
To investigate the molecular mechanism involved in the reduction of the adipocyte response to insulin stimulation determined by oxLDL treatment, we evaluated insulin-induced IRS-1 activation, because of its central role in the insulin-induced translocation of GLUT4. First, we determined the cellular content of IRS-1 protein with the specific antibody by Western blot analysis in total cell lysates, to detect any possible alteration of its expression due to the oxLDL treatment. When we evaluated IRS-1 abundance at different time points after oxLDL treatment, we found that it gradually reached a 70% reduction at h 4 as compared with the time 0 (Fig. 3A). These results suggested that oxLDL enhanced IRS-1 protein degradation. Because serine phosphorylation, specifically at Ser307, has been reported to favor IRS-1 degradation and to affect its activity (25, 30–34, 68–70), we evaluated serine phosphorylation of IRS-1 in oxLDL-treated adipocytes. Immunoblotting on IRS-1 immunoprecipitates was done using a phospho-ser307-IRS-1 antibody. OxLDL treatment induced a remarkable increase of Ser307 phosphorylation with respect to control cells with the maximal response after 1 h (Fig. 3B).
Fig. 3.

Immunoblotting and serine phosphorylation of insulin receptor substrate (IRS)-1 3T3-L1 adipocytes were serum-starved for 18 h and treated with 100 mg/l nLDL or oxLDL from 30 min to 4 h and then stimulated with 20 nM insulin for 15 min. A: Total cell lysates were separated by SDS-PAGE and analyzed using anti-IRS-1 antibody. Results were normalized to GAPDH protein content. B: Immunoprecipitates with anti-IRS-1 antibody were separated by 7.5% SDS-PAGE, and analyzed with antinonphospho- or antiphospho-ser307 IRS-1 antibody as described in Materials and Methods. Blots are representative of at least three independent experiments at 1 h. The data are means ± SEM of three independent experiments. * P < 0.001 compared with time 0; # P < 0.001 compared with nLDL (CTR).
Role of IKKβ and JNK in IRS-1 serine phosphorylation induced by oxLDL
Phosphorylation of Ser307 in IRS-1 can be obtained by either JNK or IKKβ (71); hence we hypothesized that the oxLDL treatment should be able to activate these two serine-kinases in 3T3-L1 cells. To verify our hypothesis, we evaluated the active phosphorylated form of IKKβ and JNK in immunoprecipitates by using the phospho-specific antibodies. OxLDL-treatment induced a significant increase of IKKβ and JNK phosphorylation with respect to the control cells (Fig. 4A, B). To further confirm the role of IKKβ and JNK in Ser307 phosphorylation of IRS-1 protein, the specific inhibitors 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and SP600125 respectively, were used. We found that Ser307 phosphorylation was completely inhibited when oxLDL-treated adipocytes were incubated with the aforementioned specific chemical inhibitors of IKKβ or JNK (Fig. 5A). Worthy of note, the treatment of the adipocytes with the inhibitors of the two serine-kinases counteracted the inhibition of GLUT4 translocation to PM observed in oxLDL-treated adipocytes (Fig. 5B). Moreover, a recovery of glucose uptake was detected (Fig. 5C). Our findings support a specific involvement of JNK, and mainly of IKKβ, in the impairment of insulin signaling induced by oxLDL treatment. In addition, because 15d-PGJ2 has been largely demonstrated to be a PPARγ agonist, we evaluated whether the observed effect of prostaglandin was dependent on PPARγ activation. Our results demonstrated that oxLDL induced a significant decrease in PPARγ activity with respect to control cells and that 15d-PGJ2 was ineffective at activating PPARγ in the presence of oxLDL (Fig. 5D).
Fig. 4.
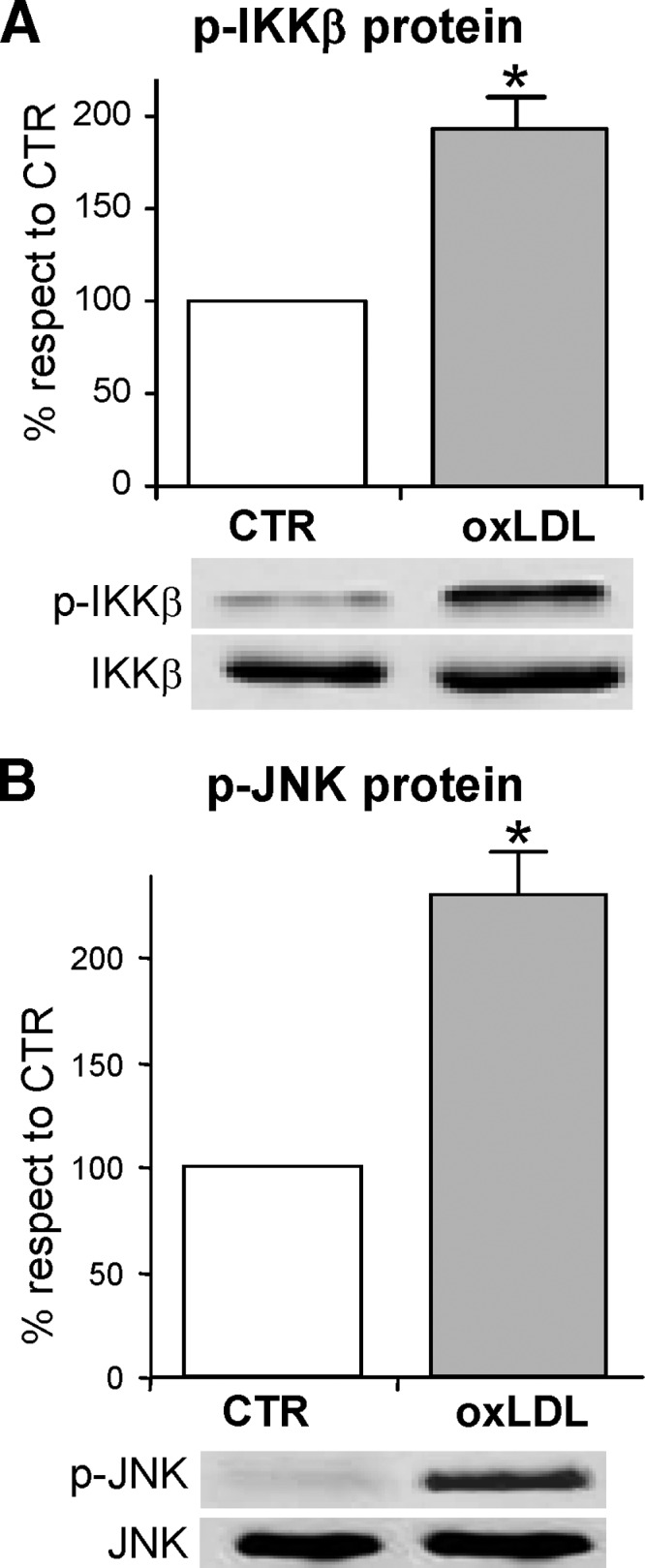
Activation of the Serine Kinases by oxLDL Activations of inhibitor of κB-kinase β (IKKβ) (A) and c-Jun NH2-terminal kinase (JNK) (B) by oxLDL are shown. 3T3-L1 adipocytes were serum-starved for 18 h, treated with 100 mg/l nLDL or oxLDL for 1 h and stimulated with 20 nM insulin for 15 min. Immunoprecipitates with anti-IKKα/β or anti-SAPK/JNK were separated by 12% SDS-PAGE and analyzed using antinonphospho- or phosphoantibodies against IKKα/β (A) or JNK (B), as described in Materials and Methods. Representative images are shown from three independent experiments. * P < 0.05 compared with nLDL (CTR).
Fig. 5.

Effects of specific kinase inhibitors on oxLDL-stimulated insulin resistance 3T3-L1 adipocytes were serum starved for 18 h, preincubated with 50 μM 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) or 50 μM SP600125 for 30 min, treated with 100 mg/l nLDL or oxLDL, and then stimulated with 20 nM insulin for 15 min. A: Effects of IKKβ and JNK inhibitors on oxLDL-induced serine phosphorylation of IRS-1. The immunoprecipitates with IRS-1 antibody were resolved by 7.5% SDS-PAGE and analyzed with antinonphospho- or antiphospho-ser307 IRS-1 antibody. Blots are representative of at least three independent experiments at 1 h. B: Immunoblotting of GLUT4 on plasma membrane fractions. Cells were washed after nLDL or oxLDL treatment for 4 h, and plasma membrane fractions were prepared as described in Materials and Methods. Representative blots from at least four independent experiments are shown. Results were normalized to GAPDH protein content. C: Effects of IKKβ and JNK inhibitors on oxLDL-inhibited glucose uptake. The rate of glucose uptake was determined upon addition of [H3]2-DG for 45 min. D: Effect of IKKβ inhibitor on PPARγ activation. The data are means ± SEM of three independent experiments. * P < 0.001 compared with nLDL (CTR); # P < 0.05 compared with oxLDL; $ P < 0.05 compared with nLDL (CTR); § P < 0.05 compared with 15-PGJ2 treated cells.
OxLDL induce the activation of NF-κB pathway in 3T3-L1 adipocytes
It is widely known that a large number of stimuli inducing stress conditions, among which oxLDL, activate the transcription factor NF-κB (58), which is involved in triggering the induction of inflammatory and proliferating genes. NF-κB is normally sequestered by IκB proteins, in particular IκBα, in the cytoplasm. Therefore, IκBα must be degraded to allow the translocation of the transcription factor to the nucleus where it binds to specific DNA sequences. The degradation of IκBα is induced by the phosphorylation of two serine residues mediated by IKKs (72), and represents the critical regulatory step in the activation of NF-κB. In the light of this knowledge and on the basis of our findings on the oxLDL-induced activation of IKKβ, we hypothesized that NF-κB could be also involved in the impairment of insulin sensitivity in our experimental model. We thus evaluated the activation of NF-κB by measuring its nuclear level in 3T3-L1 adipocytes after 4 h treatment with oxLDL. A two-fold increase in the nuclear content was observed in cells treated with modified lipoproteins with respect to the controls by using specific NF-κB p65 antibody (Fig. 6A). The activation of NF-κB was consistent with the finding that the content of IκBα in the cytoplasm was significantly decreased with respect to the control cells (Fig. 6B). This result was likely dependent on the increased degradation of the IκBα protein due to its serine phosphorylation mediated by IKKβ. This hypothesis was supported by the findings that 1) IKKβ inhibitor 15d-PGJ2 counteracted IκBα degradation and the consequent NF-κB activation, (Fig. 6A, B) while, as expected, JNK inhibitor SP600125 was ineffective (data not shown); and 2) IκBα serine phosphorylation increased by two-fold following the addition of oxLDL to the cells and was hindered by IKKβ inhibitor (Fig. 7).
Fig. 6.
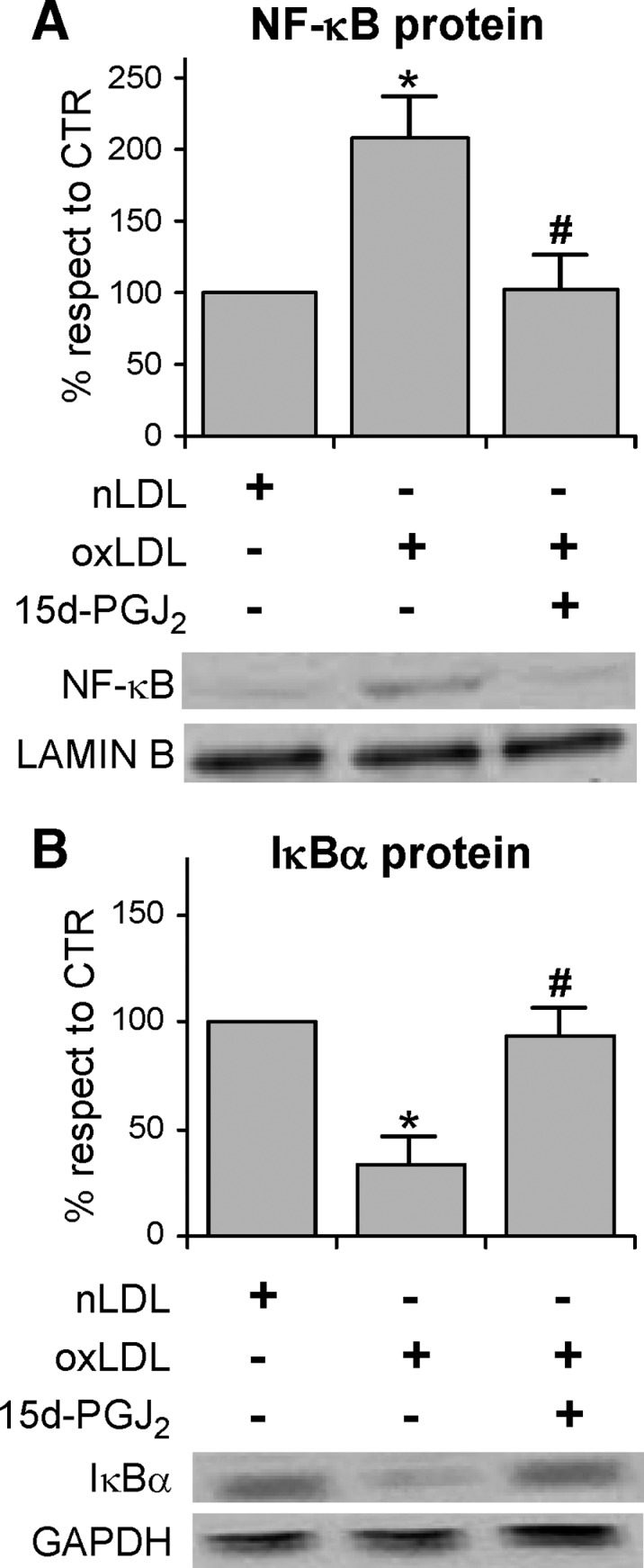
Effects of IKKβ inhibitor on nuclear factor κB (NF-κB) activation and IκBα degradation in oxLDL-induced 3T3-L1 adipocytes Cells were serum starved for 18 h, preincubated with 50 μM 15d-PGJ2 for 30 min, treated with 100 mg/l nLDL or oxLDL for 4 h, and then stimulated with 20 nM insulin for 15 min. Nuclear and cytosolic extracts were prepared as described in Materials and Methods and analyzed by immunoblotting of NF-κB p65 (A), IκBα (B). Lamin B and GAPDH antibodies were used as markers for nuclear and cytosolic extracts, respectively. Representative images are shown from three independent experiments. * P < 0.05 compared with nLDL (CTR). # P < 0.05 compared with oxLDL.
Fig. 7.
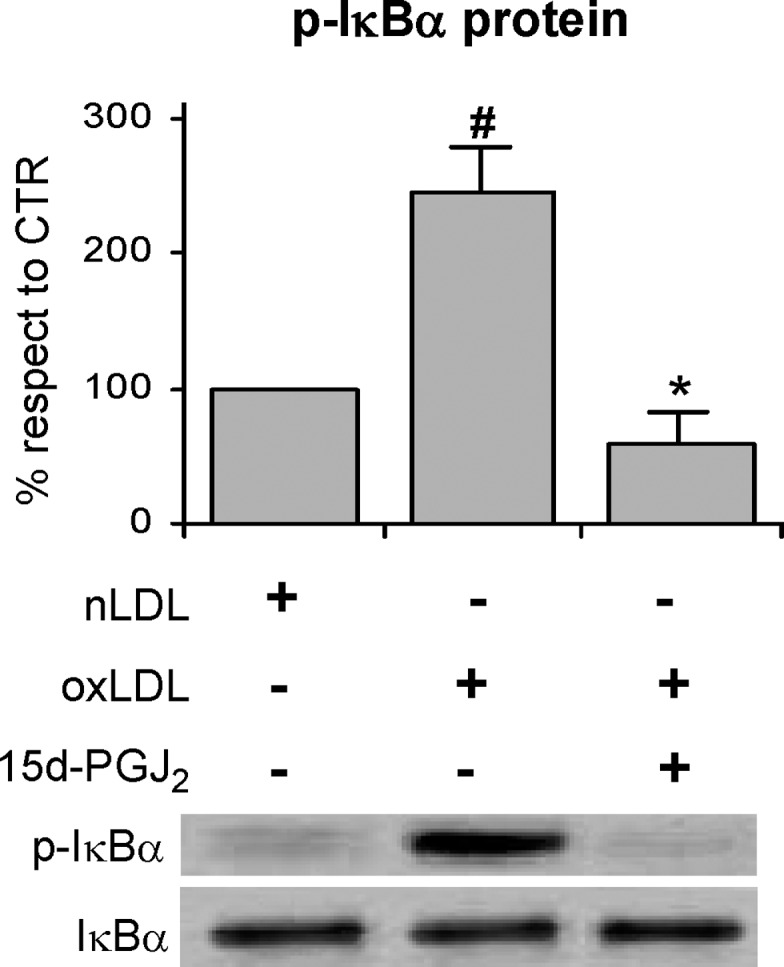
IKKβ inhibitor prevents oxLDL-induced IκBα degradation in 3T3-L1 adipocytes 3T3-L1 adipocytes were preincubated with 50 μM 15d-PGJ2 for 30 min followed by nLDL or oxLDL treatment for 4 h before stimulation with 20 nM insulin for 15 min as described in Materials and Methods. Total cell lysates were analyzed by Western blotting using anti-phospho(ser32/36)-IκBα and anti-GAPDH antibodies. Representative images are shown from three independent experiments. # P < 0.05 compared with nLDL (CTR); * P < 0.001 compared with oxLDL.
NF-κB reduces GLUT4 translocation through an IRS-1 independent mechanism
To evaluate whether activated NF-κB was implicated in impairing the response to insulin, the cells were treated with a cell-permeable peptide inhibitor of NF-κB nuclear translocation, namely NF-κB SN50, and with an inhibitor of IκBα phosphorylation, namely BAY-11-7082 before oxLDL treatment and glucose uptake determination. The results indicated that both the inhibitors managed to revert the glucose uptake to control level (Fig. 8A). Worthy of note, the treatment with NF-κB inhibitors restored the normal recruitment of GLUT4 to the plasma membrane (Fig. 8B). Our results are in agreement with the finding that fatty acid-induced insulin resistance requires nuclear translocation of NF-κB in L6 myotubes (38).
Fig. 8.

Effects of NFκB inhibitors on oxLDL treatment 3T3-L1 adipocytes were serum starved for 18 h, preincubated with 10 μM SN50 or 5μM BAY-11-7082 for 30 min, treated with 100 mg/l nLDL or oxLDL, and then stimulated with 20 nM insulin for 15 min. A: Effects of NF-κB inhibitors on oxLDL-inhibited glucose uptake. Cells were washed after 4 h of nLDL or oxLDL treatment. The rate of glucose uptake was determined upon addition of [H3]2-DG for 45 min. B: Immunoblotting of GLUT4 on plasma membrane fractions. Cells were washed after nLDL or oxLDL treatment for 4 h and plasma membrane fractions were prepared as described in Materials and Methods. Results were normalized to GAPDH protein content. Blots are representative of at least four independent experiments. The data are means ± SEM of three independent experiments. * P < 0.05 compared with nLDL (CTR); # P < 0.05 compared with oxLDL.
Finally, to clarify the relationship between NF-κB and insulin signaling, we assessed the effect of NF-κB SN50 on the impairment of insulin signaling induced by oxLDL. The NF-κB inhibitor was ineffective at counteracting the Ser307-phosphorylation of IRS-1 (Fig. 9).
Fig. 9.

Effects of NFκB inhibitor on oxLDL-induced serine phosphorylation of IRS-1. Cells were prepared after 1 h treatment with nLDL or oxLDL, and immunoprecipitates with IRS-1 antibody were resolved by 7.5% SDS-PAGE and analyzed with antinonphospho- or antiphospho-ser307 IRS-1 antibody. Blots are representative of at least four independent experiments. The data are means of four independent experiments ± SEM.* P < 0.001 compared with nLDL (CTR).
Our findings suggested that the activation of NF-κB played an important role in the development of insulin resistance by affecting insulin-stimulated glucose uptake, independently of the impairment of IRS-1 functionality in oxLDL-treated adipocytes. To elucidate the mechanism responsible for the involvement of the NF-κB signaling pathway in oxLDL-induced insulin resistance, we investigated the role of the proinflammatory cytokines TNFα and IL-6, which have been demonstrated to be specific NF-κB target genes and also NF-κB activators. Immunodetection demonstrated the absence of any cytokine release in the culture media after the 4 h treatment with oxLDL. Because the short period of time could negatively affect release detection, we investigated a possible role of the cytokines inside the cells by using specific monoclonal antibodies to neutralize TNFα and IL-6 activities. Specifically, we found that cell treatment with anti-TNFα and IL-6 neutralizing antibodies had no effect on either NF-κB activation or GLUT4 translocation (Fig. 10).
Fig. 10.

Effects of anti-TNFα and anti-IL-6 neutralizing antibodies on GLUT4 membrane translocation and NF-κB activation in oxLDL-treated 3T3-L1 adipocytes. 3T3-L1 adipocytes were serum starved for 18 h, preincubated with monoclonal anti-TNFα (2 μg/ml) or anti-IL-6 (0.5 μg/ml) antibodies for 30 min, treated with 100 mg/l nLDL or oxLDL, and then stimulated with 20 nM insulin for 15 min. A: Immunoblotting of GLUT4 on plasma membrane fractions. Plasma membrane fractions were prepared as described in Materials and Methods. Results were normalized to GAPDH protein content. B: Immunoblotting of NF-κB p65 on nuclear extracts. Nuclear extracts were prepared as described in Materials and Methods and analyzed by immunoblotting of NF-κB p65. Lamin B antibody was used as marker for nuclear extracts.
DISCUSSION
In the present study, we used 3T3-L1 adipocytes as an in vitro model system to assess the effect, and the underlying molecular mechanisms, of oxLDL on the glucose uptake and the response of adipocytes to insulin. As such our study provided insight into the mechanisms of insulin resistance induced by oxLDL. Although positive correlations have been demonstrated among visceral fat accumulation, circulating levels of oxLDL, and insulin resistance (73, 74), only few studies have explored the link between oxLDL and insulin resistance, in particular regarding a possible alteration of adipose tissue response to insulin signal. OxLDL are considered major players in the pathogenesis of atherosclerosis and have been shown to be greatly increased in the plasma of patients affected by type-2 diabetes, obesity, and metabolic syndrome. In vitro and in vivo experiments seem to suggest interactions between adipocytes and oxLDL as well as a physiological role of adipose tissue in the clearance of circulating oxLDL (51, 53, 75). In the present study we provide the first evidence that oxLDL induced insulin resistance by impairing insulin signal at multiple levels as shown in Fig. 11. As a consequence, oxLDL impaired the insulin-stimulated translocation of GLUT4 to the plasma membrane and consequently decreased glucose transport. Under basal conditions the majority of GLUT4 is sequestered intracellularly within insulin-responsive storage compartments not well biochemically or structurally identified yet. Insulin stimulates the recruitment of GLUT4 from intracellular compartments to plasma membrane and the increased level of the transporter at the cell surface appears to largely account for the concomitant increase in transport activity (76, 77). Insulin increases the concentration of the transporter in plasma membrane through complex mechanisms, which involve a cascade of scaffolding proteins downstream of the activated IR (78). Among these proteins, there are the four members of the IRS family proteins IRS-1, -2, -3, -4. IRS-1 contains several potential tyrosine and serine/threonine phosphorylation sites that can be phosphorylated in response to different signals, modulating IRS-1 activity. Our results provided evidence that oxLDL increased serine307-phosphorylation of IRS-1 and enhanced its degradation in 3T3-L1 adipocytes, revealing a potential role in insulin action. In this regard the involvement of serine phosphorylation of IRS-1 has been pointed out as one of the mechanisms known for attenuating IR signaling because of the lower efficiency of serine-phosphorylated IRS-1 to couple the IR (18), thus decreasing its effectiveness in insulin signal transmission (70, 79, 80). The serine phosphorylation of IRS-1 can, in fact, induce conformational changes, steric hindrance, and cellular relocalization (81), leading to the decrease in tyrosine phosphorylation induced by IR and consequently to the impairment of GLUT4 functionality (82). Worthy of note, we provided the first evidence that in 3T3-L1 adipocytes the oxLDL are able to activate IKKβ and JNK. Furthermore, the inhibition of both IKKβ and JNK by their specific inhibitors 15d-prostaglandin J2 and SP600125 prevented the phosphorylation at Ser307 of IRS-1 and the functional alterations induced in adipocytes by oxLDL. Therefore the effects on glucose uptake and the impairment of insulin signaling were largely mediated by the activation of the two serine/threonine kinases, which have been previously demonstrated to be activated by the proinflammatory conditions associated with insulin resistance (26, 83, 84). The involvement of IKKβ and JNK in our experimental model was in agreement with several published papers, which have suggested serine kinases activation as an intervening mechanism in inducing insulin resistance (25, 30–34, 85–87). As a point of fact, high doses of aspirin or salicylate, which inhibit IKKβ activity (88), have been shown to reverse hyperglycemia, hyperinsulinemia, and dyslipidemia in obese rodents by sensitizing the animals to insulin signaling (89, 90). Furthermore, IKKβ and JNK might activate other pathways. Specifically, IKKβ has been shown to be involved in the activation of NF-κB signaling pathway (91–93). In this regard, we demonstrated that NF-κB nuclear content was significantly increased in 3T3-L1 adipocytes treated with oxLDL. This finding was in agreement with the well-known effect of low concentrations of oxLDL that rapidly activate NF-κB in other cell types, under conditions inducing mitogenic and proinflammatory response (94). The activation of NF-κB normally requires the phosphorylation and the subsequent degradation of the inhibitory κB proteins (IκBs), which sequester the NF-κB dimer in the cytoplasm. It is noteworthy that the increased nuclear level of NF-κB was associated with the strong decrease in NF-κB inhibitor IκBα as we found in oxLDL treated adipocytes. We thus hypothesized that the observed activation of IKKβ, responsible for the serine phosphorylation of IRS-1, was also able to inactivate IκBα producing the NF-κB activation observed in oxLDL treated adipocytes. In fact, by phosphorylating IκBα, IKKβ triggers its polyubiquitination and proteosomal degradation, which brings about the release of unbound NF-κB (35, 36) and its translocation to the nucleus, where it induces gene transcription. Our hypothesis was confirmed by showing the serine-phosphorylation of IκBα in the adipocytes treated with oxLDL. As further support we demonstrated that IκBα degradation was counteracted by IKKβ inhibitor, as shown by the nonoccurrence of its phosphorylation.
Fig. 11.

Schematic representation of the oxLDL effects on insulin sensitivity in 3T3-L1 adipocytes. OxLDL increase serine307-phosphorylation of IRS-1 by IKKβ and JNK. The impairment of insulin signaling leads to reduced uptake of glucose via GLUT4. IKKβ is also involved in the activation of NF-κB signaling pathway by phosphorylating IκBα. NF-κB downregulates GLUT4 translocation and glucose uptake. OxLDL, oxidized lipoproteins; IRS-1, insulin receptor substrate 1; GLUT4, glucose transporter 4; p-tyr, phosphotyrosine; p-ser, phosphoserine; IKKβ, inhibitory protein κB kinase β; JNK, c-Jun NH2-terminal kinase; IκBα, inhibitory κB protein α; NF-κB, nuclear factor κB.
The involvement of NF-κB in oxLDL-induced insulin resistance was further supported by treating the adipocytes with the specific inhibitors of NF-κB translocation or IκBα-mediated activation. The NF-κB inhibitors counteracted the impaired recruitment of GLUT4 to the plasma membrane and the decrease in glucose uptake.
Moreover, we demonstrated that the prevention of NF-κB nuclear translocation had no effect on the Ser307-phosphorylation of IRS-1. This finding thus demonstrated, in agreement with Sinha et al. (38), that nuclear translocation of NF-κB was required to affect insulin-stimulated glucose uptake and GLUT4 translocation, but not to impair insulin signaling.
Taken together, the results obtained by inhibiting IKKβ or NF-κB suggested that the oxLDL-induced activation of IKKβ could affect glucose uptake by activating NF-κB. This finding could also suggest that product(s) of NF-κB-regulated gene(s) could be responsible for the impairment of GLUT4 functionality induced by oxLDL. In the attempt to address this possibility we evaluated the role of two proinflammatory cytokines, namely TNFα and IL-6, which have been demonstrated to be specific NF-κB target genes (95, 96) and to induce insulin-resistance by activating NF-κB (97, 98). The absence of cytokines release as well as the inefficacy of cytokines neutralizing antibodies at counteracting GLUT4 dysfunction, suggested that these cytokines were not involved in the mediation of oxLDL effects under our experimental conditions. However, the large number of NF-κB target genes that could be potentially involved in this process require further study.
Finally, it should be considered that the effect of oxLDL on the sensitivity of adipocytes to insulin could be associated with the induction of intracellular oxidative stress by the modified lipoproteins. It has been reported that the oxidative stress induced by hydrogen peroxide impairs insulin-stimulated GLUT4 translocation through the impairment of the cellular response to insulin (99, 100). However, we can exclude the implication of oxidative stress in oxLDL-treated 3T3-L1. In fact, although oxLDL have been demonstrated to induce an intracellular redox imbalance in several cell kinds (101, 102), in our experimental system this did not occur as supported by the lack of ROS production and of GSH consumption following oxLDL treatment, and by the inability of the antioxidant vitamin E to restore glucose uptake. Finally, cell treatment with anti-CD36 antibody, which blocks the main scavenger receptor for oxLDL present on the cell membrane, counteracted the effect of lipoproteins, clearly demonstrating that oxLDL must be internalized to affect glucose uptake.
The results herein reported could appear in contrast with a report that showed the effect of thiazolidinedione treatment on 3T3-L1 (54). In this study the authors demonstrated that thiazolidinedione improved adipocyte insulin sensitivity and, at the same time, strongly increased the cellular uptake of oxLDL by the up-regulation of the scavenger receptor OLR1. This was related to the PPARγ activation due to thiazolidinedione. What makes the present study different is that in our system PPARγ was not activated, but oxLDL decreased its level also in agreement with our previous papers (56). Summarizing, we provided evidence that oxLDL lowered the sensitivity of adipocytes to insulin by impairing GLUT4 translocation and glucose uptake. Specifically, we showed for the first time that oxLDL were able to activate IKKβ, which enhanced serine phosphorylation of IRS-1 and IκBα, thus impairing the cell response to insulin. Worth of note, we also demonstrated that IKKβ−induced nuclear translocation of NF-κB affected GLUT4 recruitment through a mechanism independent of the impairment of IRS-1 functionality and the secretion and activity of TNFα and IL-6.
In conclusion, this study further sheds light on the complex mechanisms involved in the onset of insulin resistance in adipocytes. A better understanding of the cell-signaling pathways playing a crucial role in the modulation of insulin action and how they integrate with each other provides an opportunity for the effective exploitation of these pathways as a potential target for efficient drugs in the treatment of insulin resistance and type 2 diabetes.
Acknowledgments
The author thanks Prof. Gabriella Girelli, Director of Centro Trasfusionale, University of Rome “La Sapienza,” for providing human plasma, and Ms. Monica Brocco for the linguistic revision of the manuscript.
Abbreviations
15d-PGJ2, 15-deoxy-Δ12,14-prostaglandin J2
CTR, control
GLUT4, glucose transporter 4
IκBs, inhibitory κB proteins
IKKβ, inhibitor of kappa B kinase β
IR, insulin receptor
IRS, insulin receptor substrate
JNK, c-Jun NH2-terminal kinase
NF-κB, nuclear factor κB
oxLDL, oxidized LDL
PM, plasma membranes
2-DG, 2-deoxyglucose
ROS, reactive oxygen species
Published, JLR Papers in Press, January 9, 2009.
References
- 1.Pradhan A. 2007. Obesity, metabolic syndrome, and type 2 diabetes: inflammatory basis of glucose metabolic disorders. Nutr. Rev. 65 S152–S156. [DOI] [PubMed] [Google Scholar]
- 2.Guilherme A., J. V. Virbasius, V. Puri, and M. P. Czech. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegele R. A. 2000. Familial partial lipodystrophy: a monogenic form of the insulin resistance syndrome. Mol. Genet. Metab. 71 539–544. [DOI] [PubMed] [Google Scholar]
- 4.Kim J. K., O. Gavrilova, Y. Chen, M. L. Reitman, and G. I. Shulman. 2000. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J. Biol. Chem. 275 8456–8460. [DOI] [PubMed] [Google Scholar]
- 5.Gan S. K., K. Samaras, A. Carr, and D. Chisholm. 2001. Anti-retroviral therapy, insulin resistance and lipodystrophy. Diabetes Obes. Metab. 3 67–71. [DOI] [PubMed] [Google Scholar]
- 6.Weigert C., K. Brodbeck, H. Staiger, C. Kausch, F. Machicao, H. U. Haring, and E. D. Schleicher. 2004. Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J. Biol. Chem. 279 23942–23952. [DOI] [PubMed] [Google Scholar]
- 7.Bays H., L. Mandarino, and R. A. DeFronzo. 2004. Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J. Clin. Endocrinol. Metab. 89 463–478. [DOI] [PubMed] [Google Scholar]
- 8.Peraldi P., and B. Spiegelman. 1998. TNF-alpha and insulin resistance: summary and future prospects. Mol. Cell. Biochem. 182 169–175. [PubMed] [Google Scholar]
- 9.Saltiel A. R., and C. R. Kahn. 2001. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 414 799–806. [DOI] [PubMed] [Google Scholar]
- 10.Mauvais-Jarvis F., R. N. Kulkarni, and C. R. Kahn. 2002. Knockout models are useful tools to dissect the pathophysiology and genetics of insulin resistance. Clin. Endocrinol. (Oxf.). 57 1–9. [DOI] [PubMed] [Google Scholar]
- 11.Charron M. J., E. B. Katz, and A. L. Olson. 1999. GLUT4 gene regulation and manipulation. J. Biol. Chem. 274 3253–3256. [DOI] [PubMed] [Google Scholar]
- 12.Petersen K. F., and G. I. Shulman. 2002. Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am. J. Cardiol. 90 11G–18G. [DOI] [PubMed] [Google Scholar]
- 13.Bryant N. J., R. Govers, and D. E. James. 2002. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 3 267–277. [DOI] [PubMed] [Google Scholar]
- 14.Pessin J. E., D. C. Thurmond, J. S. Elmendorf, K. J. Coker, and S. Okada. 1999. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J. Biol. Chem. 274 2593–2596. [DOI] [PubMed] [Google Scholar]
- 15.Zierath J. R., A. Krook, and H. Wallberg-Henriksson. 2000. Insulin action and insulin resistance in human skeletal muscle. Diabetologia. 43 821–835. [DOI] [PubMed] [Google Scholar]
- 16.van Dam E. M., R. Govers, and D. E. James. 2005. Akt activation is required at a late stage of insulin-induced GLUT4 translocation to the plasma membrane. Mol. Endocrinol. 19 1067–1077. [DOI] [PubMed] [Google Scholar]
- 17.White M. F. 2002. IRS proteins and the common path to diabetes. Am. J. Physiol. Endocrinol. Metab. 283 E413–E422. [DOI] [PubMed] [Google Scholar]
- 18.Sesti G., M. Federici, M. L. Hribal, D. Lauro, P. Sbraccia, and R. Lauro. 2001. Defects of the insulin receptor substrate (IRS) system in human metabolic disorders. FASEB J. 15 2099–2111. [DOI] [PubMed] [Google Scholar]
- 19.Tanti J. F., S. Grillo, T. Gremeaux, P. J. Coffer, E. Van Obberghen, and Y. Le Marchand-Brustel. 1997. Potential role of protein kinase B in glucose transporter 4 translocation in adipocytes. Endocrinology. 138 2005–2010. [DOI] [PubMed] [Google Scholar]
- 20.Hill M. M., S. F. Clark, D. F. Tucker, M. J. Birnbaum, D. E. James, and S. L. Macaulay. 1999. A role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol. 19 7771–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohn A. D., S. A. Summers, M. J. Birnbaum, and R. A. Roth. 1996. Expression of a constitutively active Akt Ser/Thr kinase in 3T3–L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 271 31372–31378. [DOI] [PubMed] [Google Scholar]
- 22.Cong L. N., H. Chen, Y. Li, L. Zhou, M. A. McGibbon, S. I. Taylor, and M. J. Quon. 1997. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol. Endocrinol. 11 1881–1890. [DOI] [PubMed] [Google Scholar]
- 23.Ducluzeau P. H., L. M. Fletcher, G. I. Welsh, and J. M. Tavare. 2002. Functional consequence of targeting protein kinase B/Akt to GLUT4 vesicles. J. Cell Sci. 115 2857–2866. [DOI] [PubMed] [Google Scholar]
- 24.Sano H., S. Kane, E. Sano, C. P. Miinea, J. M. Asara, W. S. Lane, C. W. Garner, and G. E. Lienhard. 2003. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278 14599–14602. [DOI] [PubMed] [Google Scholar]
- 25.Aguirre V., E. D. Werner, J. Giraud, Y. H. Lee, S. E. Shoelson, and M. F. White. 2002. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 277 1531–1537. [DOI] [PubMed] [Google Scholar]
- 26.He J., I. Usui, K. Ishizuka, Y. Kanatani, K. Hiratani, M. Iwata, A. Bukhari, T. Haruta, T. Sasaoka, and M. Kobayashi. 2006. Interleukin-1alpha inhibits insulin signaling with phosphorylating insulin receptor substrate-1 on serine residues in 3T3–L1 adipocytes. Mol. Endocrinol. 20 114–124. [DOI] [PubMed] [Google Scholar]
- 27.Zick Y. 2004. Uncoupling insulin signalling by serine/threonine phosphorylation: a molecular basis for insulin resistance. Biochem. Soc. Trans. 32 812–816. [DOI] [PubMed] [Google Scholar]
- 28.Garvey W. T., L. Maianu, J. A. Hancock, A. M. Golichowski, and A. Baron. 1992. Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 41 465–475. [DOI] [PubMed] [Google Scholar]
- 29.Rondinone C. M., L. M. Wang, P. Lonnroth, C. Wesslau, J. H. Pierce, and U. Smith. 1997. Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. USA. 94 4171–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguirre V., T. Uchida, L. Yenush, R. Davis, and M. F. White. 2000. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 275 9047–9054. [DOI] [PubMed] [Google Scholar]
- 31.Gao Z., D. Hwang, F. Bataille, M. Lefevre, D. York, M. J. Quon, and J. Ye. 2002. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J. Biol. Chem. 277 48115–48121. [DOI] [PubMed] [Google Scholar]
- 32.Gao Z., A. Zuberi, M. J. Quon, Z. Dong, and J. Ye. 2003. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J. Biol. Chem. 278 24944–24950. [DOI] [PubMed] [Google Scholar]
- 33.Hirosumi J., G. Tuncman, L. Chang, C. Z. Gorgun, K. T. Uysal, K. Maeda, M. Karin, and G. S. Hotamisligil. 2002. A central role for JNK in obesity and insulin resistance. Nature. 420 333–336. [DOI] [PubMed] [Google Scholar]
- 34.Rui L., V. Aguirre, J. K. Kim, G. I. Shulman, A. Lee, A. Corbould, A. Dunaif, and M. F. White. 2001. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J. Clin. Invest. 107 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L. F., and W. C. Greene. 2004. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5 392–401. [DOI] [PubMed] [Google Scholar]
- 36.Shoelson S. E., J. Lee, and M. Yuan. 2003. Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int. J. Obes. Relat. Metab. Disord. 27 (Suppl 3): S49–S52. [DOI] [PubMed] [Google Scholar]
- 37.Chung S., J. M. Brown, J. N. Provo, R. Hopkins, and M. K. McIntosh. 2005. Conjugated linoleic acid promotes human adipocyte insulin resistance through NFkappaB-dependent cytokine production. J. Biol. Chem. 280 38445–38456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sinha S., G. Perdomo, N. F. Brown, and R. M. O'Doherty. 2004. Fatty acid-induced insulin resistance in L6 myotubes is prevented by inhibition of activation and nuclear localization of nuclear factor kappa B. J. Biol. Chem. 279 41294–41301. [DOI] [PubMed] [Google Scholar]
- 39.Berliner J. A., M. Navab, A. M. Fogelman, J. S. Frank, L. L. Demer, P. A. Edwards, A. D. Watson, and A. J. Lusis. 1995. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 91 2488–2496. [DOI] [PubMed] [Google Scholar]
- 40.Robbesyn F., R. Salvayre, and A. Negre-Salvayre. 2004. Dual role of oxidized LDL on the NF-kappaB signaling pathway. Free Radic. Res. 38 541–551. [DOI] [PubMed] [Google Scholar]
- 41.Ogihara T., T. Asano, H. Katagiri, H. Sakoda, M. Anai, N. Shojima, H. Ono, M. Fujishiro, A. Kushiyama, Y. Fukushima, et al. 2004. Oxidative stress induces insulin resistance by activating the nuclear factor-kappa B pathway and disrupting normal subcellular distribution of phosphatidylinositol 3-kinase. Diabetologia. 47 794–805. [DOI] [PubMed] [Google Scholar]
- 42.Chisolm G. M., and D. Steinberg. 2000. The oxidative modification hypothesis of atherogenesis: an overview. Free Radic. Biol. Med. 28 1815–1826. [DOI] [PubMed] [Google Scholar]
- 43.Tsuzura S., Y. Ikeda, T. Suehiro, K. Ota, F. Osaki, K. Arii, Y. Kumon, and K. Hashimoto. 2004. Correlation of plasma oxidized low-density lipoprotein levels to vascular complications and human serum paraoxonase in patients with type 2 diabetes. Metabolism. 53 297–302. [DOI] [PubMed] [Google Scholar]
- 44.Myara I., C. Alamowitch, O. Michel, D. Heudes, J. Bariety, B. Guy-Grand, and J. Chevalier. 2003. Lipoprotein oxidation and plasma vitamin E in nondiabetic normotensive obese patients. Obes. Res. 11 112–120. [DOI] [PubMed] [Google Scholar]
- 45.Sigurdardottir V., B. Fagerberg, and J. Hulthe. 2002. Circulating oxidized low-density lipoprotein (LDL) is associated with risk factors of the metabolic syndrome and LDL size in clinically healthy 58-year-old men (AIR study). J. Intern. Med. 252 440–447. [DOI] [PubMed] [Google Scholar]
- 46.Wallenfeldt K., B. Fagerberg, J. Wikstrand, and J. Hulthe. 2004. Oxidized low-density lipoprotein in plasma is a prognostic marker of subclinical atherosclerosis development in clinically healthy men. J. Intern. Med. 256 413–420. [DOI] [PubMed] [Google Scholar]
- 47.Ehara S., M. Ueda, T. Naruko, K. Haze, A. Itoh, M. Otsuka, R. Komatsu, T. Matsuo, H. Itabe, T. Takano, et al. 2001. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 103 1955–1960. [DOI] [PubMed] [Google Scholar]
- 48.Holvoet P., D. H. Lee, M. Steffes, M. Gross, and D. R. Jacobs, Jr. 2008. Association between circulating oxidized low-density lipoprotein and incidence of the metabolic syndrome. JAMA. 299 2287–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krause B. R., and A. D. Hartman. 1984. Adipose tissue and cholesterol metabolism. J. Lipid Res. 25 97–110. [PubMed] [Google Scholar]
- 50.Hofmann S. M., L. Zhou, D. Perez-Tilve, T. Greer, E. Grant, L. Wancata, A. Thomas, P. T. Pfluger, J. E. Basford, D. Gilham, et al. 2007. Adipocyte LDL receptor-related protein-1 expression modulates postprandial lipid transport and glucose homeostasis in mice. J. Clin. Invest. 117 3271–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuniyasu A., S. Hayashi, and H. Nakayama. 2002. Adipocytes recognize and degrade oxidized low density lipoprotein through CD36. Biochem. Biophys. Res. Commun. 295 319–323. [DOI] [PubMed] [Google Scholar]
- 52.Kuniyasu A., N. Ohgami, S. Hayashi, A. Miyazaki, S. Horiuchi, and H. Nakayama. 2003. CD36-mediated endocytic uptake of advanced glycation end products (AGE) in mouse 3T3–L1 and human subcutaneous adipocytes. FEBS Lett. 537 85–90. [DOI] [PubMed] [Google Scholar]
- 53.Zhao S. P., J. Wu, D. Q. Zhang, H. J. Ye, L. Liu, and J. Q. Li. 2004. Fenofibrate enhances CD36 mediated endocytic uptake and degradation of oxidized low density lipoprotein in adipocytes from hypercholesterolemia rabbit. Atherosclerosis. 177 255–262. [DOI] [PubMed] [Google Scholar]
- 54.Chui P. C., H. P. Guan, M. Lehrke, and M. A. Lazar. 2005. PPARgamma regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J. Clin. Invest. 115 2244–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masella R., R. Vari, M. D'Archivio, C. Santangelo, B. Scazzocchio, M. T. Maggiorella, L. Sernicola, F. Titti, M. Sanchez, U. Di Mario, et al. 2006. Oxidised LDL modulate adipogenesis in 3T3–L1 preadipocytes by affecting the balance between cell proliferation and differentiation. FEBS Lett. 580 2421–2429. [DOI] [PubMed] [Google Scholar]
- 56.D'Archivio M., B. Scazzocchio, C. Filesi, R. Vari, M. T. Maggiorella, L. Sernicola, C. Santangelo, C. Giovannini, and R. Masella. 2008. Oxidised LDL up-regulate CD36 expression by the Nrf2 pathway in 3T3–L1 preadipocytes. FEBS Lett. 582 2291–2298. [DOI] [PubMed] [Google Scholar]
- 57.Maziere C., P. Morliere, R. Santus, V. Marcheux, C. Louandre, M. A. Conte, and J. C. Maziere. 2004. Inhibition of insulin signaling by oxidized low density lipoprotein. Protective effect of the antioxidant Vitamin E. Atherosclerosis. 175 23–30. [DOI] [PubMed] [Google Scholar]
- 58.Schreck R., K. Albermann, and P. A. Baeuerle. 1992. Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 17 221–237. [DOI] [PubMed] [Google Scholar]
- 59.Ishiki M., and A. Klip. 2005. Minireview: recent developments in the regulation of glucose transporter-4 traffic: new signals, locations, and partners. Endocrinology. 146 5071–5078. [DOI] [PubMed] [Google Scholar]
- 60.Bouzakri K., and J. R. Zierath. 2007. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. J. Biol. Chem. 282 7783–7789. [DOI] [PubMed] [Google Scholar]
- 61.Masella R., R. Vari, M. D'Archivio, R. Di Benedetto, P. Matarrese, W. Malorni, B. Scazzocchio, and C. Giovannini. 2004. Extra virgin olive oil biophenols inhibit cell-mediated oxidation of LDL by increasing the mRNA transcription of glutathione-related enzymes. J. Nutr. 134 785–791. [DOI] [PubMed] [Google Scholar]
- 62.Lowry O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 265–275. [PubMed] [Google Scholar]
- 63.Yagi K. 1987. Lipid peroxides and human diseases. Chem. Phys. Lipids. 45 337–351. [DOI] [PubMed] [Google Scholar]
- 64.Holvoet P., J. Vanhaecke, S. Janssens, F. Van de Werf, and D. Collen. 1998. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 98 1487–1494. [DOI] [PubMed] [Google Scholar]
- 65.Braiman L., A. Alt, T. Kuroki, M. Ohba, A. Bak, T. Tennenbaum, and S. R. Sampson. 1999. Protein kinase Cdelta mediates insulin-induced glucose transport in primary cultures of rat skeletal muscle. Mol. Endocrinol. 13 2002–2012. [DOI] [PubMed] [Google Scholar]
- 66.McKeel D. W., and L. Jarett. 1970. Preparation and characterization of a plasma membrane fraction from isolated fat cells. J. Cell Biol. 44 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ge X., Q. Yu, W. Qi, X. Shi, and Q. Zhai. 2008. Chronic insulin treatment causes insulin resistance in 3T3–L1 adipocytes through oxidative stress. Free Radic. Res. 42 582–591. [DOI] [PubMed] [Google Scholar]
- 68.Takano A., I. Usui, T. Haruta, J. Kawahara, T. Uno, M. Iwata, and M. Kobayashi. 2001. Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol. Cell. Biol. 21 5050–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haruta T., T. Uno, J. Kawahara, A. Takano, K. Egawa, P. M. Sharma, J. M. Olefsky, and M. Kobayashi. 2000. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14 783–794. [DOI] [PubMed] [Google Scholar]
- 70.Paz K., R. Hemi, D. LeRoith, A. Karasik, E. Elhanany, H. Kanety, and Y. Zick. 1997. A molecular basis for insulin resistance. Elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J. Biol. Chem. 272 29911–29918. [DOI] [PubMed] [Google Scholar]
- 71.Bloch-Damti A., R. Potashnik, P. Gual, Y. Le Marchand-Brustel, J. F. Tanti, A. Rudich, and N. Bashan. 2006. Differential effects of IRS1 phosphorylated on Ser307 or Ser632 in the induction of insulin resistance by oxidative stress. Diabetologia. 49 2463–2473. [DOI] [PubMed] [Google Scholar]
- 72.Siebenlist U., G. Franzoso, and K. Brown. 1994. Structure, regulation and function of NF-kappa B. Annu. Rev. Cell Biol. 10 405–455. [DOI] [PubMed] [Google Scholar]
- 73.Couillard C., G. Ruel, W. R. Archer, S. Pomerleau, J. Bergeron, P. Couture, B. Lamarche, and N. Bergeron. 2005. Circulating levels of oxidative stress markers and endothelial adhesion molecules in men with abdominal obesity. J. Clin. Endocrinol. Metab. 90 6454–6459. [DOI] [PubMed] [Google Scholar]
- 74.Sjogren P., S. Basu, M. Rosell, A. Silveira, U. de Faire, B. Vessby, A. Hamsten, M. L. Hellenius, and R. M. Fisher. 2005. Measures of oxidized low-density lipoprotein and oxidative stress are not related and not elevated in otherwise healthy men with the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 25 2580–2586. [DOI] [PubMed] [Google Scholar]
- 75.Wu Z. H., and S. P. Zhao. 2006. Adipocyte: a potential target for the treatment of atherosclerosis. Med. Hypotheses. 67 82–86. [DOI] [PubMed] [Google Scholar]
- 76.Malide D., G. Ramm, S. W. Cushman, and J. W. Slot. 2000. Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells. J. Cell Sci. 113 4203–4210. [DOI] [PubMed] [Google Scholar]
- 77.Holman G. D., I. J. Kozka, A. E. Clark, C. J. Flower, J. Saltis, A. D. Habberfield, I. A. Simpson, and S. W. Cushman. 1990. Cell surface labeling of glucose transporter isoform GLUT4 by bis-mannose photolabel. Correlation with stimulation of glucose transport in rat adipose cells by insulin and phorbol ester. J. Biol. Chem. 265 18172–18179. [PubMed] [Google Scholar]
- 78.Watson R. T., M. Kanzaki, and J. E. Pessin. 2004. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr. Rev. 25 177–204. [DOI] [PubMed] [Google Scholar]
- 79.Griffin M. E., M. J. Marcucci, G. W. Cline, K. Bell, N. Barucci, D. Lee, L. J. Goodyear, E. W. Kraegen, M. F. White, and G. I. Shulman. 1999. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 48 1270–1274. [DOI] [PubMed] [Google Scholar]
- 80.Liu Y. F., K. Paz, A. Herschkovitz, A. Alt, T. Tennenbaum, S. R. Sampson, M. Ohba, T. Kuroki, D. LeRoith, and Y. Zick. 2001. Insulin stimulates PKCzeta -mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J. Biol. Chem. 276 14459–14465. [DOI] [PubMed] [Google Scholar]
- 81.Mothe I., and E. Van Obberghen. 1996. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J. Biol. Chem. 271 11222–11227. [DOI] [PubMed] [Google Scholar]
- 82.Lim J. H., J. I. Lee, Y. H. Suh, W. Kim, J. H. Song, and M. H. Jung. 2006. Mitochondrial dysfunction induces aberrant insulin signalling and glucose utilisation in murine C2C12 myotube cells. Diabetologia. 49 1924–1936. [DOI] [PubMed] [Google Scholar]
- 83.Yi P., F. E. Lu, L. J. Xu, G. Chen, H. Dong, and K. F. Wang. 2008. Berberine reverses free-fatty-acid-induced insulin resistance in 3T3–L1 adipocytes through targeting IKKbeta. World J. Gastroenterol. 14 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo H., W. Ling, Q. Wang, C. Liu, Y. Hu, and M. Xia. 2008. Cyanidin 3-glucoside protects 3T3–L1 adipocytes against H2O2- or TNF-alpha-induced insulin resistance by inhibiting c-Jun NH2-terminal kinase activation. Biochem. Pharmacol. 75 1393–1401. [DOI] [PubMed] [Google Scholar]
- 85.Kuan C. Y., D. D. Yang, D. R. Samanta Roy, R. J. Davis, P. Rakic, and R. A. Flavell. 1999. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 22 667–676. [DOI] [PubMed] [Google Scholar]
- 86.Rincon M., A. Whitmarsh, D. D. Yang, L. Weiss, B. Derijard, P. Jayaraj, R. J. Davis, and R. A. Flavell. 1998. The JNK pathway regulates the In vivo deletion of immature CD4(+)CD8(+) thymocytes. J. Exp. Med. 188 1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zandi E., D. M. Rothwarf, M. Delhase, M. Hayakawa, and M. Karin. 1997. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 91 243–252. [DOI] [PubMed] [Google Scholar]
- 88.Yin M. J., Y. Yamamoto, and R. B. Gaynor. 1998. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 396 77–80. [DOI] [PubMed] [Google Scholar]
- 89.Yuan M., N. Konstantopoulos, J. Lee, L. Hansen, Z. W. Li, M. Karin, and S. E. Shoelson. 2001. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 293 1673–1677. [DOI] [PubMed] [Google Scholar]
- 90.Kim J. K., Y. J. Kim, J. J. Fillmore, Y. Chen, I. Moore, J. Lee, M. Yuan, Z. W. Li, M. Karin, P. Perret, et al. 2001. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Invest. 108 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Castrillo A., M. J. Diaz-Guerra, S. Hortelano, P. Martin-Sanz, and L. Bosca. 2000. Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-delta(12,14)-prostaglandin J(2) in activated murine macrophages. Mol. Cell. Biol. 20 1692–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rossi A., P. Kapahi, G. Natoli, T. Takahashi, Y. Chen, M. Karin, and M. G. Santoro. 2000. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 403 103–108. [DOI] [PubMed] [Google Scholar]
- 93.Boyault S., A. Bianchi, D. Moulin, S. Morin, M. Francois, P. Netter, B. Terlain, and K. Bordji. 2004. 15-Deoxy-delta(12,14)-prostaglandin J(2) inhibits IL-1beta-induced IKK enzymatic activity and IkappaBalpha degradation in rat chondrocytes through a PPARgamma-independent pathway. FEBS Lett. 572 33–40. [DOI] [PubMed] [Google Scholar]
- 94.Maziere C., M. Auclair, M. Djavaheri-Mergny, L. Packer, and J. C. Maziere. 1996. Oxidized low density lipoprotein induces activation of the transcription factor NF kappa B in fibroblasts, endothelial and smooth muscle cells. Biochem. Mol. Biol. Int. 39 1201–1207. [DOI] [PubMed] [Google Scholar]
- 95.Lappas M., K. Yee, M. Permezel, and G. E. Rice. 2005. Sulfasalazine and BAY 11–7082 interfere with the nuclear factor-kappa B and I kappa B kinase pathway to regulate the release of proinflammatory cytokines from human adipose tissue and skeletal muscle in vitro. Endocrinology. 146 1491–1497. [DOI] [PubMed] [Google Scholar]
- 96.Li Y., M. A. Reddy, F. Miao, N. Shanmugam, J. K. Yee, D. Hawkins, B. Ren, and R. Natarajan. 2008. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 283 26771–26781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nieto-Vazquez I., S. Fernandez-Veledo, C. de Alvaro, and M. Lorenzo. 2008. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes. 57 3211–3221. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Nieto-Vazquez I., S. Fernandez-Veledo, D. K. Kramer, R. Vila-Bedmar, L. Garcia-Guerra, and M. Lorenzo. 2008. Insulin resistance associated to obesity: the link TNF-alpha. Arch. Physiol. Biochem. 114 183–194. [DOI] [PubMed] [Google Scholar]
- 99.Tirosh A., R. Potashnik, N. Bashan, and A. Rudich. 1999. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3–L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J. Biol. Chem. 274 10595–10602. [DOI] [PubMed] [Google Scholar]
- 100.Shimoyama T., S. Yamaguchi, K. Takahashi, H. Katsuta, E. Ito, H. Seki, K. Ushikawa, H. Katahira, K. Yoshimoto, H. Ohno, et al. 2006. Gliclazide protects 3T3L1 adipocytes against insulin resistance induced by hydrogen peroxide with restoration of GLUT4 translocation. Metabolism. 55 722–730. [DOI] [PubMed] [Google Scholar]
- 101.Ermak N., B. Lacour, T. B. Drueke, and S. Vicca. 2008. Role of reactive oxygen species and Bax in oxidized low density lipoprotein-induced apoptosis of human monocytes. Atherosclerosis. 200 247–256. [DOI] [PubMed] [Google Scholar]
- 102.Giovannini C., B. Scazzocchio, P. Matarrese, R. Vari, M. D'Archivio, R. Di Benedetto, S. Casciani, M. R. Dessi, E. Straface, W. Malorni, et al. 2008. Apoptosis induced by oxidized lipids is associated with up-regulation of p66Shc in intestinal Caco-2 cells: protective effects of phenolic compounds. J. Nutr. Biochem. 19 118–128. [DOI] [PubMed] [Google Scholar]