

Abstract
Lipid rafts are membrane microdomains involved in many cellular functions, including transduction of cellular signals and cell entry by pathogens. Lipid rafts can be enriched biochemically by extraction in a nonionic detergent at low temperature, followed by floatation on a sucrose density gradient. Previous proteomic studies of such detergent-resistant membranes (DRMs) are in disagreement about the presence of mitochondrial proteins in raft components. Here, we approach the status of mitochondrial proteins in DRM preparations by employing stable isotope labeling by amino acids in cell culture to evaluate the composition of differentially purified subcellular fractions as well as high-resolution linear density gradients. Our data demonstrate that F1/F0 ATPase subunits, voltage-dependent anion selective channels, and other mitochondrial proteins are at best partially copurifying contaminants of raft preparations.
Keywords: detergent-resistant membranes, membrane proteins, stable isotope labeling by amino acids in cell culture, quantitative proteomics, mass spectrometry, methyl-β-cyclodextrin, protein correlation profiling
Biological membranes form barriers, compartmentalizing cells into organelles or separating cells from their outside environment. They are composed of lipids and proteins at ratios ranging from 1:4 to 4:1 by mass, with the proteins conferring several capabilities, including ion transport, energy storage, and information transduction. The original fluid mosaic model (1) of membranes suggested a homogenous distribution of proteins and lipids across the two-dimensional surface, but more recent evidence suggests that membranes themselves are compartmentalized by uneven distributions of specific lipids and/or proteins into various microdomains (2). Lipid rafts are one such class of microdomains that were originally defined biochemically as the low density detergent-resistant membrane (DRM) fraction of cells but that are now recognized as a subset of DRMs enriched in cholesterol and sphingolipids (3–7). Cholesterol is thought to intercalate between the rigid hydrophobic tails of sphingolipids and saturated phospholipids, allowing a very tightly packed structure with unique biophysical characteristics compared with surrounding membranes. Lipid raft theory (8) proposes that certain proteins preferentially cluster into this unique environment, forming reaction centers essential for many cellular processes, such as cell signaling and trafficking (8, 9). The diverse array of vital processes that rafts are implicated in make these membrane microdomains an interesting subject for proteomic characterization.
At least two dozen proteomic investigations of DRMs have been reported since 2001 [reviewed elsewhere (10)], and without exception, all have found certain mitochondrial proteins to be present in the preparations, particularly mitochondrial ATP synthase subunits and the voltage-dependent anion selective channels (VDACs). Mitochondria are quite dense, however, so they should not migrate upwards in the standard DRM preparation. Thus, there are two possible explanations for the observation of mitochondrial proteins in DRMs: 1) mitochondria themselves contain bona fide lipid rafts or another detergent-resistant membrane microdomain, or 2) the localization of proteins such as the ATP synthase subunits or VDACs is not restricted to the mitochondria. We have addressed an aspect of the former possibility previously using stable isotope labeling by amino acids in cell culture (SILAC) to encode the sensitivity to cholesterol disruption into a proteomic analysis of DRMs (11). We were able to demonstrate that the “mitochondrial” proteins in DRMs are not sensitive to cholesterol disruption by methyl-β-cyclodextrin (MβCD), the standard test applied to putative lipid raft components (12, 13). While these findings suggested that ATP synthase subunits and VDACs are not in rafts, several other more recent studies have claimed otherwise (14–16). Here, we use quantitative proteomics and multiple subcellular fractionation procedures to approach the issue of mitochondrial proteins being in lipid rafts from several angles in three different cell types to conclude that there are no rafts in mitochondria and that there are no mitochondrial proteins in cell surface rafts.
MATERIALS AND METHODS
Materials
The following materials were obtained from the indicated commercial sources: normal DMEM, Roswell Park Memorial Institute (RPMI)-1640 medium, L-glutamine, penicillin/streptomycin, SuperSignalWest PicoChemiluminescent detection system and BCA assay kit, HEPES, sodium pyruvate, and cell culture trypsin (ThermoFisher, Nepean, Ontario, Canada); FBS, both qualified and dialyzed forms (Invitrogen, Burlington, Ontario, Canada); L-lysine and l-arginine-deficient DMEM and RPMI-1640 (Caisson Labs, North Ogden, UT); L-lysine, l-arginine, methyl-β-cyclodextrin, Triton X-100, sodium deoxycholate (SDC), DTT, iodoacetamide, and Percoll (Sigma-Aldrich, St. Louis, MO); 2H4-lysine, 13C6-arginine, 13C615N2-lysine, and 13C615N4-arginine (Cambridge Isotope Laboratories, Cambridge, MA); sequencing grade modified porcine trypsin solution (Promega, Madison, WI) and protease inhibitor cocktail tablets with EDTA (Roche Diagnostics, Mannheim, Germany). Antibodies used and their commercial sources were as follows: α-flotillin-2 (BD Transduction, San Jose, CA), α-ATP synthase subunit β (Molecular Probes, Burlington, CA), and horseradish-peroxidase-conjugated anti-mouse secondary (Bio-Rad, Hercules, CA). The three cell lines used here, HeLa, swiss-3T3, and Jurkat, were all obtained from the American Type Culture Collection (Manassas, VA).
Cell culture and SILAC
HeLa and 3T3 cells were maintained in DMEM supplemented with 10% FBS (v/v), 1% L-glutamine (v/v), and 1% penicillin/streptomycin (v/v) at 5% CO2 and 37°C. Jurkat cells were maintained suspended in cell culture flask in RPMI-1640 supplemented with 10% FBS (v/v), 1% L-glutamine (v/v), 1% penicillin/streptomycin (v/v), 10 mM HEPES, and 1 mM sodium pyruvate at 5% CO2 and 37°C. Double and triple SILAC labeling was conducted as described (17), allowing a 200-fold increase in the cell population during labeling. We will henceforth refer to the different labels as 0/0 for the normal isotopic abundance Lys and Arg, 4/6 for 2H4-Lys and 13C6-Arg, and 8/10 for 13C615N2-Lys and 13C615N4-Arg. To obtain enough material for effective proteomic analysis, six 15 cm plates of confluent adherent cells (HeLa and 3T3) or 1.6 × 108 suspension cells (Jurkat) were used for each of the 0/0 and 4/6 conditions for isolation of rafts from whole cells. In the triple label experiment used for determining the presence of mitochondrial rafts, we used fifteen 15 cm plates or 4.0 ×108 cells for the 0/0 and 4/6 conditions and three 15 cm plates or 0.8 × 108 cells for the 8/10 condition. All cells were serum starved (18 h for HeLa, 9 h for 3T3, and 20 h for Jurkat) to deplete free cholesterol before MβCD treatment, mitochondria isolation, or DRM isolation.
To determine the optimal MβCD concentration for each cell type, serum-starved HeLa, 3T3, and Jurkat cells in six-well plates were treated with the compound for 1 h at several concentrations between 5 and 20 mM. Cell viability was assessed after the treatment by visual inspection, and the maximum ([MβCD]max) dose that did not cause detectable cell death was used in all further experiments (HeLa, 10 mM; 3T3, 5 mM; Jurkat, 5 mM). As the effects of MβCD on some raft proteins can often be subtle, our goal in this optimization was to maximize the concentration used for each cell type.
Whole-cell membrane preparation
Three 15 cm plates of 0/0 labeled HeLa or 3T3 cells were washed three times with ice-cold PBS and then scraped into homogenization buffer (250 mM sucrose, 10 mM Tris-HCl, and 0.1 mM EGTA, pH 7.4) with protease inhibitor cocktail added fresh separately. Cells were lysed by forcing them through a 25 G syringe. Unbroken cells and large pieces of debris were pelleted down for 10 min, 4°C at 600 relative centrifugal force (r.c.f.), and the supernatant was saved by spinning down for 30 min, 4°C at 166,000 r.c.f.
Mitochondria isolation
Mitochondria were isolated as described with some modifications (18). Briefly, serum starved cells were washed three times with PBS and then scraped or resuspended into homogenization buffer (250 mM sucrose, 10 mM Tris-HCl, and 0.1 mM EGTA, pH 7.4) with protease inhibitor cocktail added fresh. Cells were lysed by forcing them through a 25 G syringe; cell breakage was tracked using phase contrast microscopy and was continued until at least 95% of the cells were broken. Lysates were then centrifuged for 10 min at 600 r.c.f. to pellet unbroken cells and nuclei. The supernatant from this step was collected and centrifuged for a further 10 min at 5,000 r.c.f. to pellet crude mitochondria. The pellet was resuspended in homogenization buffer and recentrifuged under the same conditions. Following this, the pellet was resuspended and mitochondria were resolved in 20% Percoll (in 10 mM Tris-HCl and 0.1 mM EGTA, pH 7.4) as a white band near the top of the tube after centrifugation for 60 min at 65,000 r.c.f. The band was extracted by puncturing the site of the centrifuge tube with a 22 G syringe and drawing solution out. The extracted band was then diluted 3-fold in PBS, and mitochondria were pelleted by centrifugation for 30 min at 65,000 r.c.f. The final mitochondria pellet was washed once with ice-cold PBS. All isolation steps were carried out at 4°C.
Detergent-resistant membrane preparation
Detergent-resistant membranes (DRMs) were extracted from isolated mitochondria (0/0 condition treated with [MβCD]max for 30 min at 4°C where indicated) or serum starved cells (0/0 condition treated with [MβCD]max for 1 h at 37°C where indicated) or 4/6 labeled cells as described (11) with minor modifications. Briefly, cells were solubilized in lysis buffer [1% Triton X-100, 25 mM MES, pH 6.5, and protease inhibitor cocktail] by end-over-end rotation for 1 h. Total protein concentrations of cell lysates were determined using the Coomassie Plus kit (Pierce, Nepean, Ontario, Canada), and equal masses of protein from each SILAC condition were mixed together. The combined lysates were mixed with an equal volume of 90% sucrose (in 25 mM MES, 150 mM NaCl, pH 6.5, MES-buffered saline [MBS]) and transferred into the bottom of an ultracentrifuge tube. On to this was layered 5 ml of 35% sucrose in MBS and then enough 5% sucrose in MBS to fill the tube. These gradients were then centrifuged for 18 h at 166,000 r.c.f. The white, light-scattering band appearing between 35% and 5% sucrose after centrifugation corresponded DRMs, and this was extracted using a 22 G syringe. The sucrose was diluted out approximately 3-fold with MBS and membranes were further pelleted by centrifugation at 166,000 r.c.f. for 2 h. Finally, the DRM pellet was washed once with ice-cold MBS prior to further processing for proteomic analysis. All isolation steps were carried out at 4°C.
Protein correlation profiling with SILAC and DRM preparation for linear sucrose gradient
A total of 8.0 ×108 Jurkat cells were used for the 0/0 linear gradient condition, and 5.0 ×108 Jurkat cells were used for the 4/6 nonlinear control condition. A crude DRM lysate was prepared from 0/0 and 4/6 Jurkat cells as above. A linear sucrose gradient was prepared by mixing 6 ml each of 30% and 10% sucrose in MBS into a centrifuge tube with a linear gradient mixer. A 1 ml 5% sucrose cushion was layered on top of the linear gradient, followed by the 0/0 extracted DRMs. The nonlinear control condition was prepared by mixing 4/6 cell lysate with an equal volume of 90% sucrose in MBS and then layering 5 ml 35% and 5 ml 5% sucrose on top as above. After centrifuging the gradients for 18 h at 166,000 r.c.f., 12 1 ml fractions were extracted from the bottom curvature of the linear gradient tube, and one 3 ml fraction was collected at the 35–5% interface of the nonlinear condition. Fractions were diluted and pelleted as above and then resuspended in 1% SDC. The seven final linear samples were single fractions or combinations as follows: A (fraction 1 to fraction 2), B (3, 4), C (5), D (6), E (7), F (8, 9), and G (10–12), with fraction 1 being the bottom (most dense) fraction and 12 being the top fraction. The protein concentrations were measured by BCA assay. Equal amounts of protein from the 0/0 linear samples and 4/6 nonlinear samples were mixed for the protein correlation profiling (PCP) with SILAC (19).
Western blotting
Equal volumes of the seven linear fractions and of the nonlinear fraction (on average 15 μg protein) were combined with protein sample buffer, separated by 12% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and blocked with 5% milk powder. Primary antibodies were used as follows: α-flotillin-2, diluted 1 in 200 for 1 h; and α-ATP synthase subunit β, diluted 1/250 for 18 h. Horseradish-peroxidase-conjugated anti-mouse secondary was used at 1/4,000 and signal detected with the SuperSignalWest PicoChemiluminescent detection system.
Liquid chromatography-tandem mass spectrometry, database searching, and data analysis
Most analyses described here involved direct analysis of an in solution digestion of the samples in question. In solution digestions in SDC were carried out exactly as described (17) with protein pellets being solubilized directly in SDC and then subjected to trypsin digestion. For each sample, ∼5 μg of digested peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on a LTQ-Orbitrap (ThermoFisher, Bremen, Germany) exactly as described (17). For the DRM versus whole-cell membrane (WCM) comparison experiment, DRM pellets were resuspended in Triton lysis buffer. Protein concentrations were measured by Bradford assay for both DRM and WCM samples; equal amounts of protein were mixed and then pelleted down. In this experiment only, digested peptides were then further fractionated by strong cation exchange chromatography into five fractions using 0, 20, 50, 100, and 500 mM NH4CH3COO as described (20) and analyzed as above on an LTQ-Orbitrap.
MS/MS were extracted using the Extract_MSN.exe tool (v3.0; ThermoFisher) at its default settings, and the spectra were searched against the International Protein Index human (v3.37; 69,164 sequences) or mouse (v3.35; 51,490 sequences) databases using Mascot (v2.2; Matrix Science) using the following criteria: tryptic specificity with up to one missed cleavage; ±5 parts-per-million and ±0.6 kDa accuracy for MS and MS/MS measurements, respectively; electrospray ionization-ion trap fragmentation characteristics; cysteine carbamidomethylation as a fixed modification; N-terminal protein acetylation, methionine oxidation, 2H4-Lys, 13C6-Arg, 13C615N2-Lys, and 13C615N4-Arg as variable modifications as necessary. Proteins were considered identified if we observed at least two unique peptides with mass errors <3 parts-per-million, at least seven amino acids in length, and with Mascot IonsScore >25. These criteria yielded an estimated false discovery rate of ∼1% using a reversed database search. Quantitative ratios were extracted from the raw data using MSQuant (http://msquant.sourceforge.net), which calculates an intensity-weighted average of within-spectra ratios from all spectra across the chromatographic peak of each peptide ion. For automatic quantitation, only those proteins with a coefficient of variation (CV) <50% were accepted with no further verification. For proteins with high CVs or with only one quantified peptide, the chromatographic peak assignment was manually verified or rejected. Analytical variability of SILAC data in the types of experiments performed here is typically <20% in our hands, and biological variability was addressed in these experiments by performing at least three independent replicates of each experiment.
For the linear gradient experiments, spectra were extracted using MaxQuant (21) and searched against the International Protein Index Human database using the same parameters as above except for a ±0.5 kDa requirement for MS/MS accuracy. MaxQuant was then employed again to extract quantitative data, either SILAC ratios or PCPs. The resulting ratios for 0/0 linear fractions A to G relative to the 4/6 nonlinear control (light/heavy) were corrected for the volume of sample used and normalized to the greatest intensity fraction to view individual protein profiles. Principal component analysis (PCA) was performed on this seven-dimensional data set in MatLab as follows: the data set was converted into its corresponding covariance matrix, and the eigenvalues and eigenvectors were obtained, the eigenvectors corresponding to the greatest and second greatest eigenvalues (vectors PC1 and PC2) were used to define a plane, and protein data points were projected onto the plane to generate a two-dimensional plot. Data were then subjected to complete linkage hierarchical clustering using Cluster software (22), and the results were viewed using MapleTree (http://mapletree.sourceforge.net).
RESULTS
The cholesterol-dependent DRM proteome is similar across cell types
The DRM proteomes of at least a dozen different cells and tissues are now available (11, 14–16, 23–41). However, it is extremely difficult, if not impossible, to purify any organelle to homogeneity (42), and the single-step centrifugation used to enrich rafts is no exception, so without rigorous controls a DRM proteome cannot be equated with a raft proteome. In an effort to distinguish resident lipid raft proteins from other proteins copurifying in DRMs, we previously developed a quantitative approach to mass encode the cholesterol dependence of DRM proteins from HeLa human cervical carcinoma cells prior to LC-MS/MS analysis (11). In this way, a more accurate raft proteome can be measured because the two defining characteristics of raft proteins are measured in a single experiment: sensitivity to cholesterol disruption and enrichment in a low density membrane fraction. To estimate the similarity among raft proteomes from different cell types, we applied this method to two additional cell types commonly used in lipid raft and other cell biology investigations, 3T3 mouse fibroblasts and human Jurkat T lymphocytes.
Cell lines differ in their sensitivity to the cholesterol disrupting agent MβCD, so we initially determined the maximum sublethal dose of the drug for each cell line, [MβCD]max: 10 mM for HeLa and 5 mM for 3T3 and Jurkat. For each cell type, we then labeled two populations of cells with normal isotopic abundance (0/0) or stable isotope-enriched (4/6) forms of lysine and arginine (see Materials and Methods) prior to treating one of the two populations with MβCD. For these experiments, we chose to treat the unlabeled cells so that any proteins sensitive to the treatment would present mostly in the labeled form rather than the reverse because then any exogenous protein (e.g., keratins, trypsin, and serum components) would have the same SILAC spectral signature as a true raft component. The low density DRM fraction from each of the three cell types appeared quite different at the macroscopic level, with widely varying amounts of material isolated from each (data not shown). Despite this, 200 to 300 DRM proteins could typically be identified from <5 μg of total protein loaded into the mass spectrometer from each of the cell types (see Supplementary Tables I to III), with SILAC ratios measurable for the large majority of these. By sorting the control:MβCD ratios in decreasing order, we found that the overall distribution of cholesterol sensitivity (Fig. 1) in all three cell types was similar to our previous observations for HeLa DRMs alone (11). Namely, there was a portion of the proteins displaying extreme sensitivity to cholesterol disruption, a group with intermediate sensitivity, and a group that did not appear to be sensitive to MβCD at all. Where they were identified, known raft proteins all fell into the group with high ratios, including heterotrimeric guanine nucleotide binding proteins (G-proteins), YES tyrosine kinase, Src tyrosine kinase, SNAP-23, and aminopeptidase N. According to the known sensitivity of lipid raft proteins to this drug (13), we previously classified the three differentially sensitive groups into raft proteins, raft-associated proteins, and copurifying proteins or contaminants (11).
Fig. 1.
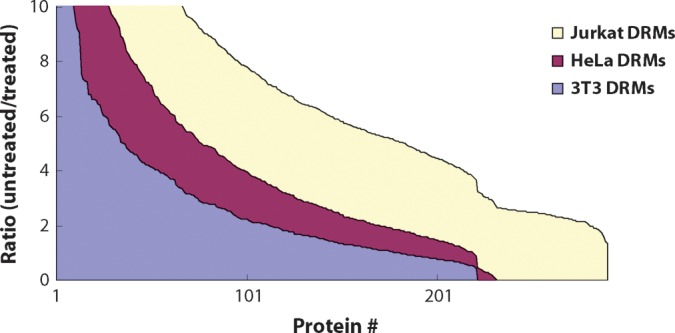
Ratios of control:MβCD treated in decreasing order. Ratios for detergent-resistant proteins identified and quantified by MSQuant were plotted from largest to smallest for all three cell types: HeLa, 3T3, and Jurkat.
Our application of the same method across multiple cell types now allows us to address the issue of how similar raft proteins are among different cell types. There are numerous reports of DRM proteomes from different cell types, but without the unbiased classification of DRM proteins enabled by our SILAC approach, comparison among those studies is difficult. In general, signaling enzymes, such as protein kinases, protein phosphatases, and G-proteins, were enriched among the proteins with high ratios relative to DRMs as a whole. Structural proteins of microfilaments, such as actin, myosin, and vimentin, were identified with intermediate ratios, suggesting a possible dependence of raft structure on the cytoskeleton consistent with recent studies of hemagglutinin dynamics at subdiffraction resolution (43) (Table 1). Likewise, the proteins with lower SILAC ratios were consistent across all cell types and generally included nuclear proteins and highly abundant cytosolic components, as would be expected. With few exceptions (e.g., mitochondrial trifunctional enzyme subunits, mitochondrial heat shock proteins, and prohibitin), all proteins classically considered to be localized to mitochondria (we will henceforth refer to this group of proteins as “mitochondrial proteins” even though the localization being discussed may not be in a mitochondrion per se) also had relatively low ratios, suggesting that these are also simply copurifying contaminants and not true raft proteins. However, since previous groups (14–16, 26) have reported some of these to be true components of rafts, we were tempted to explore further the presence of these proteins in DRMs. These published studies leave open two possibilities: 1) that mitochondria themselves contain detergent-resistant microdomains or other low density structures that copurify with rafts, or 2) that conventional cell surface rafts are enriched in mitochondrial proteins. Thus, we have designed SILAC-based approaches to test these two scenarios with our null hypothesis, H0, being that the mitochondrial proteins found in DRMs are simply just contaminants and not real components of rafts.
TABLE 1.
Examples of DRM proteins identified and quantified with their ratios in three cell types tested
| Ratios (H/L) | |||
|---|---|---|---|
| Protein Names |
HeLa |
3T3 |
Jurkat |
| Guanine nucleotide binding proteins G | >2.5 | >5.5 | >11.4 |
| Proto-oncogene tyrosine protein kinase Yes | 5.5 ± 1.6 | 4.7 ± 1.3 | 12.5 ± 9.4 |
| Proto-oncogene tyrosine protein kinase Src | ND | 1.6 ± 0.6 | 6.5 ± 0.5 |
| Aminopeptidase N | 6.5 ± 2.9 | 4.8 ± 0.7 | ND |
| SNAP-23 | 3.9 ± 0.4 | 5.9 ± 3.0 | 13.3 ± 4.4 |
| Fructose-bisphosphate aldolase A | 9.4 ± 1.9 | 3.7 ± 2.1 | 6.4 ± 3.2 |
| Vacuolar ATP synthase subunits | <1.3 | <0.8 | ND |
| Vimentin | ND | 2.8 ± 0.7 | 4.6 ± 1.8 |
| ATP synthase subunits, mitochndrial precursor | <1.0 | <1.8 | <3 |
| Voltage-dependent anion selective channel proteins | <0.7 | <0.7 | <2.2 |
| Heat shock proteins, mitochondrial precursor | <2.2 | <1.7 | <2.8 |
| Caveolin 1 α, β | <1.2 | <1.0 | ND |
ND, not detected.
Do mitochondria contain lipid rafts?
Of the two possibilities, this one seems less likely since one of the hallmarks of rafts is their high cholesterol content, and mitochondria contain very little cholesterol. Despite this, the possibility has been suggested before (35), so we tested this formal possibility using a triplex version of SILAC that would allow us to evaluate whether mitochondrial proteins in DRMs are enriched when DRMs are isolated directly from purified mitochondria (Fig. 2). Mitochondria are enriched from two of the three cell populations and treated with (0/0) or without (4/6) MβCD. DRMs are then isolated from the two mitochondrial preparations and from the third untreated, unfractionated cell population (8/10). In this scheme, only proteins that are sensitive to cholesterol disruption (high 4/6:0/0 ratio) and truly coming from mitochondria (high 4/6:8/10 ratio) would be considered components of mitochondrial rafts. Such proteins would be expected to fall in the upper right quadrants of the plots in Fig. 3, but, as predicted, those areas of the graphs were essentially empty for all three cell types. In HeLa, 3T3, and Jurkat cells, only a few proteins had ratios indicating potential mitochondrial rafts. However, in each case, these numbers represented <2% of the total proteins identified and quantified, and none of them is actually a known component of the mitochondria (see Supplementary Tables IIII to VI).
Fig. 2.
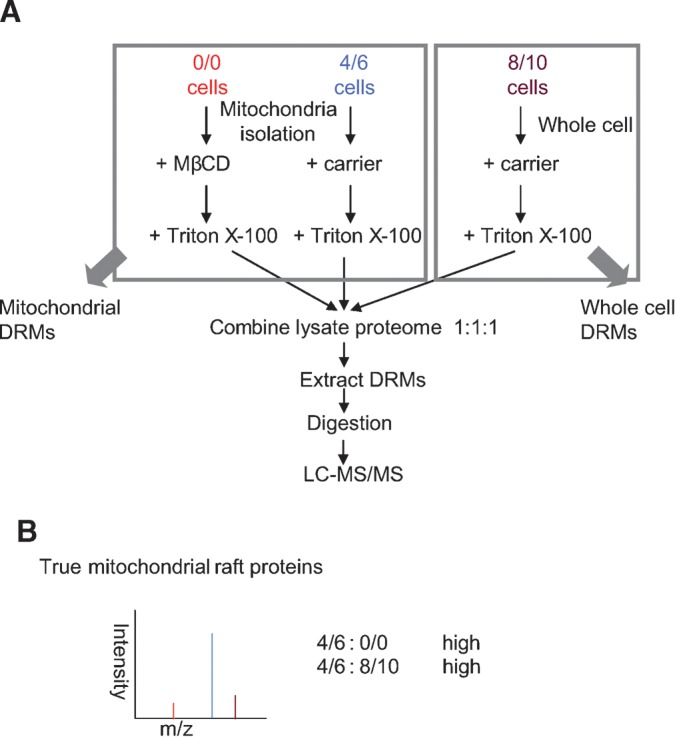
The use of SILAC to determine the origin of proteins in DRMs. A: Cells were grown in three SILAC media separately containing normal isotopic abundance Lys and Arg (0/0), 2H4-Lys and 13C6-Arg (4/6), or 13C615N2-Lys and 13C615N4-Arg (8/10). Mitochondria from both 0/0 and 4/6 cell populations were isolated (see Materials and Methods), and then 0/0 mitochondria were treated with the cholesterol-disrupting drug MβCD. Afterwards, 0/0 and 4/6 mitochondria preparations were solubilized in Triton X-100. At the same time, whole 8/10 cells were also solubilized with Triton X-100. Equal amounts of protein from all three extracts were mixed prior to isolation of DRMs by floatation on a sucrose density gradient, and then the proteins were subsequently analyzed by LC-MS/MS. B: True mitochondrial raft proteins should have high 4/6:0/0 ratios indicating their sensitivity to cholesterol disruption, while high 4/6:8/10 ratios would indicate that proteins are coming from mitochondria.
Fig. 3.

Results of triple label SILAC experiments for all three cell types: HeLa, 3T3, and Jurkat. One dot corresponds to one identified and quantified protein that has both untreated/treated (4/6:0/0, abscissa) and mitochondrial DRMs/whole cell DRMs (4/6:8/10, ordinate) ratios. The graphs represent 165 proteins for HeLa, 196 for 3T3, and 294 for Jurkat.
Do plasma membrane rafts contain mitochondrial proteins?
To test the possibility that some classical mitochondrial proteins inhabit a second subcellular location, namely, plasma membrane lipid rafts, we tested the degree to which proteins are enriched in DRMs relative to WCMs in HeLa and 3T3 cells. WCMs were isolated from 0/0 cells, and DRMs were isolated from 4/6-labeled cells. As described in Materials and Methods, the 4/6-labeled cells was treated with 1% Triton X-100 for 1 h prior to floatation on a sucrose density gradient. Finally, this DRM preparation was mixed with the oppositely labeled WCM, the mixed membranes were pelleted, solubilized in deoxycholate, and the proteins were heat denatured and digested to peptides. To probe more deeply into the DRM versus WCM proteome, we used strong cation exchange chromatography to fractionate digested peptides before LC-MS/MS. In this scheme, proteins enriched by the DRM procedure should display a high DRM:WCM ratio, including any and all lipid raft proteins.
Several hundred quantifiable proteins were identified in this analysis (see Materials and Methods for details of protein identification criteria), with DRM:WCM ratios ranging from essentially zero to three, four, or even ten. We were reassured to find that many typical plasma membrane, nonraft proteins were not enriched in the DRM preparation, including large transmembrane proteins and the sodium/potassium-transporting ATPase (Fig. 4). Interestingly, without exception all mitochondrial membrane proteins identified were also not enriched in DRMs. These included the mitochondrial components previously claimed to be in lipid rafts, including voltage-dependent anion-selective channel proteins and ATP synthase subunits. Furthermore, in this system all the MβCD-sensitive proteins we have observed previously (Fig. 1; see Supplementary Tables I to III) (11) were enriched in DRMs (see Supplementary Tables VII and VIII).
Fig. 4.

The enrichment of DRM over WCM. Abundance ratios for proteins in DRMs versus WCMs were plotted in decreasing order. Proteins with higher ratios are enriched in DRM relative to WCMs. All previously known or identified raft proteins fell into the group with high ratios, whereas mitochondrial proteins, especially mitochondrial membrane proteins, are not enriched in DRMs. Some examples of raft, mitochondrial, and plasma membrane marker proteins are indicated here as red, blue, and green lines respectively. GNG12, guanine nucleotide binding protein G(I)/G(S)/G(O) subunit γ-12 precursor; GNAI3, guanine nucleotide binding protein G; FLOT1, Flotillin-1; FLOT2, Flotillin 2; FOLR1, folate receptor α precursor; SNAP23, isoform SNAP-23a of synaptosomal-associated protein 23P; ATP6V1A, vacuolar ATP synthase catalytic subunit A; IMMT, isoform 1 of mitochondrial inner membrane protein; CLTC, isoform 1 of clathrin heavy chain 1; ATP5B, ATP synthase subunit β, mitochondrial precursor; MDH2, malate dehydrogenase, mitochondrial precursor; VIM, vimentin; GLUD1, glutamate dehydrogenase 1, mitochondrial precursor; MAPBPIP, mitogen-activated protein binding protein-interacting protein; GNB2, guanine nucleotide binding protein G(I)/G(S)/G(T) subunit β-2; ANPEP, aminopeptidase N; YES1, proto-oncogene tyrosine-protein kinase Yes; ATP1A1, isoform long of sodium/potassium-transporting ATPase subunit α-1 precursor; HSPA9, stress-70 protein, mitochondrial.
Do mitochondrial proteins really comigrate with rafts?
The classical discontinuous gradient system for isolating DRMs has a very low resolution: essentially anything with a density that falls between that of 5% and 35% sucrose would appear to comigrate. Thus, while mitochondrial proteins and rafts appear to comigrate in this system, they may resolve if subjected to a higher-resolution separation on a linear density gradient. To this end, we applied detergent-solubilized Jurkat cells onto a continuous, linear sucrose gradient from 10–30% sucrose (see Materials and Methods). One white, light-scattering band was observed after ultracentrifugation, similar to that obtained with the nonlinear gradients but more diffuse and centered at ∼15% sucrose. Western blotting of fractions taken from this gradient revealed that the raft marker flotillin-2 and the mitochondrial ATP synthase β subunit showed different distribution patterns (Fig. 5A). The profile of flotillin-2 was centered at 14–17% sucrose, corresponding to the light-scattering band mentioned above, but ATP synthase β was distributed toward the higher density fractions. While only based on two proteins, these data suggest that rafts and mitochondrial proteins may not comigrate.
Fig. 5.

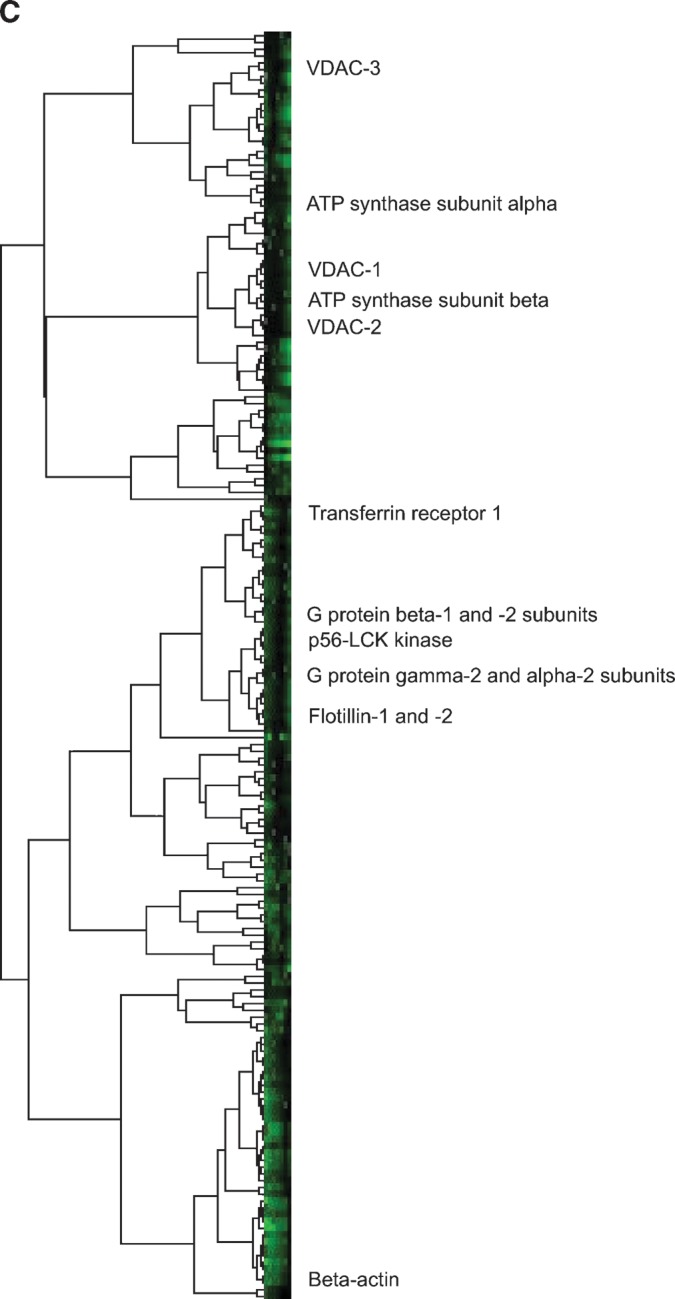

The separation of rafts and mitochondrial proteins in a linear sucrose gradient. A: Western blot of the lipid raft marker flotillin-2 and the mitochondrial ATP synthase β subunit in fractions taken from a linear sucrose gradient and a nonlinear gradient showing different distribution patterns. B: Some examples of protein profiles or protein distributions across the linear density gradient by PCP-SILAC. C: Hierarchical clustering on the PCP-SILAC data showing the separation of rafts and mitochondrial proteins into two distinct groups. D: PCA analysis on the PCP-SILAC data.
To test the comigration of rafts and mitochondrial proteins more generally, we used PCP-SILAC (19, 44) to measure the distribution of proteins across a linear density gradient (Fig. 5B). As expected, profiles of known raft proteins (flotillin-1, flotillin-2, p56-LCK kinase, and G-protein subunit β-1 as examples) peaked in the same fraction as flotillin-2 measured by Western blotting (Fig. 5A). By contrast, the ATP synthase subunits β and α, as well as VDAC-1 and VDAC-2, peaked at higher densities of sucrose. The nonraft plasma membrane marker transferrin receptor 1 exhibited a peaked profile shifted toward the detergent-soluble fractions, and detergent-soluble β-actin was greatest in intensity in the low density detergent-soluble fraction.
To visualize these trends on a larger scale, rather than just looking at selected profiles, complete linkage hierarchical clustering was performed on the PCP-SILAC data using the CLUSTER program (22). The resultant tree bifurcated at its base into two groups (Fig. 5C). One group contained the common mitochondrial proteins found in DRMs, the ATP synthase subunits and VDACs, as well as many proteasome subunits, ribosomal subunits, and ribonucleoproteins. The second group contained the common raft and raft-associated proteins (including the flotillins, G-protein subunits, p56-LCK kinase, and V-ATPases). It also contained the nonraft plasma membrane and nuclear membrane proteins and all the cytosolic proteins, including cytoskeletal proteins. The raft proteins themselves clustered to one small branch separated from a larger branch containing the cytosolic proteins.
The clustering data were confirmed by another orthogonal method, PCA. PCA is a technique that can be used to reduce multidimensional data to its principal components (PCs), which are linear combinations of all the variables (45). The PCs summarize the greatest correlated variation of the data set. PC1 and PC2, representing the greatest and second greatest correlated variation, respectively, are graphed for the PCP-SILAC data in Fig. 5D, and the same trends seen in the clustering are observed. The ATP synthase subunits and VDACs cluster tightly and separately (bottom left) from the classical raft and raft-associated proteins (bottom center). The proteasome, ribosomal subunits, and ribonucleoproteins make a diffuse grouping (top left), and the cytosolic and cytoskeletal proteins mainly cluster (far right). The nuclear membrane and nonraft plasma membrane proteins appear in the transition between the raft and cytosolic proteins. These groupings confirm what was seen in the hierarchical clustering: that accepted raft proteins exhibit a different profile from the mitochondrial contaminants, ATP synthase subunits, and VDACs.
DISCUSSION
Based on the number of publications in PubMed, lipid rafts and/or caveolae have been an extremely popular subcellular domain for proteomic investigations (10), equivalent perhaps to mitochondria (46) and certainly more so than phagosomes (47). Prior to the advent of proteomics, rafts were considered to be exclusive to the plasma membrane and membranes immediately up- and downstream of the plasmalemma (i.e., late Golgi/trans-Golgi network and endosomes/phagosomes). Likewise, the protein constituents of rafts were thought to be typical plasma membrane proteins, such as glycosylphosphatidylinositol-anchored proteins and Src-family tyrosine kinases (9), so the discovery of mitochondrial proteins in DRM preparations came as a surprise. One of the great advantages of proteomics is its unbiased nature; it took proteomics to discover mitochondrial proteins in DRMs simply because no one thought to look for them previously. However, biochemical subcellular fractionations typically never yield absolutely homogenous preparations (42), so here we have sought to demonstrate whether mitochondrial proteins are bona fide components of plasma membrane lipid rafts or if they are present in DRMs for another reason. Thus, our null hypothesis was that mitochondrial proteins copurify in DRMs but are not localized in lipid rafts.
Our data certainly support the presence of mitochondrial proteins in DRMs, with several dozen of the most abundant (48) mitochondrial proteins being identified with many peptides each. Without exception, however, none of the proteins were sensitive to cholesterol disruption across three different cell types used as model systems for human epithelia (HeLa), mouse fibroblasts (swiss-3T3), and human T lymphocytes (Jurkat). The failure of any of these mitochondrial proteins to satisfy the cholesterol sensitivity test, the gold standard for a lipid raft protein (13), suggests that they simply copurify with lipid rafts in the DRM preparation. If mitochondrial proteins are specific components of rafts, then one prediction would be that they should be relatively enriched in DRMs versus whole cells or the entire membrane complement of whole cells. To test this hypothesis, we measured the degree of enrichment of proteins in DRMs versus a WCM preparation, and, indeed, mitochondrial proteins were not enriched in DRMs, again suggesting that they are simply contaminants. Furthermore, using a triplex SILAC scheme to simultaneously measure sensitivity to drug treatment and relative enrichment in a subcellular biochemical fraction, we also demonstrated that the mitochondrial proteins in question are indeed enriched in mitochondrial preparations, as one would expect, but that they are not sensitive to cholesterol disruption. And finally, by using high-resolution linear density gradients to better resolve the components of DRMs, classical lipid raft proteins and mitochondrial components showed different distribution profiles across the gradient, lending more support to the thesis that mitochondrial proteins are copurifying contaminants of the normal DRM preparation.
We are cognizant of four potential caveats that complicate the interpretation of our data. First, in trying to demonstrate the widespread lack of cholesterol sensitivity of mitochondrial proteins in DRM preparations, we were unable to reanalyze the more than two dozen different cells or tissues whose DRM proteomes have been reported [for review, see (10)]. Thus, it is conceivable, although we feel it unlikely, that mitochondrial proteins could demonstrate sensitivity to MβCD in other cell types. Second, in the triplex SILAC experiments described in Fig. 2, we were unable to obtain a completely homogenous preparation of mitochondria, although it was no worse than for other published proteomic analyses of mitochondria (48, 49). However, for our purposes, homogeneity is not essential because we are only interested in relative enrichment in mitochondrial DRMs, and having a small but detectable contamination from other membranes actually allows a more quantitative assessment of enrichment versus an “all or nothing” response. Third, while Triton X-100 insolubility is the most widely used method for enriching rafts, other detergents, as well as detergent-free methods, yield proteomes with subtly different protein complements (50). We have previously shown that the high pH/carbonate method (51) has even more cholesterol-insensitive proteins in it than Triton X-100-prepared DRMs (11) and that mitochondrial proteins are similarly insensitive to cholesterol depletion. We have not, however, shown the insensitivity to cholesterol disruption of mitochondrial proteins in DRMs prepared using detergents other than Triton X-100. Lastly, the conditions required to disrupt rafts, serum starvation followed by severe cholesterol starvation, amount to an extremely harsh environment for the cells, and this could substantially disturb intracellular organelles. Again, we believe the impact of such a perturbation on our conclusions to be minimal because we have confirmed the effects by treating isolated mitochondria with MβCD. Severe cholesterol depletion likely does have some effects on the biosynthetic and endosomal systems, but since there is very little movement of membranes from these compartments to the mitochondria, such effects in the whole-cell experiments would also not appreciably detract from our conclusions.
In summary, we find that we are unable to reject our stated null hypothesis that mitochondrial proteins identified in previous raft studies are actually contaminants of the DRM preparation. We find no evidence that rafts are in mitochondria or that mitochondrial proteins are in rafts.
Supplementary Material
Acknowledgments
The authors thank the other members of the Cell Biology Proteomics group for fruitful discussions and advice. In particular, the authors thank Nikolay Stoynov, Queenie Chan, and Erin Boyle for technical assistance. Thanks to Matthias Mann for early access to the MaxQuant software and to Rob Hocking for help with the PCA analysis.
Abbreviations
DRMs, detergent-resistant membranes
LC-MS/MS, liquid chromatography-tandem mass spectrometry
MβCD, methyl-β-cyclodextrin
MBS, MES-buffered saline
PC, principal component
PCA, principal component analysis
PCP, protein correlation profiling
r.c.f., relative centrifugal force
DMEM, Dulbecco's Modified Eagle's Medium
RPMI, Roswell Park Memorial Institute
SDC, sodium deoxycholate
SILAC, stable isotope labeling by amino acids in cell culture
VDAC, voltage-dependent anion selective channel
WCM, whole-cell membrane
Infrastructure used in this project was supported by the Canadian Foundation for Innovation, the British Columbia Knowledge Development Fund, and the Michael Smith Foundation through the British Columbia Proteomics Network. L.J.F. is the Canada Research Chair in Organelle Proteomics and a Michael Smith Foundation Scholar. K.B.B. was supported by a British Columbia Proteomics Network Training Award.
Published, JLR Papers in Press, January 9, 2009.
Footnotes
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of eight tables.
References
- 1.Singer S. J., and G. L. Nicolson. 1972. The fluid mosaic model of the structure of cell membranes. Science. 175 720–731. [DOI] [PubMed] [Google Scholar]
- 2.Pike, L. J. The challenge of lipid rafts. J. Lipid Res. Epub ahead of print. October 28, 2008; doi: 10.1194/jlr.R800040-JLR200. [DOI] [PMC free article] [PubMed]
- 3.Simons K., and G. van Meer. 1988. Lipid sorting in epithelial cells. Biochemistry. 27 6197–6202. [DOI] [PubMed] [Google Scholar]
- 4.Schroeder R., E. London, and D. Brown. 1994. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc. Natl. Acad. Sci. USA. 91 12130–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown D. A., and E. London. 1998. Structure and origin of ordered lipid domains in biological membranes. J. Membr. Biol. 164 103–114. [DOI] [PubMed] [Google Scholar]
- 6.Schroeder R. J., S. N. Ahmed, Y. Zhu, E. London, and D. A. Brown. 1998. Cholesterol and sphingolipid enhance the Triton X-100 insolubility of glycosylphosphatidylinositol-anchored proteins by promoting the formation of detergent-insoluble ordered membrane domains. J. Biol. Chem. 273 1150–1157. [DOI] [PubMed] [Google Scholar]
- 7.van Meer G., and Q. Lisman. 2002. Sphingolipid transport: rafts and translocators. J. Biol. Chem. 277 25855–25858. [DOI] [PubMed] [Google Scholar]
- 8.Simons K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387 569–572. [DOI] [PubMed] [Google Scholar]
- 9.Brown D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68 533–544. [DOI] [PubMed] [Google Scholar]
- 10.Foster L. J., and Q. W. Chan. 2007. Lipid raft proteomics: more than just detergent-resistant membranes. Subcell. Biochem. 43 35–47. [DOI] [PubMed] [Google Scholar]
- 11.Foster L. J., C. L. De Hoog, and M. Mann. 2003. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. USA. 100 5813–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christian A. E., M. P. Haynes, M. C. Phillips, and G. H. Rothblat. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38 2264–2272. [PubMed] [Google Scholar]
- 13.Ilangumaran S., and D. C. Hoessli. 1998. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem. J. 335 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae T. J., M. S. Kim, J. W. Kim, B. W. Kim, H. J. Choo, J. W. Lee, K. B. Kim, C. S. Lee, J. H. Kim, S. Y. Chang, et al. 2004. Lipid raft proteome reveals ATP synthase complex in the cell surface. Proteomics. 4 3536–3548. [DOI] [PubMed] [Google Scholar]
- 15.Man P., P. Novak, M. Cebecauer, O. Horvath, A. Fiserova, V. Havlicek, and K. Bezouska. 2005. Mass spectrometric analysis of the glycosphingolipid-enriched microdomains of rat natural killer cells. Proteomics. 5 113–122. [DOI] [PubMed] [Google Scholar]
- 16.McMahon K. A., M. Zhu, S. W. Kwon, P. Liu, Y. Zhao, and R. G. Anderson. 2006. Detergent-free caveolae proteome suggests an interaction with ER and mitochondria. Proteomics. 6 143–152. [DOI] [PubMed] [Google Scholar]
- 17.Rogers L. D., and L. J. Foster. 2007. The dynamic phagosomal proteome and the contribution of the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA. 104 18520–18525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mickelson J. R., M. L. Greaser, and B. B. Marsh. 1980. Purification of skeletal-muscle mitochondria by density-gradient centrifugation with Percoll. Anal. Biochem. 109 255–260. [DOI] [PubMed] [Google Scholar]
- 19.Andersen J. S., and M. Mann. 2006. Organellar proteomics: turning inventories into insights. EMBO Rep. 7 874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saito H., Y. Oda, T. Sato, J. Kuromitsu, and Y. Ishihama. 2006. Multiplexed two-dimensional liquid chromatography for MALDI and nanoelectrospray ionization mass spectrometry in proteomics. J. Proteome Res. 5 1803–1807. [DOI] [PubMed] [Google Scholar]
- 21.Cox J., and M. Mann. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26 1367–1372. [DOI] [PubMed] [Google Scholar]
- 22.Eisen M. B., P. T. Spellman, P. O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA. 95 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blonder J., M. L. Hale, K. C. Chan, L. R. Yu, D. A. Lucas, T. P. Conrads, M. Zhou, M. R. Popoff, H. J. Issaq, B. G. Stiles, et al. 2005. Quantitative profiling of the detergent-resistant membrane proteome of iota-b toxin induced vero cells. J. Proteome Res. 4 523–531. [DOI] [PubMed] [Google Scholar]
- 24.von Haller P. D., S. Donohoe, D. R. Goodlett, R. Aebersold, and J. D. Watts. 2001. Mass spectrometric characterization of proteins extracted from Jurkat T cell detergent-resistant membrane domains. Proteomics. 1 1010–1021. [DOI] [PubMed] [Google Scholar]
- 25.Blonder J., A. Terunuma, T. P. Conrads, K. C. Chan, C. Yee, D. A. Lucas, C. F. Schaefer, L. R. Yu, H. J. Issaq, T. D. Veenstra, et al. 2004. A proteomic characterization of the plasma membrane of human epidermis by high-throughput mass spectrometry. J. Invest. Dermatol. 123 691–699. [DOI] [PubMed] [Google Scholar]
- 26.Karsan A., J. Blonder, J. Law, E. Yaquian, D. A. Lucas, T. P. Conrads, and T. Veenstra. 2005. Proteomic analysis of lipid microdomains from lipopolysaccharide-activated human endothelial cells. J. Proteome Res. 4 349–357. [DOI] [PubMed] [Google Scholar]
- 27.von Haller P. D., E. Yi, S. Donohoe, K. Vaughn, A. Keller, A. I. Nesvizhskii, J. Eng, X. J. Li, D. R. Goodlett, R. Aebersold, et al. 2003. The application of new software tools to quantitative protein profiling via isotope-coded affinity tag (ICAT) and tandem mass spectrometry. II. Evaluation of tandem mass spectrometry methodologies for large-scale protein analysis, and the application of statistical tools for data analysis and interpretation. Mol. Cell. Proteomics. 2 428–442. [DOI] [PubMed] [Google Scholar]
- 28.von Haller P. D., E. Yi, S. Donohoe, K. Vaughn, A. Keller, A. I. Nesvizhskii, J. Eng, X. J. Li, D. R. Goodlett, R. Aebersold, et al. 2003. The application of new software tools to quantitative protein profiling via isotope-coded affinity tag (ICAT) and tandem mass spectrometry. I. Statistically annotated datasets for peptide sequences and proteins identified via the application of ICAT and tandem mass spectrometry to proteins copurifying with T cell lipid rafts. Mol. Cell. Proteomics. 2 426–427. [DOI] [PubMed] [Google Scholar]
- 29.Li N., A. R. Shaw, N. Zhang, A. Mak, and L. Li. 2004. Lipid raft proteomics: analysis of in-solution digest of sodium dodecyl sulfate-solubilized lipid raft proteins by liquid chromatography-matrix-assisted laser desorption/ionization tandem mass spectrometry. Proteomics. 4 3156–3166. [DOI] [PubMed] [Google Scholar]
- 30.MacLellan D. L., H. Steen, R. M. Adam, M. Garlick, D. Zurakowski, S. P. Gygi, M. R. Freeman, and K. R. Solomon. 2005. A quantitative proteomic analysis of growth factor-induced compositional changes in lipid rafts of human smooth muscle cells. Proteomics. 5 4733–4742. [DOI] [PubMed] [Google Scholar]
- 31.Paradela A., S. B. Bravo, M. Henriquez, G. Riquelme, F. Gavilanes, J. M. Gonzalez-Ros, and J. P. Albar. 2005. Proteomic analysis of apical microvillous membranes of syncytiotrophoblast cells reveals a high degree of similarity with lipid rafts. J. Proteome Res. 4 2435–2441. [DOI] [PubMed] [Google Scholar]
- 32.Sprenger R. R., D. Speijer, J. W. Back, C. G. De Koster, H. Pannekoek, and A. J. Horrevoets. 2004. Comparative proteomics of human endothelial cell caveolae and rafts using two-dimensional gel electrophoresis and mass spectrometry. Electrophoresis. 25 156–172. [DOI] [PubMed] [Google Scholar]
- 33.Li N., A. Mak, D. P. Richards, C. Naber, B. O. Keller, L. Li, and A. R. Shaw. 2003. Monocyte lipid rafts contain proteins implicated in vesicular trafficking and phagosome formation. Proteomics. 3 536–548. [DOI] [PubMed] [Google Scholar]
- 34.Tu X., A. Huang, D. Bae, N. Slaughter, J. Whitelegge, T. Crother, P. E. Bickel, and A. Nel. 2004. Proteome analysis of lipid rafts in Jurkat cells characterizes a raft subset that is involved in NF-kappaB activation. J. Proteome Res. 3 445–454. [DOI] [PubMed] [Google Scholar]
- 35.Bini L., S. Pacini, S. Liberatori, S. Valensin, M. Pellegrini, R. Raggiaschi, V. Pallini, and C. T. Baldari. 2003. Extensive temporally regulated reorganization of the lipid raft proteome following T-cell antigen receptor triggering. Biochem. J. 369 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen H. T., A. B. Amine, D. Lafitte, A. A. Waheed, C. Nicoletti, C. Villard, M. Letisse, V. Deyris, M. Roziere, L. Tchiakpe, et al. 2006. Proteomic characterization of lipid rafts markers from the rat intestinal brush border. Biochem. Biophys. Res. Commun. 342 236–244. [DOI] [PubMed] [Google Scholar]
- 37.Gupta N., B. Wollscheid, J. D. Watts, B. Scheer, R. Aebersold, and A. L. DeFranco. 2006. Quantitative proteomic analysis of B cell lipid rafts reveals that ezrin regulates antigen receptor-mediated lipid raft dynamics. Nat. Immunol. 7 625–633. [DOI] [PubMed] [Google Scholar]
- 38.Ledesma M. D., J. S. Da Silva, A. Schevchenko, M. Wilm, and C. G. Dotti. 2003. Proteomic characterisation of neuronal sphingolipid-cholesterol microdomains: role in plasminogen activation. Brain Res. 987 107–116. [DOI] [PubMed] [Google Scholar]
- 39.Sanders P. R., P. R. Gilson, G. T. Cantin, D. C. Greenbaum, T. Nebl, D. J. Carucci, M. J. McConville, L. Schofield, A. N. Hodder, J. R. Yates III, et al. 2005. Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J. Biol. Chem. 280 40169–40176. [DOI] [PubMed] [Google Scholar]
- 40.Borner G. H., D. J. Sherrier, T. Weimar, L. V. Michaelson, N. D. Hawkins, A. Macaskill, J. A. Napier, M. H. Beale, K. S. Lilley, and P. Dupree. 2005. Analysis of detergent-resistant membranes in Arabidopsis. Evidence for plasma membrane lipid rafts. Plant Physiol. 137 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nebl T., K. N. Pestonjamasp, J. D. Leszyk, J. L. Crowley, S. W. Oh, and E. J. Luna. 2002. Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. J. Biol. Chem. 277 43399–43409. [DOI] [PubMed] [Google Scholar]
- 42.Foster L. 2006. Mass spectrometry outgrows simple biochemistry: new approaches to organelle proteomics. Biophys. Rev. Lett. 1 163–175. [Google Scholar]
- 43.Hess S. T., T. J. Gould, M. V. Gudheti, S. A. Maas, K. D. Mills, and J. Zimmerberg. 2007. Dynamic clustered distribution of hemagglutinin resolved at 40 nm in living cell membranes discriminates between raft theories. Proc. Natl. Acad. Sci. USA. 104 17370–17375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen J. S., C. J. Wilkinson, T. Mayor, P. Mortensen, E. A. Nigg, and M. Mann. 2003. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 426 570–574. [DOI] [PubMed] [Google Scholar]
- 45.Dunkley T. P., R. Watson, J. L. Griffin, P. Dupree, and K. S. Lilley. 2004. Localization of organelle proteins by isotope tagging (LOPIT). Mol. Cell. Proteomics. 3 1128–1134. [DOI] [PubMed] [Google Scholar]
- 46.Prokisch H., C. Andreoli, U. Ahting, K. Heiss, A. Ruepp, C. Scharfe, and T. Meitinger. 2006. MitoP2: the mitochondrial proteome database–now including mouse data. Nucleic Acids Res. 34 D705–D711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rogers L. D., and L. J. Foster. 2008. Contributions of proteomics to understanding phagosome maturation. Cell Microbiol. 10 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foster L. J., C. L. de Hoog, Y. Zhang, Y. Zhang, X. Xie, V. K. Mootha, and M. Mann. 2006. A mammalian organelle map by protein correlation profiling. Cell. 125 187–199. [DOI] [PubMed] [Google Scholar]
- 49.Forner F., L. J. Foster, S. Campanaro, G. Valle, and M. Mann. 2006. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteomics. 5 608–619. [DOI] [PubMed] [Google Scholar]
- 50.Zheng, Y. Z., and L. J. Foster. Biochemical and proteomic approaches for the study of membrane microdomains. J. Proteomics. 72:12-22. [DOI] [PubMed]
- 51.Smart E. J., Y. S. Ying, C. Mineo, and R. G. Anderson. 1995. A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc. Natl. Acad. Sci. USA. 92 10104–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.